

Abstract
Chaperone-mediated autophagy (CMA) is a lysosomal pathway that participates in the degradation of cytosolic proteins. CMA is activated by starvation and in response to stressors that result in protein damage. The selectivity intrinsic to CMA allows for removal of damaged proteins without disturbing nearby functional ones. CMA works in a coordinated manner with other autophagic pathways, which can compensate for each other. Interest in CMA has recently grown because of the connections established between this autophagic pathway and human pathologies. Here we review the unique properties of CMA compared to other autophagic pathways and its relevance in health and disease.
Keywords: aging, chaperones, cellular stress, lysosomes, proteases, protein translocation
Introduction
Renewal of intracellular proteins through their continuous synthesis and degradation assures proper function of the cellular proteome [1,2]. Protein degradation is a conservative cellular process that allows recycling of cellular building blocks (amino acids) and at the same time provides a mechanism for protein quality control. Two main systems participate in intracellular proteolysis: the proteasome and the lysosome or autophagy [1,2]. While the proteasome is, for the most part, responsible for a rapid protein turnover, lysosomes preferentially degrade proteins with longer half-lives along with other intracellular components, such as whole organelles, lipid deposits, proteinaceous inclusions and aggregates, and even pathogens (once they reach the cytosolic compartment). Three different pathways deliver cytosolic cargo to the lysosome in mammalian cells: microautophagy, macroautophagy, and chaperone-mediated autophagy (CMA) [3]. In contrast to micro- and macroautophagy, where whole regions of the cytosol are sequestered and delivered to lysosomes, CMA targets single cytosolic proteins to the lysosomal surface and mediates their one-by-one translocation across the lysosomal membrane for degradation [4,5]. The selectivity of CMA is determined by the recognition of a specific amino acid motif in the substrate protein by a cytosolic chaperone/co-chaperone complex [6]. Despite the clear differences among autophagic pathways, growing evidence supports that they function in an interdependent manner and can partially compensate for each other’s dysfunction [7–9]. Like macroautophagy, CMA can be a provider of energy when nutrients are scarce, and it participates in protein quality control, as it can selectively target damaged proteins for degradation. CMA is thus an important component of the cellular response to different types of stressors [4,5]. Consequently, CMA dysfunction and its decreased activity with age have been proposed to contribute to the alterations in cellular homeostasis associated with severe pathologies, such as some neurodegenerative diseases, kidney pathologies, and metabolic disorders and to the diminished ability of old organisms to respond to stress. In this review, we first describe the unique characteristics, molecular effectors and regulators of this pathway compared to other autophagic pathways, and then comment on the cellular consequences of CMA failure and the recently described involvement of CMA dysfunction in human pathologies and in aging.
Molecular components of CMA
In contrast to the “in bulk” sequestration of portions of cytosol that include complete organelles observed in other autophagic pathways, CMA is a highly selective autophagic mechanism that targets only specific soluble proteins for lysosomal breakdown [4,5]. Two main characteristics that differentiate CMA from other autophagic processes are the fact that individual proteins can be separately selected for degradation through this pathway and the need for this autophagic cargo to undergo complete unfolding before they can reach the lysosomal lumen. The unfolding requirement stems from the fact that CMA substrates are translocated across the lysosomal membrane, rather than engulfed or sequestered through vesicular structures, as described for other autophagic pathways. Selectivity and direct translocation across the lysosomal membrane are achieved in CMA by the participation of two major protein complexes that constitute the essential machinery of this autophagic pathway: a subset of cytosolic chaperones and cochaperones that recognize cargo [10] and a group of lysosomal proteins that sit on both sides of the lysosomal membrane and mediate cargo translocation [11–14] (Figure 1a).
Figure 1. Chaperone-mediated autophagy, basic components and regulation. (a) Steps on CMA.
