

Abstract
To enhance the quantity and quality of eukaryotic transmembrane proteins (TMPs) available for structure determination by x-ray crystallography, we have optimized protocols for purification of TMPs expressed in the yeast Saccharomyces cerevisiae. We focused on a set of the highest-expressing endogenous yeast TMPs for which there are established biochemical assays. Genes encoding the target TMPs are transferred via ligation-independent cloning to a series of vectors that allow expression of reading frames fused to C-terminal His10 and ZZ (IgG-binding) domains that are separated from the reading frame by a cleavage site for rhinovirus 3C protease. Several TMP targets expressed from these vectors have been purified via affinity chromatography and gel filtration chromatography at levels and purities sufficient for ongoing crystallization trials. Initial purifications were based on expression of the genes under control of a galactose-inducible promoter, but higher cell densities and improved expression have been obtained through use of the yeast ADH2 promoter. Wide variations have been observed in the behavior of different TMP targets during purification-some can be readily purified, while others do not bind efficiently to affinity matrices, are not efficiently cleaved from the matrices, or remain tightly associated with the matrices even after cleavage of the affinity tags. The size, oligomeric state, and composition of purified protein-detergent complexes purified under different conditions were analyzed using a colorimetric assay of detergent concentration and by analytical size exclusion chromatography using static light scattering, refractive index detection, and UV absorption to monitor the elution profiles. Effective procedures were developed for obtaining high concentrations of purified TMPs without excessively concentrating detergents.
Keywords: Membrane proteins, structural genomics, yeast, crystallography, detergents, protein expression
Introduction
Determination of high resolution three dimensional structures of transmembrane proteins remains a major challenge in biochemistry. TMPs1 comprise 20-30% of the coding potential of most genomes, mediate many critical physiological processes and are important targets of a large number of clinically useful drugs. However, the number of unique high-resolution structures of different TMPs (currently approximately 200 [1]) is more than two orders of magnitude lower than the number of known soluble protein structures. Furthermore, structures of TMPs from eukaryotic sources appear to be particularly elusive. As of this writing, the majority of known TMP structures are of proteins derived from prokaryotes and there are only about a dozen known structures of eukaryotic TMPs.
Some of the reasons for the difficulty of TMP structure determination are well known: 1) TMPs generally can not be overexpressed to the same extent as soluble proteins. 2) Purification of particular TMPs away from other membrane components requires solubilization in detergent, which can denature proteins and remove critical co-factors or bound lipids. 3) Association of TMPs with detergent creates relatively large complexes that have proven difficult to characterize by NMR and difficult to crystallize for x-ray crystallography. Despite recent advances in NMR of TMPs [2, 3], the overwhelming majority of available TMP structures have been obtained using crystallography.
The basis for the increased difficulty of structure determination for eukaryotic, compared with prokaryotic, TMPs remains unclear (see[4]). Possible explanations include: 1) Low protein abundance. Many TMPs are present in tissues at low abundances and are difficult to overexpress in heterologous systems. In fact, the first eukaryotic structures were obtained based on proteins purified from native tissues in which they were known to be particularly abundant [5-7]; 2) Incompatibility between eukaryotic TMPs and the bacterial expression systems favored by crystallographers for protein purification. Only one eukaryotic TMP structure based on bacterially expressed protein has been obtained to date [8]; 3) Complexity and heterogeneity of post-translational modifications in eukaryotes. Authentic, homogeneous modifications may be difficult to achieve in heterologous expression systems. On the other hand, the use of mutagenesis for removal of sites of modification, as performed in several structure determinations, may affect protein folding or stability and uniform enzymatic removal of modifications may be difficult to achieve. 4) Requirements for particular lipids or lipophilic co-factors. A recent structure has revealed the presence of cholesterol tightly associated with a purified TMP [9]. 5) Dynamic behavior of eukaryotic membrane proteins. Flexibility of proteins in eukaryotic membrane that may be required for their functions or subcellular targeting may render them unstable in detergent or may make crystallization difficult. Several recent structures have been obtained by generating mutated version of proteins with enhanced stabilities or reduced access to multiple conformational states [9-11].
As part of a project to develop improved approaches for structure determination of eukaryotic TMPs, we have developed a set of procedures for purification of eukaryotic TMPs in the baker's yeast Saccharomyces cerevisiae (Fig. 1). The project has focused on expression of endogenous yeast TMPs based on the following considerations: 1) Previous analysis of a genomic collection of cloned yeast genes had previously identified TMPs that could be expressed to high levels and effectively solubilized [12, 13]. 2) Purification of yeast proteins from an endogenous expression system should minimize problems arising from incorrect or missing post-translational modifications or co-factors. 3) The high degree of characterization of the proteome of S. cerevisiae, compared with other eukaryotes, allows relatively unambiguous identification of TMP targets with assayable functions that are not components of multimeric complexes. 4) S. cerevisiae as a host system for protein expression provides relatively rapid and inexpensive culture conditions, a wide variety of useful engineered host strains, and rapid creation of stably expressing strains.
Fig. 1.

Workflow for expression of yeast transmembrane proteins in yeast.
In developing these procedures for efficient purification of TMPs from yeast, we have had to overcome several classes of technical barriers, including 1) Relatively low yields of protein production and cell mass per unit volume of cultures of S. cerevisiae compared with other hosts such as E. coli or Pichia pastoris; 2) Difficulty of cell lysis arising from the need to disrupt the yeast cell wall; 3) Protein degradation arising from the presence of multiple active proteases in S. cerevisiae; 4) Inefficient affinity purification and tag cleavage for PDCs; 5) Difficulty in minimizing detergent concentrations while maintaining high protein concentrations required for crystallization.
Materials and Methods
The overall work flow of the pipeline for protein production is shown in Fig. 1.
Plasmids and Yeast Strains
Vectors for rapid cloning and expression of TMPs (Fig. 2) were created using a plasmid backbone derived from BG1766 [12]. To create the vector pSGP36, expressing targets under control of a galactose-inducible promoter and containing a consisting of a cleavable RGS-His10 sequence, an insert region shown in Fig. 3A was assembled from overlapping synthetic oligonucleotides, then ligated into BG1766 that had been digested with PmlI-Asp718. This led to loss of the Asp718 site. To provide a G-free region of adequate length for LIC cloning, a 5′ region from the highly expressed yeast PGK1 gene was included as part of the upstream LIC cloning site. Vector pSGP37, expressing proteins under galactose control and containing a ZZ-His6 tag, was created by ligation of a modified insert region assembled from overlapping primers, shown in Fig. 3C to plasmid BG1766 that had been cleaved with PmlI and EagI. A region of BG1766 containing the ZZ tag was then amplified by PCR using a 5′ primer that exactly matched a region of BG1766 upstream of the ZZ domain (CTTCTTACAACAAATATACCAAAATTGTGGTTTGGAGGTTTTGTTTCAAGGTCC) and a 3′ primer (AAAAGGTACCTCAGTGATGGTGATGGTGATGAGAACCTCTACCCTGATGATTCGCGTCT ACTTTCGGC) that added an RGS-His6 tag at the carboxyl terminal of the ZZ region. The product of this PCR reaction was then cleaved with EagI and Asp718 and ligated into similarly cut pSGP37 to create pSGP38 (retaining the Asp718 site).
Fig. 2.

Insert regions of yeast vectors used for expression of transmembrane proteins. Note that the indicated regions are not drawn to scale.
Fig. 3.

