

Abstract
Recent studies have shown that both innate and adaptive immunity contribute to hypertension. Inflammatory cells, including macrophages and T cells accumulate in the vessel wall, particularly in the perivascular fat, and in the kidney. Mice lacking lymphocytes are resistant to development of hypertension, and adoptive transfer of T cells restores hypertensive responses to angiotensin II and DOCA-salt challenge. Immune modulating agents have variable, but often-beneficial effects in ameliorating end-organ damage and blood pressure elevation in experimental hypertension. The mechanisms by which hypertension stimulates an immune response remain unclear, but might involve formation of neo-antigens that activate adaptive immunity. Identification of these neo-antigens and understanding how they form might prove useful in prevention and treatment of this widespread and devastating disease.
INTRODUCTION
Despite intensive study, there remains substantial debate regarding the etiology of hypertension and most research has focused on the roles of the kidney, the vasculature or the central nervous system. Recent data however, have suggested that components of the innate and adaptive immune system also contribute to hypertension. Traditionally, atherosclerosis has been considered an inflammatory disease, however increasing evidence suggests that inflammation also contributes to hypertension, and if efforts are taken to block inflammation, the end-organ damage and severity of blood pressure elevation can be reduced. Surprisingly, thymus-derived lymphocytes (T cells) seem to be involved in hypertension, indicating that the adaptive immune system might contribute to this disease. This is an emerging area of investigation, and the exact manner by which T cells and other inflammatory cells are activated and contribute to hypertension is far from understood. In this review, we will consider the existing data and speculate as to possible mechanisms responsible for immune cell activation and their contribution to blood pressure elevation.
Inflammation and atherosclerosis
It is useful to begin this review with a brief discussion of atherosclerosis, which is clearly an inflammatory disease and thus has parallels to hypertension. In early atherosclerotic lesions, macrophages take up oxidized low-density lipoprotein and become foam cells [1]. In more complex, established lesions, macrophages accumulate in the shoulder region and contribute to plaque instability [2]. Thus, these cells of the innate immune system are important in several phases of atherogenesis. More surprisingly, activated T cells are also found in human atherosclerotic lesions and genetic manipulation of T cells reduces lesion formation in mice [3]. In addition, dendritic cells, which are critical for presenting antigen and promoting T cell activation, are present in atherosclerotic lesions of humans and experimental animals [4,5]. Observations such as these have led investigators to look for potential antigens that contribute to atherosclerosis [6], and oxidized low density lipoproteins, heat shock proteins, various bacterial proteins and platelet glycoprotein B1 have all been implicated in atherosclerosis [7], although no specific protein has been definitively identified as the antigen responsible for either human or experimental atherosclerosis. Cytokines released from T cells are thought to contribute to various stages of lesion development and destabilization. Importantly, cells of the vessel wall, including endothelial and vascular smooth muscle cells, contribute to the inflammatory cascade involving T cells, by presenting antigen and responding to cytokines from T cells [8]. This paradigm, while likely incomplete, has provided a guidebook for understanding how adaptive immunity could contribute to other common diseases such as hypertension.
The role of adaptive immunity in hypertension
While a role of adaptive immunity in atherosclerosis is rather well accepted, it has also been implicated as a contributing factor in hypertension, perhaps with less fanfare. More than 25 years ago, Svendson showed that the delayed phase of DOCA salt hypertension was blunted in thymectomized animals [9]. Pre-eclampsia is associated with an increase in lymphocyte markers and the cytokine profile of natural killer lymphocytes in the uterus [10,11]. Interestingly, a recent analysis of almost 6000 people with AIDS (with reduced CD4+ cells) showed that the incidence of hypertension was significantly lower than the general population matched non-infected individuals. Treatment with highly active anti-retroviral therapy for 2 years restored the incidence of hypertension to that of the control population [12]. This finding might reflect a need for functioning helper T cells in hypertension development.