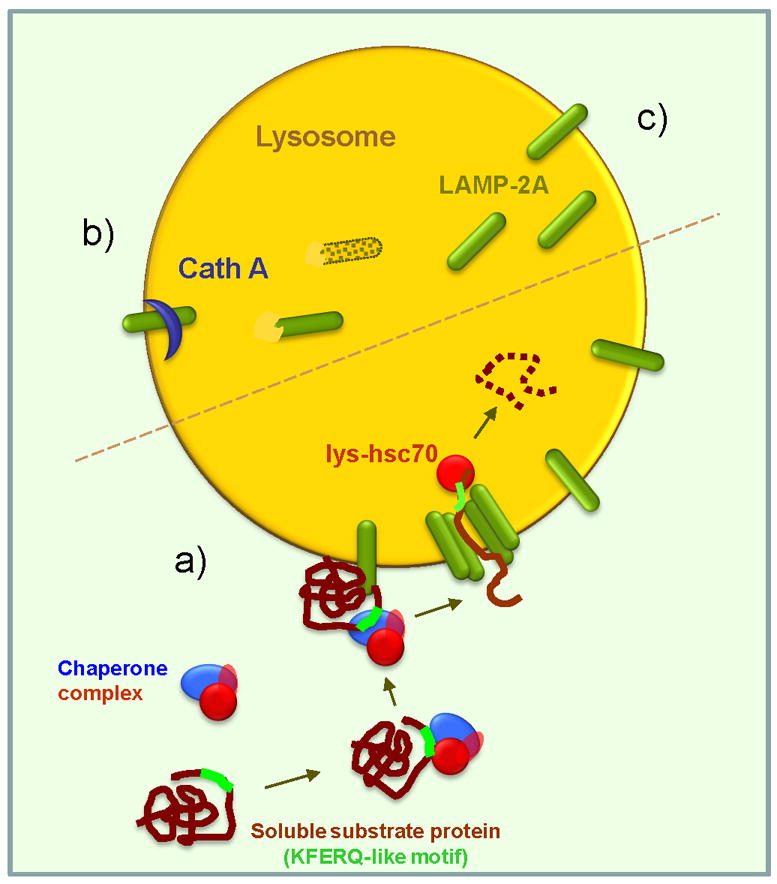
The two essential components of CMA are the cargo recognition complex (in the cytosol) and the cargo translocation complex (at the lysosomal membrane). The CMA targeting motif (KFERQ-like) in the substrate protein is recognized by cytosolic hsc70, main component of the cargo recognition complex, and this delivers it to the surface of the lysosomes where it binds to LAMP-2A. This single span membrane protein and a luminal form of hsc70 (lys-hsc70) are the major components of the cargo translocation complex. Whereas substrate proteins bind to monomeric forms of LAMP-2A, organization of LAMP-2A into high order multimeric complexes is required for substrate translocation. Once in the lumen the substrate is rapidly degraded. (b–c) Regulation of CMA. CMA activity is directly dependent on the amount of LAMP-2A molecules available at the lysosomal membrane. Levels of LAMP-2A are regulated by de novo synthesis (not depicted here), through changes in their regulated Cathepsin A-dependent degradation at the lysosomal membrane (b) and by the direct insertion of a luminal resident pool of LAMP-2A into the lysosomal membrane (c).
The Cargo Recognition Complex
All CMA substrates contain in their amino acid sequence a motif biochemicaly related to the pentapeptide KFERQ required for their selective recognition by the CMA cytosolic chaperone complex [6]. Sequence analysis revealed that 30% of cytosolic proteins carry these type of motifs making them putative CMA substrates [15]. However, very often, many of these potential CMA substrates also carry targeting signals for other proteolytic systems. This multiplicity of degradative pathways for a single protein, initially perceived as an exception, has become a common feature of many intracellular proteins. Although the current challenge resides in understanding how these degradative signals are prioritized, growing evidence supports the notion that each protein’s fate likely depends on factors such as cellular conditions, activity and accessibility of the different proteolytic systems, and reasons for its degradation (i.e. damage vs. as further supply of amino acids). Consequently, the presence of a CMA-targeting motif is necessary but not sufficient to determine its degradation through this autophagic pathway. In fact, the targeting motif needs to be exposed for recognition by the heat shock cognate protein of 70 kDa, hsc70, a cytosolic member of the hsp70 family of chaperones, and the main component of the CMA cargo recognition complex [10]. As for many other cellular processes involving chaperones, the interaction of hsc70 with the substrate proteins is modulated by co-chaperones that determine the kinetics of substrate binding/release [16]. Once the substrate-chaperone complex is formed, this complex is rapidly targeted toward the CMA translocation complex at the lysosomal membrane, by still unknown mechanisms (Figure 1a).
The CMA Translocation Complex
The only known lysosomal transmembrane protein involved in CMA substrate binding and uptake is the lysosome-associated membrane protein type 2A (LAMP-2A), a splice variant of the lamp2 gene [13]. This single-span membrane protein participates in both substrate binding to the lysosomal membrane and its translocation into the lumen. Studies, using blocking antibodies and synthetic peptide homologues to the 12 amino acid tail, which LAMP-2A exposes at the cytosolic side of the lysosomal membrane, revealed that CMA substrates bind to a positive charged stretch of amino acids in this tail [17,18]. Binding of substrates to the LAMP-2A tail occurs while still in their native confirmation, but complete substrate unfolding is required for a successful transport [19]. Although the exact mechanism leading to substrate unfolding is currently unknown, it has been proposed that some of the components of the chaperone/co-chaperone targeting complex may also contribute to substrate unfolding once at the lysosomal membrane [16]. The minimal components required for translocation of CMA substrates across the lysosomal membrane have been identified as LAMP-2A and a form of hsc70 that resides in the lysosomal lumen (lys-hsc70) [11,20], however, none of these proteins display the membrane multispan properties typical of most transporter systems. A longstanding search for a CMA-related translocon-like protein has failed to identify a “stable” channel or pore at the lysosomal membrane. Instead, organization of LAMP-2A molecules at the lysosomal membrane with other yet unknown proteins to form a 700kDa molecular weight protein complex is necessary for delivery of CMA substrate proteins to the lysosomal lumen [14] (Figure 1a). Both lys-hsc70 and a recently identified lysosome-associated hsp90 are required for stabilization, assembly and disassembly of the LAMP-2A-enriched multimeric complexes [14]. Once the unfolded substrate reaches the lysosome, it is degraded within minutes by the wide array of luminal proteases. CMA activity in a given cell is directly dependent on the lysosomal levels of these two components, LAMP-2A at the lysosomal membrane and lys-hsc70 in the lysosomal lumen [12,21,22]. In fact, lysosomes lacking lumenal hsc70, despite containing LAMP-2A in the membrane, are unable to perform CMA [23]. Likewise, if LAMP-2A is knocked down, the presence of hsc70 in the lysosomal lumen is not sufficient to achieve CMA [7]. In fact, changes in levels of LAMP-2A at the lysosomal membrane (through modifications in its de novo synthesis or degradation in this compartment) directly determine rates of CMA in most cells [21]. In contrast, conditions in which levels of the cytosolic targeting complex become limiting have not been described until date, maybe due to the multiplicity of functions in addition to CMA that the chaperones constituent of these complexes are involved in.