Sequences of insertion sites of expression vectors. A) Sequence of insertion region of vector pSGP36 expressing target proteins under galactose control fused to a cleavable C-terminal His10 tag. The thin arrows indicate the cleavage sites of BstXI. The thick arrow shows the site of insertion of assembled insert region into the PmlI site of BG1766 during creation of the vector. 5′ sequences from PGK1 are shown in bold. One base internal to the PGK1 sequences was altered to reduce secondary structure in the synthetic oligonucleotides during assembly. B) Sequence of pSGP36 and pSGP46 modified by insertion of a cloned reading frame. The open arrowhead shows the site of rhinovirus 3C cleavage. C) Sequence of insertion region of pSGP40 expressing proteins under galactose control fused to a C-terminal cleavable ZZ-His10 tag. The thick arrow shows the site of insertion of assembled insert region into the PmlI site of BG1766 during creation of the vector. D) Sequence of the pSGP38 and pSGP40 and pSGP46 following insertion of a reading frame using LIC cloning. The open arrowhead shows the site of rhinovirus 3C cleavage.
Vector pSGP40 containing both ZZ and His10 tags was created by first PCR amplifying a 0.5 kb bp region of pSGP38 using the 5′ primer (CTTCTTACAACAAATATACCAAAATTGTGGTTTGGAGGTTTTGTTTCAAGGTCC) and the 3′ primer (AAAAGGTACCTCAGTGATGGTGATGGTGATGGTGATGGTGATGAGAACCTCTACCCTGA TGATTCGCGTCTACTTTCGGC) that replaces the His6 sequence with 10 histidine codons. The PCR product was digested with Asp718 and EagI, then ligated into similarly cut pSGP37, creating pSGP40.
Vectors pSGP46 and pSGP47 for expressing targeted reading frames under control of the ADH2 promoter (Fig. 2) were created by removing the GAL1 promoter from vectors pSGP40 and pSGP36, respectively, by digestion with SacI and EcoRI. The ADH2 sequence (571 bp) was amplified from S. cerevisiae genomic DNA utilizing a 5′ primer that introduced a Sac1 site (GACATCAGAGCTCAAAACGTAGGGGCAAACAAAC) and a 3′ primer that introduces an EcoR1 site (GAGTCGTGAATTCGTATTACGATATAGTTAATAGTTGATA), followed by ligation of the SacI and EcoRI-cut PCR product into similarly cut pSGP36 and pSGP40.
Yeast host strains used for TMP expression were BJ2168 (ATCC® 208277) MATa prc1-407 prb1-1122 pep4-3 leu2 trp1 ura3-52 gal2, BJ5460 (ATCC® 208285) MATa ura3-52 trp1 lys2-801 leu2-Δ1 his3-Δ200 pep4∷HIS3 prb1-Δ1.6R can1 GAL+, and YM317 MATx leu2 trp1 ura3 prb1-112 pep4-1 his3Δ∷pGAL10-GAL4 [14], and Y258C MATa pep4-3 his4-580, ura3-52, leu2-3,112 [12, 15].
Growth of Yeast for Protein Expression
Vectors expressing targeted reading frames were transformed into yeast strains, maintained on SD-ura selective media [16] and frozen until ready for growth. Prior to expression, yeast cultures were expanded in liquid SD-ura then moved into YPD media [16] for protein expression. Growth of yeast for expression in fermentor cultures was conducted in 9-liter volumes in a BioFlo 3000 fermentor (New Brunswick Scientific). The starting OD600 was between 0.1 to 0.3. After glucose depletion to below 0.35 g/l (based on determination of glucose levels using a Lifescan One Touch Ultra Smart medical blood glucose monitor), protein expression was induced by the addition of 20 % w/v galactose solution to a final concentration of 2% (for reading frames expressed under galactose control) or allowed to progress in the absence of glucose (when the reading frame is expressed under control of the ADH2 promoter). Expression times (from induction) were optimized for the different proteins but were typically in the range of 16-24 hrs. Growth temperatures ranged from 26-30 °C. For ORFs with low expression, cell density and resulting protein production were increased by inoculating final cultures at higher cell densities in the range 1-3 OD600. This was accomplished by growing larger initial cultures on selective media and concentrating cells by centrifugation at 4000 × g for 10 min. before inoculating into YPD medium for expression.
LIC Cloning
For cloning of ORFs into expression vectors, the relevant ORFs were amplified using Phusion™ High-Fidelity DNA Polymerase (NEB) from S. cerevisiae genomic DNA or plasmids from the MORF library [12] as template. To introduce the overlap sequences for LIC cloning, the 5′ primer used for amplification contained the sequence AACACCTTCTTACAACAAATATACCAAAATG-ORF (which includes the ATG) and the 3′ primer contained the sequence AAACAAAACCTCCAAACCACT-ORF. Sufficient sequence complementary to the reading frame was introduced to yield a predicted melting temperature of 63 °C.
Expression vectors, described above, were digested with BstXI, then single stranded regions were created using T4 DNA polymerase, in 20 μl reactions containing 0.037 pmol digested vector, 5 mM DTT, 2.5 mM dGTP, 1× T4 polymerase buffer (Novagen) and 0.2 units of T4 polymerase (LIC certified, Novagen (#70099). Amplified DNAs containing the ORFs to be inserted were gel purified, then similarly treated with T4 DNA polymerase in 20 μl reactions containing 0.12-0.48 pmol PCR product, 5 mM DTT, 2.5 mM dCTP, 1× T4 polymerase buffer and 5 units of polymerase per pmol of DNA. T4 DNA polymerase treatments were allowed to proceed at 22°C for 40 min., then were incubated at 75°C for 15 min. to inactivate the T4 polymerase. T4 polymerase-treated vector and insert were then annealed at 22°C for 5 min. in a reaction containing 5 μl of each, followed by addition of 2.5 μl of 25 mM EDTA and incubation for an additional 5 min. at 22°C. 3 μl of the annealing reaction were transformed into 25 μl Novagen NovaBlue competent cells.
Detergents, Lipids
All detergents were purchased from Anatrace, including Anagrade® dodecyl-β-D-maltopyranoside (DDM), Anapoe® dodecyloctaethylene (C12E8), octyltetraoxyethylene (C8E4), and dodecylnonaethylene (C12E9), Anagrade FOS-choline®-16 (FC-16), N,N-dimethyldodecylamine-N-oxide (LDAO), and N-octyl-β-d-glucopyranoside (OG). Detergents were maintained as 10% (w/v) stocks in water. Polyoxyethylene detergents were purchased as 10 or 25% solutions in water in sealed amber glass ampules under argon and stored in amber glass screw capped vials under argon. All lipids were purchased as chloroform solutions from Avanti Polar Lipids Inc. Prior to addition to purified membrane proteins, the chloroform was removed by evaporation under argon and the lipid was resuspended in the appropriate buffer using sonication by a probe-type sonicator (Misonix Sonicator 3000 with a microtip probe) at the lowest power setting. After lipid addition (to 0.1 mg/ml final concentration) the protein/lipid/detergent mix was incubated at 4 °C for 1 hr.