Recently, our group has provided additional evidence that experimental hypertension is dependent on adaptive immunity. We found that RAG-1−/− mice, which lack both T and B cells, have very blunted hypertensive responses to prolonged angiotensin II infusion or DOCA-salt challenge [13]. Adoptive transfer of T cells, but not B cells, led to a complete restoration of the hypertensive response to these stimuli, strongly implicating T cells in the genesis of hypertension. We also found that endothelium-dependent vasodilatation and vascular superoxide production, which are generally abnormal in hypertension, were not affected in RAG-1−/− mice given prolonged angiotensin II infusion, but became abnormal after adoptive transfer of T cells. These studies have provided reasonably definitive evidence that T cells are important in hypertension and the vascular dysfunction caused by this disease.
In these studies, we sought to determine if T cells entered the vessel to mediate these abnormalities. Surprisingly, and unlike the condition in atherosclerosis, few if any T cells are found in hypertensive vessels per se, but analysis of perivascular tissues showed that the perivascular fat, immediately adjacent to the adventitia of the aorta and mesenteric vessels, became infested with T cells in the setting of hypertension. These bear markers of effector T cells and are accompanied by other leukocytes, including natural killer cells and macrophages. Importantly, in the absence of T cells, perivascular accumulation of other leukocytes does not occur, supporting a key role of T cells in mediating the overall inflammatory response.
The kidney is also a target of inflammatory cells in hypertension. Hypertension stimulates lymphocytic infiltration in the kidney, and immunosuppressive therapy prevents this and reduces renal damage while lowering blood pressure in some cases [14,15], but not in others [16,17]. In particular, the T cell modulating agent mycophenolate mofetil prevents renal T cell accumulation and hypertension in some experimental models [15,18] and in at least one small study in hypertensive humans [19]. Hypertension due to nitric oxide synthase inhibition and high salt diet promotes the renal accumulation of macrophages and T cells and causes proteinuria in mice. These events are prevented by genetically inhibiting the proinflammatory transcription factor nuclear factor kappa B (NFκB) [20]. In keeping with this, Curcumin, which inhibits NFκB, also prevents renal inflammation, progressive renal failure and proteinuria in rats with 5/6th nephrectomy [21]. Of note, commonly employed drugs such as statins, ACE inhibitors, angiotensin receptor blockers and spironolactone blunt renal inflammation in hypertensive models [22,23], potentially providing additional benefit beyond lipid or blood pressure lowering.
Mechanisms underlying T cell promotion of hypertension
The studies described above clearly show that T cells contribute to blood pressure elevation, vascular dysfunction and renal disease in hypertension. A major question however is what T cells are doing to cause these effects. A likely mechanism involves release of cytokines that influence vascular and renal function directly or indirectly by stimulating other cells to release cytokines and recruiting additional inflammatory cells. Traditionally, it is believed that activation of CD4+ T cells leads to either a T helper-1 (TH1) or a TH2 phenotype. TH1 polarized cells produce the signature cytokines IFN-γ, IL-2, TNFα and TNFβ, while TH2 cells produce IL4, IL5, IL10, and transforming growth factor β. Cells polarized to the TH1 profile are thought to participate in inflammatory diseases such as rheumatoid arthritis, psoriasis and bacterial infections, while TH2 cells contribute to allergies and responses to helminths. Of note, the T cells in atherosclerotic lesions seem to be TH1-polarized. We found that the TNFα antagonist etanercept prevents hypertension and vascular dysfunction in angiotensin II-induced hypertension [13], and others have shown it prevents hypertension in fructose-fed rats [24]. This agent also prevents renal injury in salt-dependent hypertension [25]. Interleukin-6 has also been implicated in angiotensin II-induced hypertension [26,27], while endogenous IL-10 has been found to have protective effects [27]. Taken together, these findings suggest that TH1 cytokines contribute to hypertension.