CMA Regulation and Physiologic Relevance
Selective targeting of cytosolic proteins for degradation through CMA contributes to both cellular “housekeeping” and to the cellular ability to respond to stress [4,5]. Although initially identified as a stress-induced pathway, it has become evident that certain level of CMA activity is detectable in all cells even under basal conditions. Basal CMA activity varies from cell type to cell type and may contribute to selective degradation of certain regulatory proteins and to specialized functions. For example, CMA has been proposed to contribute to delivering of specific intracellular antigens to the lysosomal compartment for processing and presentation upon binding to MHC class II molecules [24]. Activation of CMA occurs in response to two main cellular events: when an alternative source of amino acids and energy is needed, or when return to homeostasis requires removal of damaged or malfunctioning intracellular proteins. During prolonged nutritional deprivation, CMA can selectively recycle the constituent amino acids of no longer needed proteins, in order to maintain synthesis of essential proteins and to provide an additional fueling source until access to nutrients is regained [22]. Conditions that promote accumulation of misfolded or oxidized proteins also often induce activation of CMA as part of the quality control cellular system to eliminate these dysfunctional proteins [25].
Activation of CMA during starvation is paralleled by an increase of the levels of LAMP-2A at the lysosomal membrane and of luminal lys-hsc70 [21,23]. LAMP-2A is present in a large subset of lysosomes, whereas lys-hsc70 can only be detected in a discrete pool of cellular lysosomes – those able to perform CMA [23]. However, the percentage of CMA active lysosomes is not fixed but changes to accommodate to the cellular requirements. Thus, although, for example, in liver under basal conditions only 20–30% of lysosomes contain lys-hsc70, as starvation persists, lys-hsc70 can be detected in up to 80% of lysosomes [23]. Lysosome levels of hsc70 are modulated through changes in the stability of this luminal chaperone, which are tightly dependent on lysosomal pH [23]. In hsc70-containing lysosomes, availability and dynamics of the LAMP-2A receptor at the lysosomal membrane become the rate-limiting step in CMA [21]. The levels of LAMP-2A are regulated differently, depending on the type of the intracellular environment and the type of stimuli inducing CMA activation. The increase in LAMP-2A levels at the lysosomal membrane observed during prolonged nutrient deprivation does not require de novo synthesis but instead is attained by decreasing the rates of degradation of this receptor at the lysosomal membrane [21,26]. LAMP-2A degradation is a tightly regulated process that involves the recruitment of this protein to discrete lipid microdomains on the lysosomal membrane and a sequential cleavage by two proteases in these regions [26] (Figure 1b). Conditions leading to activation of CMA decrease LAMP-2A degradation by preventing its association to the lipid-rich microdomains and favoring its organization into the multimeric translocation complexes [27]. Further increase of LAMP-2A molecules at the lysosomal membrane is often attained through recruitment toward this compartment of the pool of LAMP-2A molecules normally present in the lysosomal lumen [21] (Figure 1c). In contrast, activation of CMA during mild oxidative stress is mainly achieved by increasing de novo synthesis of LAMP-2A [25]. The actual signaling mechanisms that modulate this transcriptional activation of LAMP-2A are still undetermined. It is likely that activation of CMA in response to different intra- and extracellular stressors is regulated through separate signaling cascades.
