

Abstract
This Letter describes the synthesis and SAR, developed through an iterative analog library approach, of a novel series of selective M1 mAChR antagonists, based on an N-(4-(4-alkylpiperazin-1-yl)phenyl)benzamide scaffold for the potential treatment of Parkinson's disease, dystonia and other movement disorders. Compounds in this series possess M1 antagonist IC50s in the 350 nM to >10 μM range with varying degrees of functional selectivity versus M2-M5.
There are five subtypes of muscarinic acetylcholine receptors (mAChR1-5 or M1-M5), members of the G Protein-Coupled Receptor (GPCR) family A, that mediate the metabotropic actions of the neurotransmitter acetylcholine.1,2 M1, M3 and M5 activate phospholipase C and calcium mobilization through Gq whereas M2 and M4 block the action of adenylyl cyclase through Gi/o.1,2 The cholinergic system, mediated by mAChRs, plays a critical role in a wide variety of CNS and peripheral functions including memory and attention mechanisms, motor control, nociception, regulation of sleep wake cycles, cardiovascular function, renal and gastrointestinal function to mention only a few.1-4 As a result, agents that can selectively modulate the activity of mAChRs have the potential for therapeutic use in multiple peripheral and central pathological states. Due to high sequence conservation within the orthosteric binding site of the five mAChR subtypes, it has been historically difficult to develop mAChR subtype-selective ligands.1-5 Based on brain expression and cellular localization, data from mAChR knock-out mice and clinical trials with muscarinic agents, the M1 subtype is an attractive molecular target for the treatment of CNS disorders. M1 has been implicated in the pathologies of Alzheimer's disease (AD), Parkinson's disease (PD) and dystonia due to its role in cognition and motor control.6
The majority of reported muscarinic antagonists are unselective, such as a scopolamine, 1.7 Recently, pirenzapine, 2 has emerged as a relatively selective M1 receptor antagonist (20- to 50-fold versus M2-M5) and there are numerous reports of moderately selective M3 antagonists (20- to 50-fold versus M2) such as 3.8 Interestingly, the most selective M1 antagonist, MT7, 4, the 65 amino acid peptide, (>1,000-fold versus M2-M5) was derived from venom extracts of the green mamba snake (Fig. 1).9 From an M1 functional screen within the MLSCN, we identified M1 antagonists such as 5 (M1 IC50 of 441 nM and with >340-fold selectivity versus M4, but modest selectivity versus M2, M3 and M5 (7.9-fold, 7-fold, and 2.4-fold, respectively)) and 6 (M1 IC50 of 5.0 μM and with >30-fold selective versus M2-M5).10-12 Based on the M1 selectivity of 6, attractive physiochemical properties (MW < 350, clogP 3.6) and the fact that it was the only benzamide-containing analog in the series, we initiated a library synthesis effort13 to develop SAR around 6.
Figure 1.
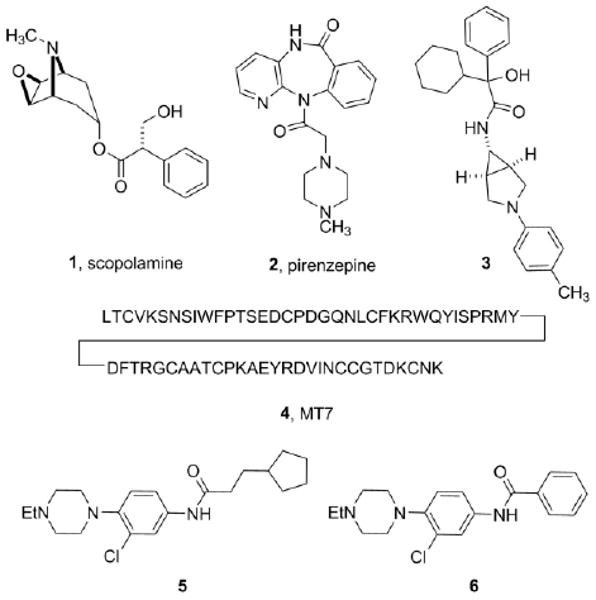
Structures of representative mAChR antagonists.
As shown in Scheme 1, the first round of library synthesis focused on benzamide analogs of 6. Commercially available 3-chloro-(4-(4-ethylpierazin-1yl)aniline 7 was acylated under standard conditions employing polymer-supported reagents and scavengers13 to afford a 24-member library of analogs 8, along with resynthesized 6. All analogs were then purified by mass-guided HPLC to analytical purity.14 To effectively screen small libraries of potential mAChR ligands, we have adopted a strategy to triage compounds in single-point screens (at 10 μM) at M1, M3 and M5 – the Gq-coupled mAChRs – to identify active and selective compounds prior to running full concentration-response curves (CRCs).15 Figure 2 shows the 10 μM single-point screens for the first 25-member library of benzamide analogs 8.