Recently, we have provided support for the role of a non-TH1 cytokine in hypertension. The cytokine IL-17 is a novel cytokine produced by TH17 cells, cytotoxic T cells, mast cells, neutrophils and natural killer T cells [28]. TH17 cells are thought to develop independently of the TH1 or TH2 lineages, and seem to both promote and in some cases inhibit inflammation [29,30]. We found that chronic angiotensin II infusion increased the percent of circulating TH17 cells by 2- to 3-fold and caused accumulation of vascular levels of IL17 [31]. The hypertension and vascular dysfunction caused by angiotensin II was reduced in IL-17−/− mice. Importantly, IL-17 has been shown to induce chemokines and adhesion molecules in tissues that promote tissue accumulation of other inflammatory cells. In keeping with this, we found that vascular accumulation of leukocytes that accompanies angiotensin II-infusion was virtually eliminated in IL-17−/− mice. Thus, IL-17 seems to be an important cytokine that participates in hypertension. We propose that T cells residing in the perivascular fat release cytokines such as IL-17, that diffuse to the adjacent vascular smooth muscle cells where they enhance superoxide production, reduce endothelium-dependent vasodilatation and promote vasoconstriction. It is quite likely that other cytokines, such as TNFα and IL-6 also contribute by creating a cytokine milieu that promotes hypertension.
Mechanisms of T cell activation in hypertension – potential role of the central nervous system
Hypertension has variously been attributed to alterations of renal function, vascular function and altered CNS signaling. Moreover, oxidative stress in the brain, the vasculature and the kidney clearly contribute to hypertension via mechanisms that are incompletely understood. How these various systems are involved in T cell activation and how oxidative events contribute to T cell activation remain unclear. Recently, we have found that oxidative stress in the circumventricular organs (CVO) of the brain, and in particular in the subfornical organ, likely participates in this process. These regions lack a well-formed blood brain barrier and are therefore affected by circulating signals like angiotensin II. Seminal work by Davisson and coworkers has shown that reactive oxygen species produced in neurons of the CVO contribute to hypertension, in part by promoting sympathetic outflow. We produced mice in which loxP sites were placed on either side of the coding region of the extracellular superoxide dismutase (ecSOD or SOD3). This permitted deletion of SOD3 in adult animals by intracerebroventricular (ICV) injection of an adenovirus expressing Cre-recombinase (AdCre). Acute deletion of SOD3 caused an oxidative insult in the CVO, raised systemic blood pressure by 20 mmHg and markedly sensitized the mice to infusion of low-dose angiotensin II. More importantly, we observed that this central manipulation caused an increase in the percent of T cells with an activated phenotype, and markedly increased the vascular inflammation associated with angiotensin II infusion. Analysis of heart rate and blood pressure variability indicated that deletion of SOD3 in the CVO markedly enhanced sympathetic outflow [32]. These data are in keeping with prior studies by Ganta et al, suggesting that sympathetic outflow can promote T cell activation [33].
Potential role of neoantigens in hypertension
It is interesting to speculate that an important mechanism underlying T cell activation in hypertension is formation of antigens that are not identified as self. These “neoantigens” could be produced in response to oxidative modification of proteins, lipids or nucleic acids, exposure of components of the basement membrane, by induction of cell death and release of intracellular antigens that normally are immune privileged or by other unknown mechanisms. Of interest, several small molecular weight heat shock proteins, which have been proposed to serve as neoantigens in atherosclerosis are increased in the kidneys of hypertensive animals [34]. It is conceivable that low levels of blood pressure elevation could induce these changes, perhaps via mechanical trauma to peripheral tissues. In RAG-1−/− mice, angiotensin II causes an increase of systolic pressure to approximately 135 mmHg. This is similar in p47phox−/− mice [35], and in animals treated with antioxidants [36]. Thus, it seems that in the absence of oxidative signaling or inflammation, even maximal doses of angiotensin II or other stimuli only increase pressure to pre-hypertensive levels.