Recent studies also support that CMA activity is directly interdependent with the activity of other autophagic pathways, and in particular macroautophagy. Both macroautophagy and CMA are often upregulated in response to similar stimuli (nutritional deprivation, oxidative stress, etc.) but with a different time-course of activation. In the case of nutritional deprivation, for example, in cells, such as fibroblasts and hepatocytes, activation of macroautophagy occurs first (during the first 4–6 hours of starvation), whereas CMA activation does not occur until macroautophagic activity starts to decrease (about 10 hours into starvation) [7,22,28]. The existence of a close relationship between both autophagic pathways has been further supported by the fact that experimental blockage of one of them results in compensatory upregulation of the other, thus revealing that the two autophagic pathways cross-talk in both directions. Chronic blockage of CMA in cultured cells via lentiviral knock-down of LAMP-2A leads to constitutive upregulation of macroautophagy [7]. However, both pathways are not completely redundant, and even though basal cellular homeostasis is preserved in these cells through this compensatory activation of macroautophagy, they become highly sensitive to stressors that induce CMA, such as oxidative stress and exposure to UV light [7]. Likewise, blockage of macroautophagy in different cell types leads to constitutive activation of CMA [8], which confers higher resistance to oxidative stress but makes them vulnerable to other stressors where engagement of macroautophagy is essential for cell survival [9]. The molecular mechanisms that modulate cross-talk between macroautophagy and CMA are currently under investigation. Further studies are necessary to determine whether this cross-talk also transcends to other proteolytic systems, such as the ubiquitin/proteasome system. Acute and chronic blockage of the proteasome have been shown to upregulate or deregulate macroautophagy, respectively [29,30], but the effect of these interventions on CMA is unknown. However, the fact that during the early stages of CMA inhibition the proteosomal proteolytic activities are impaired [31] and that specific subunits of the proteasome undergo selective degradation via CMA [32] provide the evidence that cross-talk between these two proteolytic pathways may also occur. For example, CMA degradation of proteasome subunits could be behind the observed decrease in proteasome activity during prolonged nutrient deprivation that leads to the stabilization of important regulatory proteins under these conditions.
In addition to activation of CMA to selectively degrade a pool of cytosolic proteins to provide cellular fueling during prolonged starvation or to selectively eliminate damaged protein molecules, CMA also contributes to modulating cellular functions directly dependent on certain CMA substrates. Thus, for example, the proposed specialized function of CMA in antigen presentation in professional antigen presenting cells, or a recently identified role of CMA as modulator of neuronal survival through the selective degradation of the transcription factor myocyte enhancer factor D [33]. Further characterization of CMA substrate proteins may reveal involvement of CMA in other fundamental cellular functions.
CMA and Pathology
Our current understanding of the involvement of CMA in different aspects of cellular physiology, the identification of a close interrelation between CMA activity and that of other proteolytic pathways, as well as the discovery of new CMA substrates, have all contributed to an increased awareness of the role of CMA in certain pathologies. In some of these conditions a primary CMA defect is likely to be behind the pathogenesis, whereas in others, changes in CMA activity are secondary to the main defect but add up to the pathology and may contribute to the course of the disease.
Lysosomal Storage Diseases
Altered CMA in this group of diseases is often secondary to the primary impairment in the lysosomal compartment common to all of them. Lysosomal storage diseases (LSD) is the common name for disorders that lead to improper degradation of certain substrates in lysosomes. Although the causes for defective lysosomal activity vary (they include mutations in specific enzymes or disrupted trafficking of these enzymes to lysosomes, etc.), the consequences are similar: abnormal accumulation of the substrate leads to the expansion of the lysosomal compartment, dysfunction of other lysosomal processes, eventual leakage of lysosomal contents, and often cell death [34]. This global lysosomal dysfunction is thus likely to disrupt CMA activity along with other autophagic pathways. However, there are also several LSDs in which defective CMA activity may precede global failure of the lysosomal system. Abnormal CMA upregulation has been observed in galactosialidosis, a disorder resulting from defective function of protective protein/Cathepsin A, a lysosomal protein required for proper lysosomal targeting of two major glycosidases. The observed changes in CMA are independent of the defect in these glycosidases but result from the fact that Cathepsin A is one of the two proteases required for the regulated degradation of LAMP-2A [26]. The abnormally high levels of LAMP-2A, observed in galactosialidosis, and the consequent increase in CMA activity, could be normalized when Cathepsin A levels were restored in culture by overexpression or in galactosialidosis mouse models by gene therapy [26].
A primary defect in CMA has been proposed to contribute to pathogenesis of mucolipidosis type IV (MLIV) [35]. The transient receptor potential mucolipin 1 (TRPML1), mutated in this disorder, has been found to associate with hsc70 and hsp40 at the lysosomal membrane and might be a novel regulator of the CMA translocation channel. Fibroblasts from the MLIV patients have decreased CMA activity and, consequently, increased levels of oxidized proteins [35].
Changes in CMA are also likely to occur in Danon disease, a lysosomal glycogen storage disorder with normal acid maltase activity, recently linked to mutation in the lamp2 gene [36]. However, due to the multiplicity of functions of the different LAMP-2 protein variants and the poor condition of the lysosomal system in these patients (defects in lysosomal biogenesis due to altered intracellular trafficking of essential lysosomal enzymes have been described), further studies are required to dissect whether or not CMA activity is altered in these patients and how it contributes to the disease.
Nephropathies
As described in the previous section, recent findings have linked CMA activity to tubular kidney cell growth through the degradation of Pax2 [37]. Hypertrophic cellular growth can be achieved when protein synthesis overweighs protein degradation. In nephropathies like diabetes, acidosis, and chronic kidney disease, the kidney undergoes hypertrophy. Recent studies have demonstrated that CMA is inhibited in such pathologies, thus contributing to a decrease in proteolysis along with a progressive increase in the cytosolic pool of CMA substrate proteins [38]. Among them, the increased levels of the transcription factor Pax2 may contribute to maintenance of the hypertrophic state through upregulation of tubular cell growth [37].