Scheme 1.
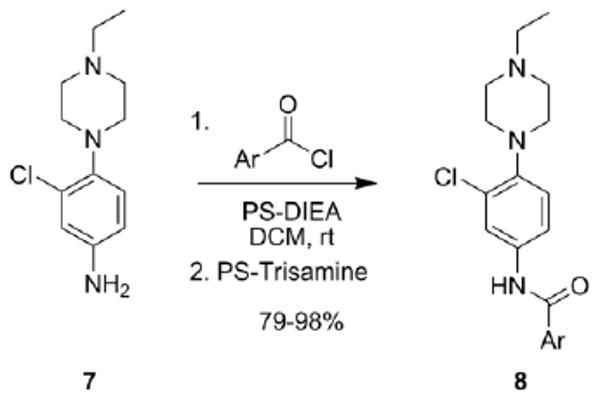
Library synthesis of first generation analogs 8. All library compounds were purified by mass-guided HPLC to >98% purity.14
Figure 2.
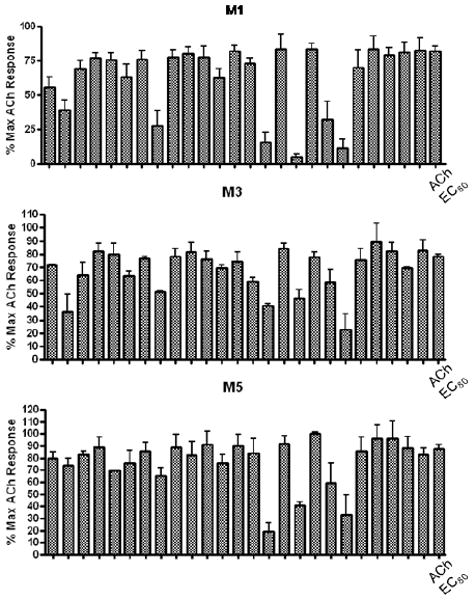
Single-point EC80 plus 10 μM compound triage screen at M1, M3 and M5 to select compounds for full CRCs.
As Shown in Table 1, re-synthesized 6 displayed comparable potency and mAChR selectivity to the original sample (M1 IC50 = 3.2 μM, IC50 ≫10 μM for M2-M5). Functionalized benzamide analogs 8 possessed a wide range of M1 potency and mAChR selectivity, and we initially evaluated anlaogs 8 against M1, M3 and M5. Substitution in the 2-position, 8a (2-Cl) and 8b (2-OMe) possessed submicromolar M1 IC50s (960 nM and 820 nM, respectively), but also showed low micromolar activity at M3 and M5. A pentafluorophenyl congener 8e (Fig. 3A) proved to be a submicromolar antagonist of both M1 and M5 (IC50s of 350 nM and 830 nM, respectively). Substitution at the 4-position, as with the 4-OMe derivative 8f, was comparable to the original 6. Interestingly, a 2,5-bisCF3 analog 8h had an M1 IC50 of 490 nM, with ∼ 9-fold functional selectivity versus M3 and M5 (Fig. 3B). Intrigued by this potent and selective M1 antagonist, we screend against M2 and M4 as well, but found that 8h possessed only 3- to 4-fold selectivity versus the Gi/o-coupled mAChRs (Table 1). 8i, a 3,5-bisCF3 analog possessed a unique profile as a dual M1/M4 antagonist (IC50s of 2.6 μM and 3.7 μM, respectively), with little effect on an ACh EC80 at 10 μM on M2, M3 or M5. Finally, a 3,4-difluoro 8j derivative was also comparable to the original 6. While this library afforded interesting results, further optimization was required.
Table 1.
Structures and mAChR activities of analogues 8.