Cell Lysis
For small scale purifications (from <500 ml of culture), cell lysis was performed by disruption with zirconium/silica beads. Cells were initially pelleted at 4000 × g and resuspended in lysis buffer (1 ml buffer/250 OD×mls culture). The composition of the lysis buffer composition was protein-specific, consisting of 20-50 mM Hepes pH 7.5 or 50 mM Tris-HCl pH 8.0, 150-500 mM NaCl, 5-15 % glycerol, 2 mM β-mercaptoethanol, and protease inhibitors (4 mM pepstatin, 5.2 mM leupeptin, 1mM AEBSF, 3 μM chymostatin. (All protease inhibitors were purchased from Roche Applied Sciences.) 1 ml of resuspended cells was then transferred to 2 ml screw capped tubes and the remaining volume of the tube was filled with zirconium/silica beads (Biospec products, Inc. Cat. # 11079105z). Cells were then disrupted using a BioSpec Products Mini Bead-Beater™ for ten cycles of 20 sec. each with cooling on ice for 1 min. between cycles. For larger scale purifications (>500 ml), cells were lysed using an Avestin EmulsiFlex C3 homogenizer (Avestin International Inc.) fitted with an exit cooling coil immersed in ice water. Cells were pelleted at 4,000 × g, resuspended in ice-cold lysis buffer (3 mls buffer/1 g wet pellet weight), passed through a wire mesh kitchen strainer (to prevent particulate blockage of the homogenizer) and disrupted by one pass through the homogenizer at pressures of 20,000-30,000 psi, maintaining exit lysate temperature below 20 °C. After the first pass, the lysate was centrifuged at 4,000 × g to pellet unbroken cells and cell debris. The pellet from the 4000 × g spin was resuspended in cold lysis buffer (2 mls buffer/1 g wet pellet weight), then subjected to another round of homogenization, followed by an additional centrifugation at 4,000 × g. To prevent accumulation of particulate matter, the Avestin homogenizer was rinsed with 0.5 N NaOH and then water after each use.
Membrane Isolation and Solubilization
Following cell disruption, lysate was centrifuged at 120,000 × g for 1 hr. at 4 °C to isolate cell membranes. The membrane pellets were resuspended in lysis buffer (0.5 mls/g wet pellet weight) using a VWR Power MAX Advanced Homogenizing System at lowest power. If a KCl wash of the membranes was required, the membrane pellets were additionally resuspended in 20 pellet volumes of lysis buffer with 1.2 M KCl using the homogenizer and the membranes were re-centrifuged at 120,000 × g. For detergent solubilization, membrane pellets were first homogenized in lysis buffer with no KCl, then detergent was added at levels greater than the CMC. For detergents with low cmcs such as DDM and C12E8, the final concentration was 1%; for high cmc detergents such as octylglucoside and C8E4, the final concentration was 2%. The membranes were solubilized at room temperature for 1- 2 hrs with agitation using a nutator. Insoluble material was then removed by centrifugation at 16,000 × g for 30 min. at 4°C [17].
Purification of Protein Detergent Complexes
Conditions for binding to affinity matrices were optimized for specific proteins during small scale purification trials, using manufacturers' specifications as initial conditions. Affinity matrices were pre-equilibrated in wash buffer with 0.1% detergent (low cmc detergents) or 0.5% detergent (high cmc detergents) according to manufacturers instructions prior to use. The volumes of affinity matrices used in purifications were generally three times the amounts calculated to be required based on the manufacturers specifications for the particular batch of affinity matrix and the expected amount of target protein in lysates. Buffers used for affinity chromatography contained 20-50 mM Hepes pH 7.5 or 50 mM Tris-HCl pH 8.0, 150 mM NaCl, 5-10 % glycerol, 2 mM β-mercaptoethanol plus detergent (see below) and protease inhibitors (1 mM PMSF, 3 μM chymostatin).
For purifications on IgG-Sepharose resin, detergent-solubilized membranes were bound to pre-equilibrated affinity matrix (GE Healthcare IgG Sepharose 6 Fast Flow) at 4°C for 2-12 hrs. The IgG-Sepharose with bound protein was washed four times with 10 bed volumes of buffer, then the IgG-binding tag was cleaved overnight in one bed volume of wash buffer containing rhinovirus 3C protease that was, itself, tagged with either glutathione S-transferase or 6-His[18]. The 3C protease was purified as described previously [12]. To prevent excess detergent foaming during incubation the suspensions were mixed on a Stuart SRT6D Roller Mixer. After cleavage, additional protein was eluted from the resin by rinsing with four additional bed volumes of buffer. The 3C protease was removed by incubation of eluted fractions with pre-equilibrated Glutathione Sepharose™ 4B resin (GE Healthcare) for 3 hrs at 4°C using twice the bead volume specified by the manufacturer based on protein binding capacity.
For affinity purification using immobilized metal affinity chromatography, imidazole (from a 1 M stock pH 7.7) was added to the solubilized membranes to a final concentration of 5-20 mM prior to resin binding. Detergent-solubilized membranes were then incubated with 0.5-1ml of pre-equilibrated TALON® Metal Affinity Resin (BD Biosciences Clontech) or Ni-NTA His-Bind® resin (Novagen) per mg of target protein in the lysate for 2-16 hours at 4° C. The resin was then washed 2-4 times with 10-25 bed volumes of wash buffer. Protein was eluted in tag-less form using one bed volume of His6-tagged rhinovirus 3C protease in wash buffer so that the protease remained associated with the resin. Alternatively, protein was eluted in tagged form using 4 bed volumes of wash buffer containing 0.2-0.5 M imidazole.
Residual affinity resin was removed from the purified protein by filtration (Corning 0.22 μm cellulose acetate 50 ml tube-top filter). The protein was then concentrated (typically 8-10-fold) using a Millipore Amicon Ultra Regenerated Cellulose concentrator prior to preparative size exclusion chromatography.
Purified and partially purified proteins were quantified by SDS-PAGE, comparing the intensity of Coomassie staining against a standard of known concentration (rhinovirus 3C protease) determined by Bradford analysis (Bio-Rad Protein Assay).
Preparative Size Exclusion Chromatography and Concentration of Samples
Concentrated eluates from affinity purification were subjected to further purification by size exclusion chromatography using a GE Healthcare HiLoad 16/60 Superdex ™ 200 Preparative Grade column in conjunction with a BioRad Biologic DuoFlow chromatography system. The column was pre-equilibrated with 2 bed volumes of size exclusion buffer buffer. Buffers used for size exclusion chromatography were the same as those used for affinity chromatography except that they contained 0.025% sodium azide and detergent at levels that were 2-3 times the cmc for detergents with low cmcs, and approximately equal to the cmc for detergents with high cmcs. The volume of sample loaded was maintained at less than 5% of total column volume. The column was eluted at a flow rate of 0.8 ml/min, collecting 0.8 ml fractions. Protein was detected based on absorbance at 280 nm. Fractions containing the purified protein were combined and concentrated (typically 8-10-fold to 4-10 mg/ml) using Millipore Amicon Ultra Regenerated Cellulose concentrators.
Analytical Triple Detection Size Exclusion Chromatography
Analytical size exclusion chromatography was conducted using a Postnova Analytics PN1122 HPLC pump in conjunction with Shodex KW-803 or 804 size exclusion columns preceded by a Shodex KW-G guard column. Eluate from the columns flowed through a Postnova PN3210 UV/VIS detector, followed by a Precision EnterpriseMDP system containing static light scattering and refractive index and detectors (Precision Detectors Inc.). Protein standards used for calibration were as follows: Carbonic anhydrase (Sigma C7025), 29,000 Da; ε1% 1.83 [19]; aldolase (Sigma A2714), 161,000 Da, ε1% 0.938 [20]; alcohol dehydrogenase (Sigma A8656), 141,000 Da, ε1% 1.26 [21]; chicken albumin (Sigma A5503), 45,000 Da, ε1% 0.735 [22]; bovine serum albumin (Sigma A0281), 66,430 Da; ε1% = 0.67 [23]; apo-transferrin (Sigma T0178), 80,000 Da; ε1% 1.12 [24]. Extinction coefficients at 280 nm for target TMPs were calculated based on amino acid sequences as described previously [25]. Buffers used for TDSEC were the same as for preparative SEC, but generally contained detergent at concentrations near the cmc for the particular detergent.