Neurodegenerative Disorders
The hallmark of most neurodegenerative disorders is the accumulation of pathogenic proteins that often organize into intra- or extracellular aggregates [39]. Alterations in the cellular quality control mechanisms have been proposed to contribute to the neuronal inability to handle the pathogenic proteins. Dysfunction of macroautophagy, often revealed as a marked increase in the number of autophagic vacuoles in the affected neurons, and the consequent loss in the autophagic cytoprotective effect, has been described in a growing number of neurodegenerative diseases. However, alteration of macroautophagy is not necessarily the primary or unique autophagic defect in many of these diseases. Many of the pathogenic proteins involved in neurodegenerative disorders, such as APP and Tau in Alzheimer’s (AD), α-synuclein, Parkin, UCH-L1, Pink-1, DJ-1 and LRRK2 in Parkinson’s (PD) and huntingtin in Huntington’s (HD) disease, contain KFERQ motifs in their amino acid sequences and could be consequently putative CMA substrates. Below we review the recent findings regarding the involvement of CMA in the pathogenesis of these disorders.
Pathogenic forms of α-synuclein were the first ones for which a connection with CMA dysfunction was established [40]. Wild-type α-synuclein can indeed be translocated through the lysosomal membrane to undergo degradation via CMA [40] (Figure 2a). However, although mutant forms of α-synuclein, described in familial forms of PD, interact with the cytosolic chaperone-complex and bind to LAMP-2A with higher binding affinity than the wild-type protein, their translocation is inefficient and their association to the membrane ends up “clogging” the CMA translocation channels. (Figure 2b) This inhibitory effect on CMA activity contributes to neurotoxicity, as disrupting the CMA targeting motif in mutant forms of α-synuclein prevents their delivery to lysosomes and increases cellular viability [40,41]. Not only mutations, but also post-translational modifications of α-synuclein could modify CMA of this protein. Phosphorylation, ubiquitination, nitration and oxidation have been detected in the α-synuclein that accumulates in cytosolic inclusions in the affected neurons. While these forms often display reduced CMA, they do not alter degradation of other CMA substrates [42]. In contrast, dopamine-modified α-synuclein clogs the translocation complex and prevents CMA of other proteins, thus making cells more vulnerable to stressors [42]. The inhibitory effect of dopamine-modified α-synuclein on CMA could be an underlying explanation for the higher rates of degeneration observed in dopaminergic neurons in PD. Altered CMA in cells expressing mutant UCH-L1, another PD pathogenic protein, have also been recently described [43]. Whether the other PD proteins, containing CMA targeting motifs, may also contribute to pathogenesis by inducing CMA dysfunction requires further investigation.
Figure 2. Contribution of CMA dysfunction to pathogenesis in neurodegeneration.
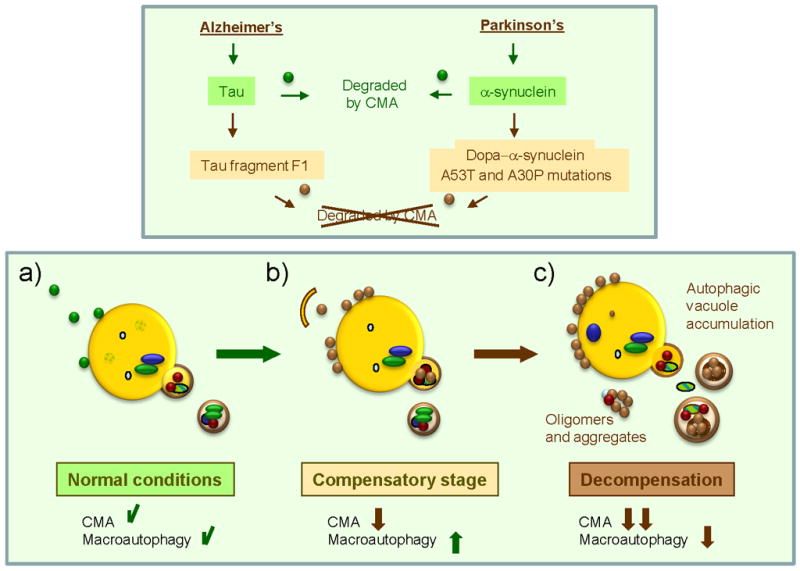
Different pathogenic proteins (α-synuclein and Tau) depicted here, have been shown to have a primary detrimental effect on CMA activity. Whereas wild-type or unmodified forms of these proteins (green circles) can be targeted and taken up by lysosomes via CMA (a), their pathogenic counterparts (brown circles) are delivered to the CMA translocation complex at the lysosomal membrane but fail to translocate into the lumen (b). These pathogenic proteins often organize into irreversible oligomeric complexes at the lysosomal membrane that further block CMA activity. Cells respond to CMA blockage by upregulating macroautophagy, the only proteolytic pathway that can directly degrade proteinaceous inclusions and aggregates (b). Failure of macroautophagy with time – due to exhaustion or primary damage by the pathogenic proteins – could precipitate progression of disease (c).