Summary
The above findings are compatible with a scenario such as that depicted in figure 1, in which hypertensive stimuli act both on the CNS and in the periphery to cause a modest increase in blood pressure. This leads to neoantigen formation and promotion of T cell activation, which leads to inflammation in target organs such as the kidney and vasculature, resulting in a marked elevation in blood pressure. According to this scheme, the presence of mild hypertension (often referred to as pre-hypertension) sets the stage for eventual severe hypertension in the absence of a therapeutic intervention. This might explain the common progression of pre-hypertension to overt hypertension observed clinically. These considerations stress the benefit of lowering blood pressure by whatever means possible to prevent the inflammatory phase of the disease.
Figure 1.
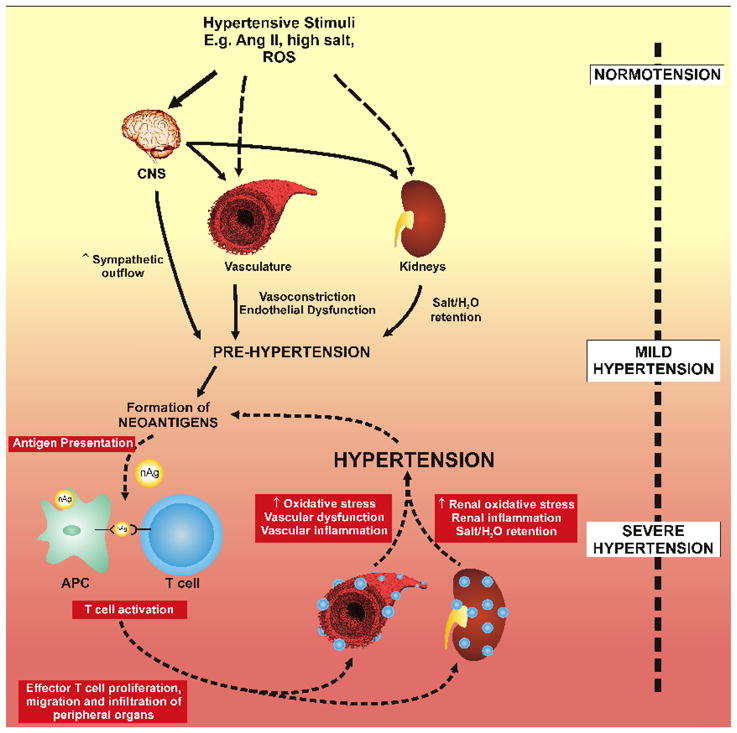
Working hypothesis describing the role of adaptive immunity in hypertension. We propose that hypertensive stimuli, such as angiotensin II, salt and reactive oxygen species affect the CNS, the kidney and the vasculature. The effects of angiotensin II and salt have predominant roles centrally, increasing sympathetic outflow and indirectly affects the kidney and vasculature, leading to salt and water retention and vasoconstriction. This promotes mild hypertension to values of approximately 135 mmHg (pre-hypertension). This promotes formation of neo-antigens that are processed by antigen presenting cells and lead to T cell activation. Activated T cells infiltrate the vasculature and kidney, promoting endothelial dysfunction, further vasoconstriction and salt and water retention and ultimately severe hypertension.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature cited
- 1.Rosenfeld ME, Khoo JC, Miller E, Parthasarathy S, Palinski W, Witztum JL. Macrophage-derived foam cells freshly isolated from rabbit atherosclerotic lesions degrade modified lipoproteins, promote oxidation of low-density lipoproteins, and contain oxidation-specific lipid-protein adducts. J Clin Invest. 1991;87:90–99. doi: 10.1172/JCI115006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galis ZS, Sukhova GK, Kranzhofer R, Clark S, Libby P. Macrophage foam cells from experimental atheroma constitutively produce matrix-degrading proteinases. Proc Natl Acad Sci U S A. 1995;92:402–406. doi: 10.1073/pnas.92.2.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 4.Bobryshev YV, Lord RS. S-100 positive cells in human arterial intima and in atherosclerotic lesions. Cardiovasc Res. 1995;29:689–696. [PubMed] [Google Scholar]
- 5.Ozmen J, Bobryshev YV, Lord RS, Ashwell KW. Identification of dendritic cells in aortic atherosclerotic lesions in rats with diet-induced hypercholesterolaemia. Histol Histopathol. 2002;17:223–237. doi: 10.14670/HH-17.223. [DOI] [PubMed] [Google Scholar]
- 6.Palinski W, Witztum JL. Immune responses to oxidative neoepitopes on LDL and phospholipids modulate the development of atherosclerosis. J Intern Med. 2000;247:371–380. doi: 10.1046/j.1365-2796.2000.00656.x. [DOI] [PubMed] [Google Scholar]