Small Scale Rapid Affinity Purifications
As an initial test of the suitability of a particular protein to purification and to optimize conditions for protein solubilization and purification, small scale affinity purifications were performed. Detergent-solubilized membranes from a total of 300 −1000 O.D.600 × ml of culture were bound to 30 μl of an affinity resin, washed, and either cleaved from the resin with 3C rhinovirus protease or stripped from the resin. Proteins were stripped either by treatment with one bed volume of a solution of 5% (w/v) SDS and 1 mM PMSF (containing no reducing agents), which leaves most of the IgG bound to Sepharose, or by a 5 min incubation at 37°C in one bed volume of SDS-urea loading buffer consisting of 9 M Urea, 5% β-mercaptoethanol, 0.1 mM EDTA, 40 mM Tris-HCl, pH 6.8, 0.02 mg/mL bromophenol blue and 1 mM PMSF, except where indicated. Treatment with this buffer also resulted in release of significant amounts of IgG from the affinity matrix. Proteins were released from IMAC resins by incubation in one bed volume of either 0.1 M EDTA or 0.5 M imidazole. Eluate was separated from residual affinity beads using 0.22 μ cellulose acetate spin filters (Corning Spin-X). Eluted protein was detected either by SDS-PAGE analysis followed by Coomassie staining or immunoblotting.
Immunoblotting
Immunoblots based on detection of RGS-His-tag sequences were used to track yields of protein at various steps of solubilization and purification. Samples were resuspended in SDS-urea loading buffer, heated at 37°C for 15 min, and electrophoresed on 8-16% or 10% Tris-HCl Criterion™ precast polyacrylamide gels (BioRad). Proteins were transferred to a nitrocellulose membrane (0.45 μm) using a wet transfer system (transfer buffer: 20% methanol, 0.02% SDS, 25 mM Tris Base, 0.2 M glycine, pH 8.1-8.4) overnight. Blots were washed, blocked with 5% newborn calf serum and incubated with antibody, (1:200,000 dilution of Anti-RGS-His-HRP-Conjugate antibody (Qiagen)) according to manufacturer's directions. HRP-labeled proteins were detected with ThermoScientific SuperSignal West Dura Extended Duration Substrate recorded on Kodak X-Omat film.
Colorimetric Detergent Assay
DDM levels in the purified protein were determined by a colorimetric method developed for glycosidic detergents, based on reaction of the sugar moieties with phenol and sulfuric acid [26, 27]. Briefly, 250 μl of 5% phenol were mixed with 50 μl of sample, then 600 μl of concentrated sulfuric acid were added and mixed by vortexing. As the reaction is exothermic, the samples were cooled to room temperature before spectrometric measurements were taken at 490 nm in 1.5 ml polystyrene cuvettes. Detergent standards were prepared in the same buffer used for protein purification (but otherwise lacking detergent), which was also used to dilute the samples of purified protein before the addition of the phenol and sulfuric acid solutions. Reactions were performed in 2 ml Eppendorf Safe-Lock tubes in a fume hood wearing protective gear. The target proteins selected for detailed characterization are either known to contain no glycosylation [28], or were mutated to remove known glycosylation sites [29]. In any case, protein-conjugated glycosidic groups are expected to be present at much lower concentrations than protein bound detergents (for proteins that bind approximately an equal mass of detergent) and thus are not expected to interfere in the detergent assay.
Results
Target selection
Open Reading Frames were initially selected as targets based on the following considerations: 1) High levels of expression in the MORF library, a previously characterized genomic collection of yeast genes cloned into multicopy plasmids under control of a galactose inducible promoter [12, 13]. We targeted ORFs in the “high” and “medium” expression category of Gelperin et al. [12], except in cases of interesting candidates that were missing from that clone collection or cases where there was independent evidence that a protein could be expressed at usable levels, as in the case of Bor1p [28]. Table 1 shows the initial set of targeted ORFs and their expression levels in three different previous studies [12, 30, 31]. It is evident from this table that there are significant discrepancies among the relative levels of expression detected in the different studies. Also, there are numerous examples of non-overlap between the sets of genes tested in these previous studies. 2) Number of predicted transmembrane segments. We targeted ORFs that are predicted to contain at least two transmembrane segments based on the HMMTop algorithm [32]. This was based on the reported inability of some algorithms to distinguish between single pass TMPs and secreted proteins containing signal peptides and on the concern that the transmembrane regions of even bona fide single pass TMPs might not adopt unique stable conformations. 3) Potential for assay of ORF function. We selected ORFs for which a biochemical function was known such that the protein's activity could potentially be assayed in the presence of detergent. This included proteins for which assays of protein activity in detergent had previously been established in the literature, or ORFs for which there exists the potential to adapt published assays to purified proteins in detergent solution. In some cases, ligand binding was considered as a suitable substitute for direct assay of function. Thus, correct folding of purified protein could be assayed based on binding of known inhibitors to adenine nucleotide and boron transporters, or binding of ligands to pheromone receptors. 4) Oligomeric state of the encoded protein. We targeted eliminated ORFs from consideration if there was evidence available in the Saccharomyces Genome Database [33] indicating that they exist as components of stable hetero-oligomeric complexes.
Table 1.
Transmembrane protein targets.
| Gene name | Genetic locus | Function | Relative Expression | Predicted transmembrane segments | |||
|---|---|---|---|---|---|---|---|
| (Gelperin et al., 2005)a | (Osterberg et al., 2006)b | (Ghaemmaghami et al., 2003)c | TMHMM (Krogh et al., 2001) [65] | HMMmiddle (Tusnady and Simon, 2001)[32] | |||
| AAC1 | YMR056C | Adenine nucleotide translocator | Medium | Not tested | Not detected | 4 | 4 |
| AAC3 | YBR085W | Adenine nucleotide translocator | Medium | Not tested | Not detected | 4 | 6 |
| ADP1 | YCR011C | Putative ABC transporter | Medium | 108 | 339 | 8 | 8 |
| ALG7 | YBR243C | Dolichol-P-dependent N-acetylglucosamine-1-P transferase | High | 34 | Not detected | 7 | 12 |
| BOR1 | YNL275W | Boron transport | Low | 111 | 195 | 10 | 12 |
| CHO1 | YER026C | Phosphatidylserine synthase | High | Not tested | Not detected | 2 | 5 |
| DGA1 | YOR245C | Diacylglycerol acyltransferase | Medium | Not tested | 907 | 1 | 4 |
| DPP1 | YDR284C | Diacylglycerol pyrophosphate phosphatase | High | 179 | 3,040 | 5 | 5 |
| ENA5 | YDR038C | P-type ATPase (sodium) | High | Not tested | 606 | 10 | 10 |
| EPT1 | YHR123W | sn-1,2-diacylglycerol ethanolamine-/choline-phosphotranferase | High | Not tested | 1800 | 7 | 8 |
| GPT2/GAT1 | YKR067W | Glycerol-3-phosphate acyltransferase | High | 312 | 3100 | 4 | 4 |
| LPP1 | YDR503C | Lipid phosphate phosphatases | Not tested | 89 | 300 | 4 | 6 |
| NFT1 | YKR103W | ABC multi-drug resistance transporter | Not tested | 6 | Not detected | 15 | 19 |
| PDR10 | YOR328W | Drug resistance ABC protein | Medium | 9 | Not detected | 11 | 15 |
| PIS1 | YPR113W | Phosphatidyl inositol synthase | Medium | 126 | 3,810 | 3 | 3 |
| PMT1 | YDL095W | Protein O-mannosyltransferase | Medium | 155 | 41,500 | 9 | 9 |
| PMT5 | YDL093W | Protein O-mannosyltransferase | High | 119 | Not detected | 11 | 11 |
| PMC1 | YGL006W | Calcium ATPase | High | 109 | 99 | 9 | 11 |
| RCE1 | YMR274C | CAAX protease | High | 16 | Not detected | 7 | 8 |
| RHK1/ALG3 | YBL082C | Dol-P-Man:Man5GlcNAc2-PP-Dol mannosyltransferase | Medium | 7 | Not tested | 9 | 10 |
| SAC1 | YKL212W | Lipid phosphoinositide phosphatase | Medium | 226 | 48,000 | 2 | 2 |
| SRT1 | YMR101C | Cis-prenyltransferase | Medium | Not tested | Not detected | 1 | 3 |
| STE2 | YFL026W | (α-factor receptor) | High | Not tested | Not detected | 7 | 6 |
| STE3 | YKL178C | (a-factor receptor) | High | 255 | Not detected | 7 | 7 |
| STE24 | YJR117W | CAAX protease | Medium | 204 | 19,600 | 4 | 5 |
| YCF1 | YDR135C | Vacuolar ATP dependent GSH transporter | Not tested | 27 | 396 | 14 | 14 |
Expression was characterized as being either low, medium, or high, as described by Gelperin et al., [12].