A recent study has also revealed a previously unknown connection between CMA and AD through the degradation of Tau. The amyloidogenic properties of fragments of Tau protein, identified in some AD patients, could make them the seeding mechanism for pathologic aggregate formation. Using an inducible neuronal tauopathy model, it has been shown that Tau fragmentation initiates in the cytosol, but it is completed at the lysosomal surface [44]. CMA is the mechanism behind Tau delivery to lysosomes, but the pathogenic fragment fails to get translocated, remaining on the lysosomal surface and promoting the formation of Tau oligomers – possible precursors of aggregation [44] (Figure 2b).
In light of the involvement of CMA in the maintenance of cellular homeostasis and in the response to stress, blockage of CMA in these pathologies may further contribute to creating a cellular environment favorable for protein aggregation. Blockage of CMA and often of the proteasome, which is also a target of the toxic effect of the pathogenic proteins, leads to the upregulation of macroautophagy (Figure 2b). Activation of macroautophagy under these conditions helps to maintain cellular homeostasis, by directly degrading pathogenic protein aggregates and also by reducing the pool of their soluble cytosolic forms that contribute to aggregate formation. Although it has been proposed that excessive macroautophagy may be detrimental, upregulation of macraoutophagy in response to CMA dysfunction, at least in cultured cells, has shown so far to have a protective effect. Thus, inhibition of this compensatory macroautophagy has been shown to compromise cell viability [7]. Progression of the disease may be tightly related to compromise of the macroautophagic function either by exhaustion – as it becomes often the only functional proteolytic system- or through an eventual toxic effect of the pathogenic proteins also in this system (Figure 2c)
In several of these neurodegenerative disorders, interventions aimed at upregulating macroautophagy are beneficial [45] and could have therapeutic value. Upregulation of CMA, together with the elucidation of the mechanism(s) behind the failed lysosomal translocation of pathogenic synuclein and Tau are also promising future direction for therapy.
CMA and other pathologies
Numerous recent studies support the pro- and anti-oncogenic functions of macroautophagy in cancer cells that could be exploited for therapeutic purposes [3]. In contrast, a relation between CMA and cancer biology has not been established yet. Preliminary evidence from our group has revealed a constitutive upregulation of CMA in a large number of cancer cell lines (Kon and Cuervo, in preparation), suggesting that CMA could be important for cancer cell progression and survival. Likewise, extensive information on the consequences of failed macroautophagy in innate and acquired immunity [46] has prompted the discovery of alterations of macroautophagy underlying severe immune disorders. However, despite the recently described participation of CMA in antigen presentation [24], connections between CMA and diseases of the immune system are yet to be identified.
CMA and aging
Increased half-life of numerous intracellular proteins and accumulation of damaged or altered proteins inside cells are common features to almost all aging organisms and have been directly linked to inefficiencies in the cellular quality control mechanisms with age [47]. Alterations in both the ubiquitin-proteasome system and of macroautophagy in different cellular and animal models of aging have been well reported and could further contribute to the aggravating effect that aging has in many of the above described pathologies. CMA activity also declines with aging in almost all tissues analyzed so far, mainly as consequence of decreased levels of LAMP-2A at the lysosomal membrane [48]. Reduced levels of LAMP-2A are not due to transcriptional downregulation or problems in lysosomal targeting of this protein, but instead originate from age-related changes in the lysosomal membrane that affect lysosomal distribution and turn-over of LAMP-2A and render this protein unstable [49].
Since CMA activity is directly proportional to the amount of available LAMP-2A receptor at the lysosomal membrane, it is reasonable to hypothesize that the deterioration of this proteolytic system with age could be reversed by restoring normal levels of the membrane receptor. In fact, in a recent study using a transgenic mouse model with an inducible exogenous copy of LAMP-2A (Tet-off-LAMP-2A mouse), activation of the transgene in liver of old mice was enough to restore levels of CMA close to those observed in young mice [50]. The lower levels of oxidized and aggregated proteins observed in the old transgenic mice, along with their improved response to hepatotoxic stressors and better overall liver function [50], further support the participation of CMA in cellular homeostasis and the stress response, and underscores the contribution of reduced CMA activity to the functional decline in old organisms. Future efforts should be directed to devising interventions that could stabilize LAMP-2A in old cells and hence prevent the age-dependent decline in the activity of this pathway.
Concluding Remarks
Although lacking the high capability of other autophagic systems, CMA has made room for itself in the field of protein degradation due to the high selectivity of this pathway in the degradation of cytosolic soluble proteins, comparable in many respects to that displayed by the ubiquitin/proteasome system. The recent advances in our understanding on how substrate proteins are targeted to lysosomes and cross the lysosomal membrane via CMA have offered new ways to manipulate this autophagic process and further understand its contribution to cell physiology. Contributions to homeostasis maintenance and to the cellular stress response are chief among the physiological functions of CMA. The recent link between CMA dysfunction and severe human pathologies has further increased the interest in this autophagic pathway. Future studies on CMA should aim to elucidate the molecular components of the crosstalk between CMA and other autophagic pathways, the specific roles of chaperones and co-chaperones at the targeting and translocation complexes and the missing components in this last complex. Moreover, future insights into the upstream signaling that leads to CMA activation will provide powerful tools to modulate this pathway for therapeutic purposes.