- 7 **.Gotsman I, Sharpe AH, Lichtman AH. T-cell costimulation and coinhibition in atherosclerosis. Circ Res. 2008;103:1220–1231. doi: 10.1161/CIRCRESAHA.108.182428. This is an excellent review of immune mechanisms in atherosclerosis. The potential role of neoantigens is discussed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mach F, Schonbeck U, Sukhova GK, Bourcier T, Bonnefoy JY, Pober JS, Libby P. Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for CD40-CD40 ligand signaling in atherosclerosis. Proc Natl Acad Sci U S A. 1997;94:1931–1936. doi: 10.1073/pnas.94.5.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Svendsen UG. Evidence for an initial, thymus independent and a chronic, thymus dependent phase of DOCA and salt hypertension in mice. Acta Pathol Microbiol Scand [A] 1976;84:523–528. doi: 10.1111/j.1699-0463.1976.tb00150.x. [DOI] [PubMed] [Google Scholar]
- 10.Matthiesen L, Berg G, Ernerudh J, Ekerfelt C, Jonsson Y, Sharma S. Immunology of preeclampsia. Chem Immunol Allergy. 2005;89:49–61. doi: 10.1159/000087912. [DOI] [PubMed] [Google Scholar]
- 11.Mahmoud F, Omu A, Abul H, El-Rayes S, Haines D. Lymphocyte subpopulations in pregnancy complicated by hypertension. J Obstet Gynaecol. 2003;23:20–26. doi: 10.1080/0144361021000043163. [DOI] [PubMed] [Google Scholar]
- 12.Seaberg EC, Munoz A, Lu M, Detels R, Margolick JB, Riddler SA, Williams CM, Phair JP. Association between highly active antiretroviral therapy and hypertension in a large cohort of men followed from 1984 to 2003. Aids. 2005;19:953–960. doi: 10.1097/01.aids.0000171410.76607.f8. [DOI] [PubMed] [Google Scholar]
- 13 **.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. doi: 10.1084/jem.20070657. Using genetically altered mice, the authors demonstrate an essential role of T cells in the genesis of hypertension. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nava M, Quiroz Y, Vaziri N, Rodriguez-Iturbe B. Melatonin reduces renal interstitial inflammation and improves hypertension in spontaneously hypertensive rats. Am J Physiol Renal Physiol. 2003;284:F447–454. doi: 10.1152/ajprenal.00264.2002. [DOI] [PubMed] [Google Scholar]
- 15.Rodriguez-Iturbe B, Quiroz Y, Nava M, Bonet L, Chavez M, Herrera-Acosta J, Johnson RJ, Pons HA. Reduction of renal immune cell infiltration results in blood pressure control in genetically hypertensive rats. Am J Physiol Renal Physiol. 2002;282:F191–201. doi: 10.1152/ajprenal.0197.2001. [DOI] [PubMed] [Google Scholar]
- 16.Franco M, Martinez F, Quiroz Y, Galicia O, Bautista R, Johnson RJ, Rodriguez-Iturbe B. Renal angiotensin II concentration and interstitial infiltration of immune cells are correlated with blood pressure levels in salt-sensitive hypertension. Am J Physiol Regul Integr Comp Physiol. 2007;293:R251–256. doi: 10.1152/ajpregu.00645.2006. [DOI] [PubMed] [Google Scholar]
- 17.Muller DN, Shagdarsuren E, Park JK, Dechend R, Mervaala E, Hampich F, Fiebeler A, Ju X, Finckenberg P, Theuer J, Viedt C, et al. Immunosuppressive treatment protects against angiotensin II-induced renal damage. Am J Pathol. 2002;161:1679–1693. doi: 10.1016/S0002-9440(10)64445-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bravo Y, Quiroz Y, Ferrebuz A, Vaziri ND, Rodriguez-Iturbe B. Mycophenolate mofetil administration reduces renal inflammation, oxidative stress, and arterial pressure in rats with lead-induced hypertension. Am J Physiol Renal Physiol. 2007;293:F616–623. doi: 10.1152/ajprenal.00507.2006. [DOI] [PubMed] [Google Scholar]