Expression was characterized based on immunoblotting of tagged protein expressed from a multicopy plasmid under control of a constitutive promoter, evaluated on a scale of 0-925 as described by Osterberg et al., [31].
The number of molecules per cell was characterized based on immunoblotting of tagged protein expressed from a the chromosomal locus as described by Ghaemmaghami et al., [30].
Vectors for TMP expression
To facilitate the expression of TMPs for structural biology, we created a series of expression vectors with the following properties (see Figs. 2 and 3): 1) They allow rapid insertion of a given PCR amplified ORF via Ligation Independent Cloning [34-36] into multiple vectors, following digestion of the vectors with a single restriction enzyme. Single stranded regions of overlap between vector and insert are created using the 3′ to 5′ exonuclease activity of T4 DNA polymerase acting on overlap sequences that lack one of the four nucleotide bases; 2) They allow for expression under control of the inducible GAL1 or ADH2 promoters; 3) They create fusions of target ORFs to C-terminal affinity tags consisting of RGS-His6 or His10 sequences, either alone or in conjunction with protein A ZZ domains. These tags are cleavable by rhinovirus 3C protease. Use of the RGS-His tags (containing the amino acids sequence Arg-Gly-Ser at the amino terminal of the His tag) allows use of an anti-RGS-His antibody that we find is particularly specific and sensitive for probing yeast-expressed proteins. The His6-ZZ tags are similar to those described by Gelperin et al. [12] but have been altered to remove the HA epitope that they originally contained, to allow for proteolytic removal of the polyhistidine tags, and to reduce the number of amino acids remaining after proteolytic removal of tags. Since most signals targeting proteins for secretion to the plasma membrane are found at the N-termini of proteins, we expected the use of C-terminal tags to be generally less perturbing of TMP structure, function, and intracellular targeting than N-terminal tags. In addition, our target selection process was based on existing data on expression of C-terminally tagged TMPs [12, 30, 31].
The procedures we used for LIC cloning make use of the restriction enzyme BstXI, which cleaves outside its recognition sequence. This makes it possible to use LIC cloning to insert PCR products following digestion with a single enzyme, with minimal addition of extra amino acids. As implemented in these vectors, annealing of PCR product and cleaved vector leaves short gaps in the vector-insert junctions that are repaired following transformation into E. coli. As shown in Figure 3, LIC cloning into these vectors leaves the sequence SerGlyLeuGluValLeuPheGln appended to the carboxyl terminal of the target ORF following 3C cleavage. The N-terminal serine and glycine residues in this sequence were selected as a flexible linker to facilitate LIC cloning and promote 3C protease cleavage. The remaining amino acids are the unavoidable remnants of the 3C recognition sequence following cleaving a tag at the carboxyl terminal of a target sequence. No extra amino acids are appended at the amino terminal ends of the cloned ORFs. Efficiency of inserting the desired ORFs into expression vectors via LIC cloning was greater than 90%, and often approached 100%.
Culture conditions for TMP expression
Achieving optimal expression of TMPs in yeast requires maximizing both cell density and the efficiency of induction of expression of the target gene. Our initial approach using the GAL1 promoter was to grow pre-cultures in synthetic medium lacking uracil, to use a large innoculum of this preculture to inoculate fermentor cultures on rich, nonselective medium, to allow the cells to deplete available glucose, then to add galactose for induction. There was little cell growth following induction, however the high cell density allowed efficient protein expression. As shown in Fig. 4, expression levels of many of the target TMPs under control of the GAL1 promoter were sufficient to be readily detected by Coomassie staining following rapid small scale purifications from small volumes of cultures.
Fig. 4.

Expression of targeted yeast ORFs. Yeast strains expressing ORFS fused to C-terminal His6-ZZ tags from the MORF library [12]were cultured in YPD media until glucose depletion then induced with galactose (2%). After solubilization in 0.5% FC-16, cell lysates were subjected to small scale rapid affinity purification on IgG-Sepharose as described in Materials and Methods with elution by treatment with SDS-urea loading buffer. Each lane of the Coomassie-stained gel contained the protein derived from approximately 250 O.D.600 × ml of culture, normalized based on wet pellet weight. The unfilled arrows indicated bands corresponding to expressed target proteins. The filled arrows at right indicate major bands eluted from the IgG Sepharose by SDS treatment even in the absence of loaded protein.
The ADH2 promoter has been reported to provide high levels of protein expression in yeast [37-39]. This promoter is induced by depletion of glucose from cultures. In the cases of several yeast targets, including Bor1p and Ste2p, we find that use of the ADH2 promoter provides an approximately two-fold increase in expression in large cultures, compared with levels of expression achieved using galactose induction. The growth strategy used for targets under control of the ADH2 promoter was similar to that used for galactose induciton, except that there was no need to add galactose following depletion of glucose during fermentor growths.
A significant limitation on protein expression appears to result from the loss of plasmid from cells in culture. Although cultures on rich, non-selective, medium can attain considerably higher cell densities than cultures on selective “drop-out” media, growth on rich medium does not provide selection for cells that retain TMP-encoding plasmids. We monitored the fraction of cells in a culture that retain plasmid with URA3 auxotrophic marker by comparing the number of cells from the culture that give rise to colonies on synthetic medium lacking uracil with the number of cells that form colonies on non-selective medium. Following 18 hours of growth of a strain expressing Ste24p under GAL control under inducing conditions on non-selective medium (see Materials and Methods), only about 35% of the cells retained plasmid. Culturing for shorter times on non-selective medium resulted in higher plasmid retention levels (50-60% for Ste24p after 12 hours of induction). In agreement with previous studies [40] (but contrary to common expectations), we find that a significant proportion of cells in a given culture can lose plasmids even when grown on selective medium, despite the fact that such plasmid loss leads to eventual failure of cells to grow in this medium. For example, we find that in a culture of cells expressing Ste24p under GAL control, the percentage of cells retaining a URA3 plasmid can be as low as 75% following culture to only 1 O.D.600. This is apparently the result of asymmetric plasmid segregation in dividing cells. Because of plasmid loss, as well as poor growth in inducing medium using either the GAL1 or ADH2 promoters, it is advantageous to pre-culture cells to high densities in selective medium, providing a large innoculum for final cultures under inducing conditions.