Acknowledgments
We thank Dr. Susmita Kaushik for critically reading the manuscript. Work in our laboratory is supported by NIH grants from NIA (AG021904, AG031782), NIDKK (DK041918), NINDS (NS038370), a Glenn Foundation Award and a Hirsch/Weill-Caulier Career Scientist Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol. 2005;6:79–87. doi: 10.1038/nrm1552. [DOI] [PubMed] [Google Scholar]
- 2.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;18:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 3.Mizushima N, Levine B, Cuervo A, Klionsky D. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cuervo AM. Chaperone-mediated autophagy: selectivity pays off. Trends Endocrinol Metab. 2009 doi: 10.1016/j.tem.2009.10.003. E-pub ahead of publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dice J. Chaperone-mediated autophagy. Autophagy. 2007;3:295–9. doi: 10.4161/auto.4144. [DOI] [PubMed] [Google Scholar]
- 6.Dice J. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci. 1990;15:305–309. doi: 10.1016/0968-0004(90)90019-8. [DOI] [PubMed] [Google Scholar]
- 7.Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM. Consequences of the selective blockage of chaperone-mediated autophagy. Proc Nat Acad Sci USA. 2006;103:5905–5910. doi: 10.1073/pnas.0507436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaushik S, Massey A, Mizushima N, Cuervo AM. Constitutive Activation of Chaperone-mediated Autophagy in Cells with Impaired Macroautophagy. Mol Biol Cell. 2008;19:2179–92. doi: 10.1091/mbc.E07-11-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, et al. Loss of macroautophagy promotes or prevents fibroblast apoptosis depending on the death stimulus. J Biol Chem. 2007 doi: 10.1074/jbc.M706666200. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiang H, Terlecky S, Plant C, Dice JF. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science. 1989;246:382–385. doi: 10.1126/science.2799391. [DOI] [PubMed] [Google Scholar]
- 11.Cuervo AM, Dice JF, Knecht E. A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J Biol Chem. 1997;272:5606–15. doi: 10.1074/jbc.272.9.5606. [DOI] [PubMed] [Google Scholar]
- 12.Agarraberes F, Terlecky S, Dice J. An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J Cell Biol. 1997;137:825–834. doi: 10.1083/jcb.137.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cuervo A, Dice J. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273:501–503. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- 14.Bandhyopadhyay U, Kaushik S, Vartikovsky L, Cuervo AM. Dynamic organization of the receptor for chaperone-mediated autophagy at the lysosomal membrane. Mol Cell Biol. 2008;28:5747–63. doi: 10.1128/MCB.02070-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wing S, Chiang HL, Goldberg AL, Dice JF. Proteins containing peptide sequences related to KFERQ are selectively depleted in liver and heart, but not skeletal muscle, of fasted rats. Biochem J. 1991;275:165–169. doi: 10.1042/bj2750165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agarraberes F, Dice JF. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J Cell Sci. 2001;114:2491–9. doi: 10.1242/jcs.114.13.2491. [DOI] [PubMed] [Google Scholar]
- 17.Cuervo A. Autophagy in neurons: it is not all about food. Trends Mol Med. 2006;12:461–4. doi: 10.1016/j.molmed.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 18.Cuervo A, Dice J. Unique properties of lamp2a compared to other lamp2 isoforms. J Cell Sci. 2000;113:4441–4450. doi: 10.1242/jcs.113.24.4441. [DOI] [PubMed] [Google Scholar]
- 19.Salvador N, Aguado C, Horst M, Knecht E. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J Biol Chem. 2000;275:27447–56. doi: 10.1074/jbc.M001394200. [DOI] [PubMed] [Google Scholar]
- 20.Agarraberes F, Terlecky SR, Dice JF. An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J Cell Biol. 1997;137:825–834. doi: 10.1083/jcb.137.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cuervo A, Dice J. Regulation of lamp2a levels in the lysosomal membrane. Traffic. 2000;1:570–83. doi: 10.1034/j.1600-0854.2000.010707.x. [DOI] [PubMed] [Google Scholar]
- 22.Cuervo A, Knecht E, Terlecky S, Dice J. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am J Physiol. 1995;269:C1200–C1208. doi: 10.1152/ajpcell.1995.269.5.C1200. [DOI] [PubMed] [Google Scholar]
- 23.Cuervo A, Dice J, Knecht E. A lysosomal population responsible for the hsc73-mediated degradation of cytosolic proteins in lysosomes. J Biol Chem. 1997;272:5606–5615. doi: 10.1074/jbc.272.9.5606. [DOI] [PubMed] [Google Scholar]