 | ||||||
|---|---|---|---|---|---|---|
| Cmpd | Ar | M1 IC50 (μM)a | M2 IC50 (μM)a | M3 IC50 (μM)a | M4 IC50 (μM)a | M5 IC50 (μM)a |
| 6 | 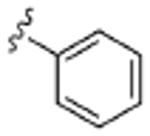 |
3.2 | >10 | >10 | >10 | >10 |
| 8a | 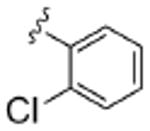 |
0.96 | ND | 8.2 | ND | 2.3 |
| 8b | 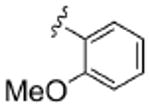 |
0.82 | ND | 5.6 | ND | 1.3 |
| 8c | 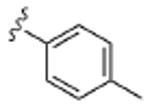 |
2.9 | 6.9 | >10 | 3.7 | >10 |
| 8d | 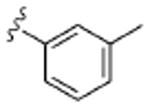 |
2.1 | ND | >10 | ND | 3.5 |
| 8e | 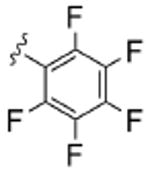 |
0.35 | ND | 3.7 | ND | 0.83 |
| 8f | 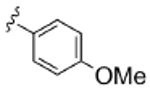 |
3.2 | ND | >10 | ND | >10 |
| 8g | 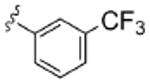 |
2.9 | >10 | 4.3 | 3.7 | 4.1 |
| 8h | 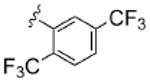 |
0.49 | 2.7 | 4.2 | 1.5 | 4.1 |
| 8i | 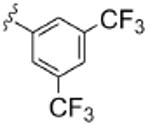 |
2.6 | >10 | >10 | 3.7 | >10 |
| 8j |  |
4.7 | >10 | >10 | >10 | >10 |
IC50s are an average of three independent experiments using mAChR (CHO) cell lines. ND = not determined.
Figure 3.
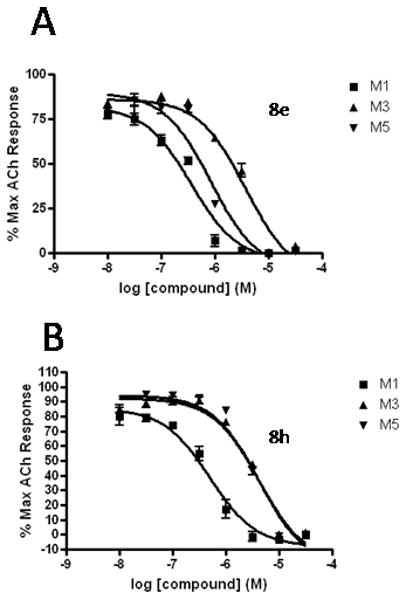
CRCs for M1, M3 and M5 for (A) compound 8e (M1 IC50 = 350 nM) and (B) compound 8h (M1 IC50 = 490 nM), showing ∼9-fold functional selectivity versus M3 and M5.
Having surveyed the amide moiety while maintaining the N-ethyl piperazine, we next generated two-dimensional libraries wherein the nature of the alkyl group was varied (9-12) while also surveying diverse benzamides to generate analogs 9a-f, 10 a-f, 11a-f and 12a-f (Scheme 2).
Scheme 2.
Library synthesis of second generation analogs 9a-f, 10a-f, 11a-f and 12a-f. All library compounds were purified by mass-guided HPLC to >98% purity.14
Application of the same strategy to triage compounds in single-point screens (at 10 μM) at M1, M3 and M5 to identify active and selective compounds prior to running full (CRCs) was employed, but >75% of these new analogs possessed no M1 antagonist activity. The SAR for this series was incredibly shallow, with only an N-propyl congener with the 3,5-dicholrobenzamide moiety 11i displaying reasonable activity (M1 IC50 = 3.7 μM, IC50 >10 μM for M3 and M5), and all other analogs possessing M1 IC50s in the 6-9 μM range.
In summary, a two-dimensional parallel synthesis library campaign was performed around 6, an M1 antagonist identified in a functional HTS screen. SAR for this series was shallow, but we were able to improve the M1 antagonist activity of 6 into the 350 to 500 nM range with analogs 8, while maintaining good mAChR selectivity. Interestingly, 8i is the first reported dual M1/M4-preferring antagonist, which compliments the prototypical M1/M4-preferring agonist xanomeline. Other chemical series from our M1 functional screen are currently under chemical optimization, and further refinements will be reported in due course.