Lysis of yeast cells
Difficulty of lysis is often considered to be a drawback of yeast expression systems. Isolation of TMPs from membrane fractions limits the choice of techniques for cell disruption to approaches that do not solubilize membranes or fragment them below a size that can be readily pelleted by centrifugation. We find that high pressure disruption using the Avestin EmulsiFlex-C3™ homogenizer is a rapid and convenient approach compatible with TMP isolation. While multiple passes through the homogenizer are required for efficient cell disruption, mechanical and thermal damage to proteins during repeated cycles of shearing can be minimized by conducting a low-speed centrifugation after each pass to separate the disrupted cells in the supernatant from unbroken cells in the pellet so that only the undisrupted cells can be cycled back through the homogenizer.
Yeast Proteases
Yeast cells contain substantial levels of endogenous protease activity. Deletion of the PEP4 gene encoding the Proteinase A of the yeast vacuole has been reported to decrease overall proteolysis because of the role of Pep4p in processing multiple additional pro-forms of proteases in the vacuole [41]. However, we find significant amounts of residual protease activity in pep4- strains that co-fractionates with membranes and solubilized TMPs during affinity chromatography (Fig. 5). While extents of proteolysis vary between preparations, as shown in Fig. 5A, the endogenous proteases in a pep4- yeast strain can be sufficient to completely degrade both the target protein and added 3C protease bound to Talon over a period of a few minutes at 4°C, even when initial lysis is performed in the presence of AEBSF. Significant degradation of target protein and 3C protease was also detected in IgG affinity purifications (Fig. 5B). To circumvent this problem, we investigated the use of a variety of protease inhibitors and the use of host strains with additional deletions of proteolytic enzymes. Initial purifications had been performed in the presence of AEBSF as a general inhibitor of serine proteases. However, we found that PMSF is actually more effective in inhibiting the yeast enzymes. To minimize the volumes of the more toxic PMSF used, we generally perform cell lysis in the presence of 1 mM AEBSF (and, in some cases 4 mM EDTA) and all subsequent steps in the presence of PMSF with no EDTA. We also find the peptide inhibitors chymostatin and antipain to provide additional inhibition of endogenous yeast proteases (results not shown). Inclusion of these protease inhibitors during purification does not significantly inhibit rhinovirus 3C protease used for cleaving of affinity tags.
Fig. 5.
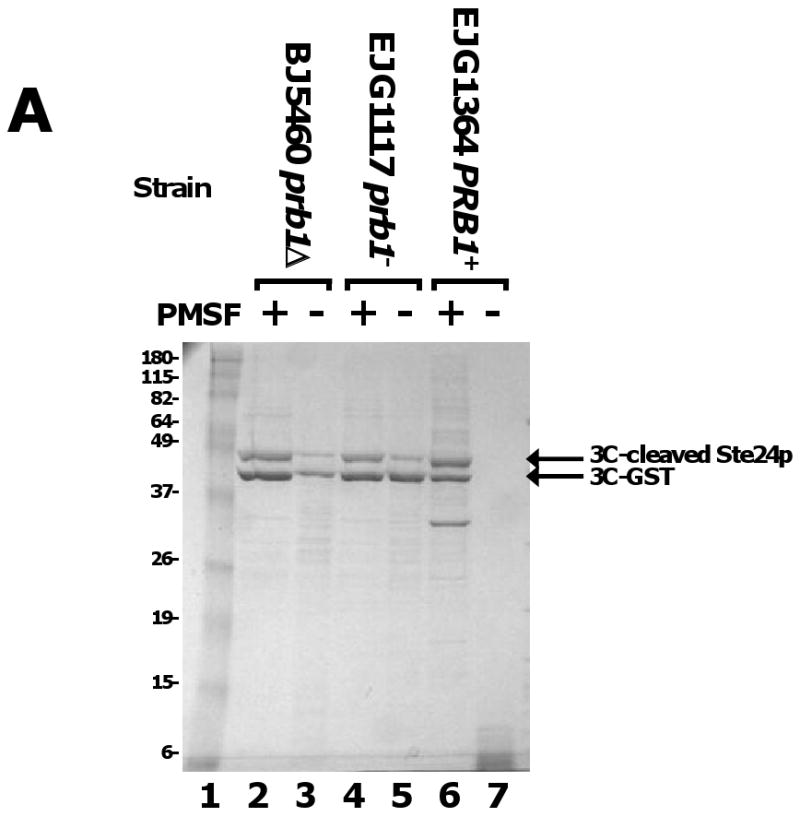
Reduction of activity of endogenous yeast proteases by PMSF and use of protease-deleted strains. A) Inhibition of proteolytic activity in purification of Ste24p. Membranes were prepared from 300 O.D.600 × ml of cultures of the indicated yeast strains expressing Ste24p with a ZZ-His10 tag (vector pSGP40) that had been lysed in the presence 1 mM AEBSF. The membranes were solubilized in 0.5% FC-16 in the presence (lanes 2,4,and 6) or absence (lanes 3,5, and 7) of 170 μg/ml PMSF, bound to 40 μl Talon, then eluted using 5 μg of GST-tagged 3C protease. B) PMSF inhibition of proteolysis in purification of Ste24p. A total of 300 O.D.600 × ml of lysate from yeast strain S258C expressing Ste24p with a ZZ-His10 tag (vector pSGP40), prepared in the presence of 1 mM AEBSF, 2.5 μg/ml leupeptin, and 2.5 μg/ml pepstatin was directly solubilized in 0.5% FC-16 in the absence of protease inhibitors (without preparing a membrane fraction), then bound to 20 μl of IgG-sepharose. The IgG-sepharose was washed in 0.1% FC-16 in the absence (lanes 3-6) or presence (lanes 9-12) of PMSF, then the protein was eluted by treatment with 25μg GST-tagged 3C protease. Following cleavage (elution 1, lanes 3 and 9)), the IgG-sepharose was washed twice with buffer containing 0.1% FC-16 (elutions 2 and 3; lanes 4, 5, 10, and 11). Material remaining on the column following treatment with 3C protease was then eluted by treatment with SDS-urea loading buffer containing (lanes 6 and 12). For reference, the figure also shows the results of SDS-urea treatment of IgG-sepharose onto which no protein had been loaded (lanes 2 and 13). Samples containing 25 μg of purified GST-tagged 3C protease are shown in lanes 7 and 14 for reference.
Use of yeast strains with deletions of protease-encoding genes other than PEP4 also provides significant reductions in proteolytic degradation of purified proteins. As shown in Fig. 5, much (but not all) degradation during purification can be prevented by use of strains with mutations in, or deletions of, the PRB1 gene encoding Proteinase B (see [41]). We have also achieved some additional improvements in yield of some, but not all, purified TMPs using host strain BJ2168 with a deletion of PRC1 encoding vacuolar carboxypeptidase Y (results not shown).