- 19.Herrera J, Ferrebuz A, Macgregor EG, Rodriguez-Iturbe B. Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J Am Soc Nephrol. 2006;17:S218–225. doi: 10.1681/ASN.2006080918. [DOI] [PubMed] [Google Scholar]
- 20 **.Henke N, Schmidt-Ullrich R, Dechend R, Park JK, Qadri F, Wellner M, Obst M, Gross V, Dietz R, Luft FC, Scheidereit C, et al. Vascular endothelial cell-specific NF-kappaB suppression attenuates hypertension-induced renal damage. Circ Res. 2007;101:268–276. doi: 10.1161/CIRCRESAHA.107.150474. Using mice transgenic for a dominant active form of I kappa B, the authors show that inflammation is essential for the renal damage and proteinuria that accompanies experimental hypertension. [DOI] [PubMed] [Google Scholar]
- 21 *.Ghosh SS, Massey HD, Krieg R, Fazelbhoy ZA, Ghosh S, Sica DA, Fakhry I, Gehr TW. Curcumin ameliorates renal failure in 5/6 nephrectomized rats: role of inflammation. Am J Physiol Renal Physiol. 2009;296:F1146–1157. doi: 10.1152/ajprenal.90732.2008. The authors used a pharmacological approach to demonstrate a role of the transcription factor nuclear factor kappa B in modulation renal inflammation in a model of renal insufficiency and hypertension. [DOI] [PubMed] [Google Scholar]
- 22 *.Park JK, Mervaala EM, Muller DN, Menne J, Fiebeler A, Luft FC, Haller H. Rosuvastatin protects against angiotensin II-induced renal injury in a dose-dependent fashion. J Hypertens. 2009;27:599–605. doi: 10.1097/HJH.0b013e32831ef369. This paper demonstrates a previously undiscovered role of an HMG CoA reductase inhibitor in modulating renal inflammation in angiotensin II-induced hypertension. [DOI] [PubMed] [Google Scholar]
- 23 *.Ikeda H, Tsuruya K, Toyonaga J, Masutani K, Hayashida H, Hirakata H, Iida M. Spironolactone suppresses inflammation and prevents L-NAME-induced renal injury in rats. Kidney Int. 2009;75:147–155. doi: 10.1038/ki.2008.507. This study shows that aldosterone likely also contributes to renal inflammation in experimental hypertension. [DOI] [PubMed] [Google Scholar]
- 24 **.Tran LT, MacLeod KM, McNeill JH. Chronic etanercept treatment prevents the development of hypertension in fructose-fed rats. Mol Cell Biochem. 2009;330:219–228. doi: 10.1007/s11010-009-0136-z. This study illustrates a role of Ttumor necrosis factor alpha (TNFα) in promoting hypertension in a model of the metabolic syndrome. [DOI] [PubMed] [Google Scholar]
- 25 **.Elmarakby AA, Quigley JE, Imig JD, Pollock JS, Pollock DM. TNF-alpha inhibition reduces renal injury in DOCA-salt hypertensive rats. Am J Physiol Regul Integr Comp Physiol. 2008;294:R76–83. doi: 10.1152/ajpregu.00466.2007. This paper demonstrates that TNFα contributes to renal injury in this salt and volume dependent form of hypertension. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schrader LI, Kinzenbaw DA, Johnson AW, Faraci FM, Didion SP. IL-6 deficiency protects against angiotensin II induced endothelial dysfunction and hypertrophy. Arterioscler Thromb Vasc Biol. 2007;27:2576–2581. doi: 10.1161/ATVBAHA.107.153080. [DOI] [PubMed] [Google Scholar]