- 24.Zhou D, Li P, Lin Y, Lott JM, Hislop AD, Canaday DH, Brutkiewicz RR, Blum JS. Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity. 2005;22:571–81. doi: 10.1016/j.immuni.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 25.Kiffin R, Christian C, Knecht E, Cuervo A. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell. 2004;15:4829–4840. doi: 10.1091/mbc.E04-06-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cuervo AM, Mann L, Bonten E, d’Azzo A, Dice J. Cathepsin A regulates chaperone-mediated autophagy through cleavage of the lysosomal receptor. EMBO J. 2003;22:12–19. doi: 10.1093/emboj/cdg002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaushik S, Massey AC, Cuervo AM. Lysosome membrane lipid microdomains: novel regulators of chaperone-mediated autophagy. EMBO J. 2006;25:3921–3933. doi: 10.1038/sj.emboj.7601283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finn P, Mesires N, Vine M, Dice JF. Effects of small molecules on chaperone-mediated autophagy. Autophagy. 2005;1:141–145. doi: 10.4161/auto.1.3.2000. [DOI] [PubMed] [Google Scholar]
- 29.Iwata A, Christianson JC, Bucci M, Ellerby LM, Nukina N, Forno LS, Kopito RR. Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc Natl Acad Sci. 2005;102:13135–13140. doi: 10.1073/pnas.0505801102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding Q, Dimayuga E, Martin S, Bruce-Keller A, Nukala V, Cuervo A, Keller J. Characterization of chronic low-level proteasome inhibition on neural homeostasis. J Neurochem. 2003;86:489–497. doi: 10.1046/j.1471-4159.2003.01885.x. [DOI] [PubMed] [Google Scholar]
- 31.Massey AC, Follenzi A, Kiffin R, Zhang C, Cuervo AM. Early cellular changes after blockage of chaperone-mediated autophagy. Autophagy. 2008;4:442–56. doi: 10.4161/auto.5654. [DOI] [PubMed] [Google Scholar]
- 32.Cuervo A, Palmer A, Rivett A, Knecht E. Degradation of proteasomes by lysosomes in rat liver. Eur J Biochem. 1995;227:792–800. doi: 10.1111/j.1432-1033.1995.tb20203.x. [DOI] [PubMed] [Google Scholar]
- 33.Yang Q, She H, Gearing M, Colla E, Lee M, Shacka J, Mao Z. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science. 2009;323:124–127. doi: 10.1126/science.1166088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wenger DA, Coppola S, Liu S. Lysosomal storage disorders: diagnostic dilemmas and prosects for therapy. Genet Med. 2002;4:412–419. doi: 10.1097/00125817-200211000-00003. [DOI] [PubMed] [Google Scholar]
- 35.Venugopal B, Mesires N, Kennedy J, Curcio-Morelli C, Laplante JM, Dice JF, Slaugenhaupt SA. Chaperone-mediated autophagy is defective in mucolipidosis type IV. J Cell Physiol. 2009;219:344–353. doi: 10.1002/jcp.21676. [DOI] [PubMed] [Google Scholar]
- 36.Nishino I, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease) Nature. 2000;406:906–910. doi: 10.1038/35022604. [DOI] [PubMed] [Google Scholar]
- 37.Sooparb S, Price S, Shaoguang J, Franch H. Suppression of chaperone-mediated autophagy in the renal cortex during acute diabetes mellitus. Kidney Int. 2004;65:2135–2144. doi: 10.1111/j.1523-1755.2004.00639.x. [DOI] [PubMed] [Google Scholar]
- 38.Franch H. Pathways of proteolysis affecting renal cell growth. Curr Opin Nephrol Hypertens. 2002;11:445–450. doi: 10.1097/00041552-200207000-00012. [DOI] [PubMed] [Google Scholar]
- 39.Martinez-Vicente M, Cuervo AM. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 2007;6:352–61. doi: 10.1016/S1474-4422(07)70076-5. [DOI] [PubMed] [Google Scholar]
- 40.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–5. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 41.Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One. 2009;4:e5515. doi: 10.1371/journal.pone.0005515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martinez-Vicente M, et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest. 2008;118:777–88. doi: 10.1172/JCI32806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kabuta T, Wada K. Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J Biol Chem. 2008;283:23731–2373. doi: 10.1074/jbc.M801918200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. 2009;18:4153–70. doi: 10.1093/hmg/ddp367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ravikumar B, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 46.Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767–77. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cuervo AM. Autophagy and Aging: keeping that old broom working. Trends Genet. 2008;24:604–612. doi: 10.1016/j.tig.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem. 2000;275:31505–31513. doi: 10.1074/jbc.M002102200. [DOI] [PubMed] [Google Scholar]
- 49.Kiffin R, Kaushik S, Zeng M, Bandyopadhyay U, Zhang C, Massey A, Martinez-Vicente M, Cuervo A. Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J Cell Sci. 2007;120:782–91. doi: 10.1242/jcs.001073. [DOI] [PubMed] [Google Scholar]
- 50.Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med. 2008;14:959–65. doi: 10.1038/nm.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]