Membrane Fractionation
Membrane proteins can be solubilized either from whole-cell lysates or from isolated membrane fractions. After initial exploration of the use of whole-cell lysates, we found that the use of membrane fractions provided higher yield of more readily purified protein (results not shown). In some cases, washing of isolated membranes in 1.2 M KCl provided additional purification that also resulted in more efficient subsequent affinity purifications, presumably because of removal of bound proteins that interfere with binding to affinity matrices as well as subsequent elution. However, this was not generally necessary.
Solubilization of TMPs
The initial choice of detergents for solubilization was based on a previous large-scale analysis that compared the effectiveness of different detergents for solubilization of yeast TMPs [13], on the set of detergents with successful histories of use for successful structure determination by x-ray diffraction (see the Membrane Protein DataBank: [42], and on literature employing specific detergents for successful purifications of functional forms of particular TMPs. The initial detergents tested in most cases were: C8E4, C12E8, DDM, LDAO, FC16, and OG. Selection of detergents for large scale purifications was based on initial small scale trials in which membrane fractions from 200-300 O.D.600 × ml of cultures were solubilized with different detergents, then subjected to small scale rapid affinity purifications. These purifications used either IgG-Sepharose, with elution either by rhinovirus 3C protease or denaturing solutions containing 5% SDS and 9 M urea, or immobilized metal matrices, with elution by 3C protease or EDTA in the presence of the detergent used for solubilization.
Purification of Protein-Detergent Complexes
Wherever possible, we have purified tagged versions of the proteins by binding to IgG-Sepharose and eluting them by cleavage with rhinovirus 3C protease, which, in some cases provided purification to near-homogeneity (see Fig. 6). However, significant difficulties were encountered in the purification of many membrane proteins that were not encountered in the purification of similarly tagged soluble proteins:
Fig. 6.
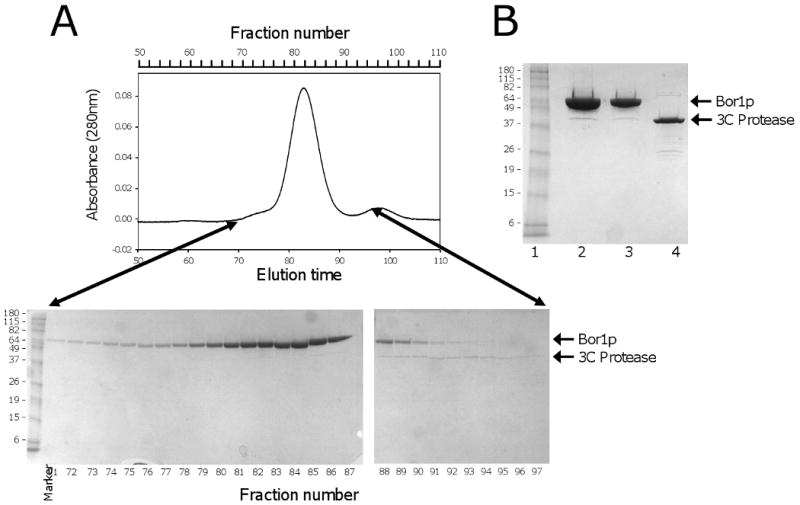
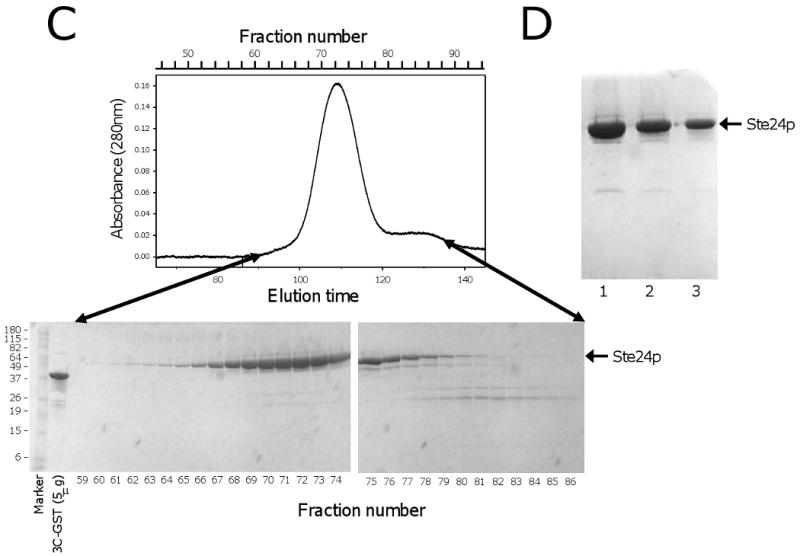
Purification of Bor1p, Ste24p. A)(top) Preparative Superdex 200 gel filtration chromatography during purification of Bor1p expressed from vector pSGP40 and cleaved from IgG Sepharose using GST-tagged 3C protease. The flow rate was 0.8 ml/min. (bottom) SDS polyacrylamide gel electrophoresis of fractions from the gel filtration separation. The molecular masses of marker proteins are indicated in kDa. B) SDS polyacrylamide gel electrophoresis of Bor1p following concentration of the purified protein. Lane 1) Molecular Weight markers; Lane 2) 10 μl purified protein; Lane 3) 5 μl purified protein; Lane 4) 5 μg purified GST-tagged 3C protease. C) Preparative Superdex 200 gel filtration chromatography during purification of Ste24p expressed from the MORF library vector [12] and cleaved from IgG Sepharose using GST-tagged 3C protease. The flow rate was 0.5 ml/min. (bottom) SDS polyacrylamide gel electrophoresis of fractions from the gel filtration separation. D) SDS polyacrylamide gel electrophoresis of Ste24p following concentration of the purified protein. Lane 1) 25 μl purified protein. Lane 2) 12.5 μl purified protein; Lane 3) 6.3 μl purified protein.
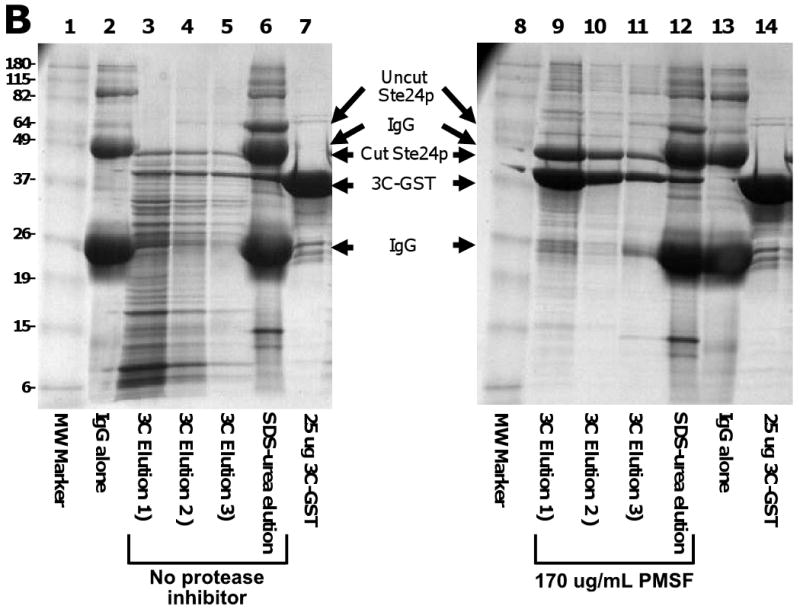
Many TMPs did not bind efficiently to IgG-Sepaharose or IMAC matrices. In fact, we were unable to efficiently bind many TMPs containing the original His6 tags from the MORF library [12]to immobilized metal columns, even though this could routinely be done for similarly-tagged soluble proteins with the same tags [12]. Much more efficient binding to these matrices was obtained using the His10 tags contained in the vectors we have created. These results are different from what has been reported for at least one bacterial TMP [17].