- 27 **.Didion SP, Kinzenbaw DA, Schrader LI, Chu Y, Faraci FM. Endogenous interleukin-10 inhibits angiotensin II-induced vascular dysfunction. Hypertension. 2009;54:619–624. doi: 10.1161/HYPERTENSIONAHA.109.137158. Using mice lacking the TH2 cytokine IL-10, the authors showed that this is important in protecting against endothelial dysfunction in angiotensin II-induced hypertension. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Witowski J, Ksiazek K, Jorres A. Interleukin-17: a mediator of inflammatory responses. Cell Mol Life Sci. 2004;61:567–579. doi: 10.1007/s00018-003-3228-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schnyder-Candrian S, Togbe D, Couillin I, Mercier I, Brombacher F, Quesniaux V, Fossiez F, Ryffel B, Schnyder B. Interleukin-17 is a negative regulator of established allergic asthma. J Exp Med. 2006;203:2715–2725. doi: 10.1084/jem.20061401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31 **.Madhur M, Lob HE, McCann LA, Iwakura Y, Biinder Y, Guzik TJ, Harrison DG. Interleukin 17 Promotes Angiotensin II-induced Hypertension and Vascular Dysfunction. Hypertension. doi: 10.1161/HYPERTENSIONAHA.109.145094. In press. This paper shows that hypertension is associated with an increase in circulating TH17 cells and that IL-17 plays an important role in sustaining hypertension in response to prolonged angiotensin II infusion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32 **.Lob HE, Marvar PJ, Guzik TJ, Sharma S, McCann LA, Weyand C, Gordon FJ, Harrison DG. Induction of Hypertension and Peripheral Inflammation by Reduction of Extracellular Superoxide Dismutase in the Central Nervous System. Hypertension. 2009 doi: 10.1161/HYPERTENSIONAHA.109.142646. The authors used a novel mouse that allowed induction of oxidative stress targeted to the circumventricular organs. This promoted hypertension and enhanced peripheral T cell activation, illustrating an important link between central stimulation and inflammation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ganta CK, Lu N, Helwig BG, Blecha F, Ganta RR, Zheng L, Ross CR, Musch TI, Fels RJ, Kenney MJ. Central angiotensin II-enhanced splenic cytokine gene expression is mediated by the sympathetic nervous system. Am J Physiol Heart Circ Physiol. 2005;289:H1683–1691. doi: 10.1152/ajpheart.00125.2005. [DOI] [PubMed] [Google Scholar]
- 34.Ishizaka N, Aizawa T, Ohno M, Usui Si S, Mori I, Tang SS, Ingelfinger JR, Kimura S, Nagai R. Regulation and localization of HSP70 and HSP25 in the kidney of rats undergoing long-term administration of angiotensin II. Hypertension. 2002;39:122–128. doi: 10.1161/hy1201.096818. [DOI] [PubMed] [Google Scholar]
- 35.Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H, Holland SM, Harrison DG. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension. 2002;40:511–515. doi: 10.1161/01.hyp.0000032100.23772.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laursen JB, Rajagopalan S, Galis Z, Tarpey M, Freeman BA, Harrison DG. Role of superoxide in angiotensin II-induced but not catecholamine-induced hypertension. Circulation. 1997;95:588–593. doi: 10.1161/01.cir.95.3.588. [DOI] [PubMed] [Google Scholar]