Acknowledgments
The authors thank the NIH, NIMH and the MLSCN for funding of the Vanderbilt Screening Center for GPCRs, Ion Channels & Transporters (3U54MH074427), the Chemistry Supplement (3U54MH074427-02S1) and the XO1 (1XO1MH077606-01) M1 antagonist screening application which enabled this work. The authors also wish to thank the second generation of the MLSCN, coined the MLPCN, and the Vanderbilt Specialized Chemistry Center (U54MH084659-01).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.(a) Bonner TI, Buckley NJ, Young AC, Brann MR. Science. 1987;237:527. doi: 10.1126/science.3037705. [DOI] [PubMed] [Google Scholar]; (b) Bonner TI, Young AC, Buckley NJ, Brann MR. Neuron. 1988;1:403. doi: 10.1016/0896-6273(88)90190-0. [DOI] [PubMed] [Google Scholar]
- 2.Felder CC, Bymaster FP, Ward J, DeLapp N. J Med Chem. 2000;43:4333. doi: 10.1021/jm990607u. [DOI] [PubMed] [Google Scholar]
- 3.Bymaster FP, McKinzie DL, Felder CC, Wess J. Neurochem Res. 2003;28:437. doi: 10.1023/a:1022844517200. [DOI] [PubMed] [Google Scholar]
- 4.Birdsall NJM, Farries T, Gharagozloo P, Kobayashi S, Lazareno S, Sugimoto M. Mol Pharmacol. 1999;55:778. [PubMed] [Google Scholar]
- 5.Eglen RM, Choppin A, Dillon MP, Hedge S. Curr Opin Chem Biol. 1999;3:426. doi: 10.1016/S1367-5931(99)80063-5. [DOI] [PubMed] [Google Scholar]
- 6.Birdsall NJM, Nathanson NM, Schwarz RD. TRENDS Pharm Sci. 2001;22:215. [Google Scholar]
- 7.(a) Burke RE. Mov Disord. 1986;1:135. doi: 10.1002/mds.870010208. [DOI] [PubMed] [Google Scholar]; (b) Buckley NJ, Bonner TI, Buckley CM, Brann MR. Mol Pharmacol. 1989;35:469. [PubMed] [Google Scholar]; (c) Walebroeck M, Tastenoy M, Camus J, Christophe J. Mol Pharmacol. 1990;36:267. [PubMed] [Google Scholar]; (d) Dei S, Bellucci C, Buccioni M, Ferraroni M, Guandalini L, Manetti L, Martini E, Marucci G, Matucci R, Nesi M, Romanelli MN, Scapecchi S, Teodori E. J Med Chem. 2007;50:1409. doi: 10.1021/jm061374r. [DOI] [PubMed] [Google Scholar]
- 8.Hammer R, Berrie CP, Birdsall NJM, Burgen ASV, Hulme EC. Nature. 1980;283:90. doi: 10.1038/283090a0. [DOI] [PubMed] [Google Scholar]
- 9.Bradley KN. Pharmacol & Therap. 2000;85:87. doi: 10.1016/s0163-7258(99)00064-9. [DOI] [PubMed] [Google Scholar]
- 10.For information on the Molecular Library Production Center Network see: http://mli.nih.gov/mli/mlpcn/
- 11.Lewis LM, Sheffler D, Williams R, Bridges TA, Kennedy JP, Brogan JT, Mulder MJ, Williams L, Nalywajko NT, Niswender C, Weaver CD, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2008;18:885. doi: 10.1016/j.bmcl.2007.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sheffler DJ, Williams R, Bridges TM, Lewis LM, Xiang Z, Zheng F, Kane AS, Byum NE, Jadhav S, Mock MM, Zheng F, Lewis LM, Jones CK, Niswender CM, Weaver CD, Conn PJ, Lindsley CW, Conn PJ. Mol Pharmacol. 2009;76:356. doi: 10.1124/mol.109.056531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kennedy JP, Williams L, Bridges TM, Daniels RN, Weaver D, Lindsley CW. J Comb Chem. 2008;10:345. doi: 10.1021/cc700187t. [DOI] [PubMed] [Google Scholar]
- 14.Leister WH, Strauss KA, Wisnoski DD, Zhao Z, Lindsley CW. J Comb Chem. 2003;5:322. doi: 10.1021/cc0201041. [DOI] [PubMed] [Google Scholar]
- 15.Details of the calcium mobilization assays: Chinese Hamster Ovary (CHO-K1) cells stably expressing human (h) M1, hM3, and hM5 were used for calcium mobilization assays. hM2 and hM4 were adapted to this assay and signaling pathway after stably transfecting Gqi5 chimeric G protein. To measure agonist-induced calcium mobilization and determine effect of novel compounds, stable muscarinic cell lines plated overnight in Costar 96-well cell culture plates (Corning) were incubated with 50μL of 2μM Fluo-4 AM diluted in assay buffer [HBSS (Invitrogen) supplemented with 20mM HEPES and 2.5mM probenecid, pH 7.4] for 45min at 37°C. Dye was then removed and replaced with assay buffer. Cells were pre-incubated with 10μM or a concentration-response curve of novel compound, followed by a sub-maximal concentration of Acetylcholine or Carbachol. The signal amplitude was first normalized to baseline and then expressed as a percentage of the maximal response to acetylcholine.