The presence of a low concentration of imidazole (∼20 mM) in the binding reaction was critical for achieving a useful yields and purities of several TMPs in IMAC affinity purifications (Fig. 7). The presence of imidazole had been expected to reduce non-specific binding to the matrix, which was observed, but imidazole also, unexpectedly, enhanced the yield of eluted target protein, perhaps by preventing non-specific binding of untagged proteins to the matrix, thereby increasing the binding capacity for the tagged protein.
Cleavage efficiency of rhinovirus 3C protease in removing C-terminal tags and releasing target proteins from affinity matrices was low. In some cases, as in Fig. 8A, the amounts of protease required for efficient cleavage approached a 1:1 molar stoichiometry with the target protein. Such high protease concentrations are not generally required for cleavage of tags from most soluble proteins with similar or identical cleavage sites [12, 34, 43]. By demonstrating efficient cleavage of soluble proteins in the presence of up to 0.5% FC-12 and FC-16 and up to 1% DDM (results not shown), we were able to determine that this is not generally an effect of detergents on the intrinsic activity of the protease, in contrast to what has been reported for other proteases [44]. Inefficient tag cleavage does not appear to be caused by degradation of rhinovirus 3C protease by endogenous yeast proteases, since the inefficiency persists in samples derived from protease-deficient strains in which the yeast proteases are effectively inhibited. 3C protease appears to be insensitive to most commonly used protease inhibitors except metal chelators. The protease is, however, inhibited by imidazole concentrations greater than 250 mM. In addition, in IMAC affinity purifications where the target protein is to be cleaved from the affinity matrix, we find that cobalt-based matrices allow higher efficiencies of 3C cleavage. Inhibition of cleavage in this case appears to result from an effect of nickel from the matrices. The improved efficiency for cobalt-containing matrices may either reflect weaker inhibition of the enzyme by cobalt or a more stable association of cobalt with the matrix under the conditions used for purification.
Inefficient removal of cleaved proteins from affinity matrices. In some cases, affinity tags could be efficiently proteolytically removed from target TMPs, but the cleaved proteins could not be eluted from the matrices without the addition of denaturing agents such as SDS (Figure 8B), suggesting that the cleaved product is precipitated onto the affinity matrix. At least in the case of Ste2p (shown in Figure 8B), this does not seem to be explained by any intrinsic insolubility of the cleaved product, since versions of the protein with short tags have been purified previously [45, 46] and versions with short tags were also effectively solubilized in our studies (results not shown). A second possible explanation for this failure to elute could be that the target protein exists in an oligomeric state that is bound to the matrix through multiple tags, so that a very high efficiency of cleavage would be required for effective elution. However, this explanation also appears unlikely, since, as shown in Fig. 8B, the actual efficiency of cleavage in these experiments is quite high. A third possibility for the failure to elute, that appears to be the most likely, is that the immobilization of the target protein on the surface of IgG-Sepharose or IMAC matrix leads to high local concentrations that cause non-specific aggregation and precipitation, despite the fact that the same forms of protein might be well-behaved in bulk solution. Preliminary attempts to reduce the density of protein bound to matrix surfaces by increasing the ration of added matrix to bound protein were not successful. This is a problem that could affect tag-based purification of many proteins with tendencies to aggregate or limited solubility under conditions of purification.
Fig. 7.
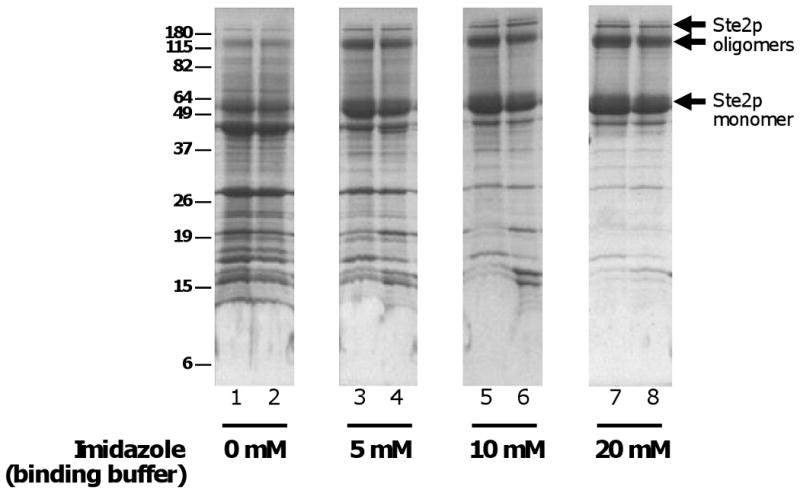
Effects of imidazole on IMAC binding. Membranes from 800 O.D.600 × ml of cells expressing Ste2p tagged with ZZ-His10 (expressed from vector pSGP46) were initially solubilized in 1% FC-16. The expressed allele of STE2 contained the substitutions N25Q and N32Q to prevent glycosylation [29]. The solubilized membranes were applied to 30 μl Talon® in 50 mM Hepes, pH 7.5, 150 mM NaCl, 15% glycerol, 2.5 μg/ml pepstatin A, 1 μg/ml chymostatin, 2.5 μg/ml leupeptin, 1 mM AEBSF, and 2 mM β-mercaptoethanol) in the presence of the indicated concentrations of imidazole, then washed four times (800 μl each time) in buffer that contained the same imidazole concentration but a reduced concentration of FC -16 (0.1%) and only 1 μg/ml chymostatin for protease inhibition. Protein was eluted using 100 μl of similar buffer containing 300 mM imidazole, then concentrated using Strataclean™ (Agilent Technologies). The entire eluate was loaded on the gel, which was stained with Coomassie Blue.
Fig. 8.
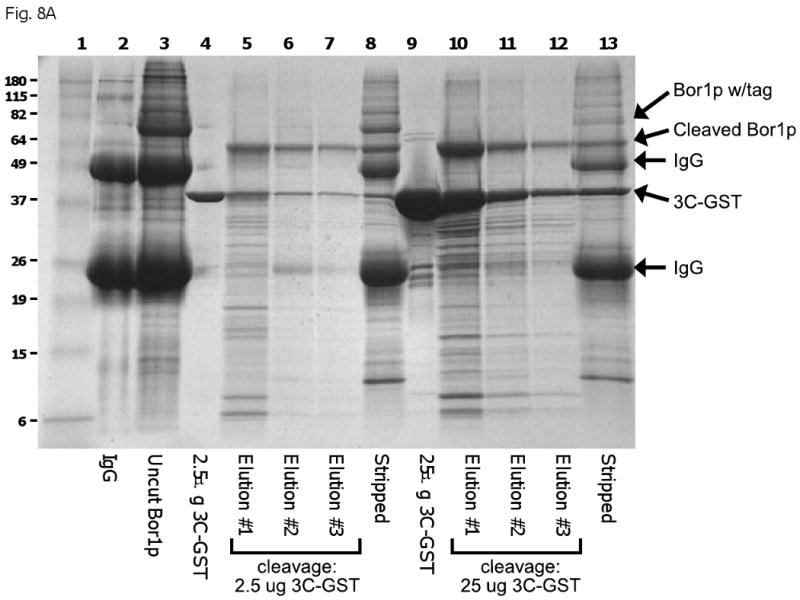
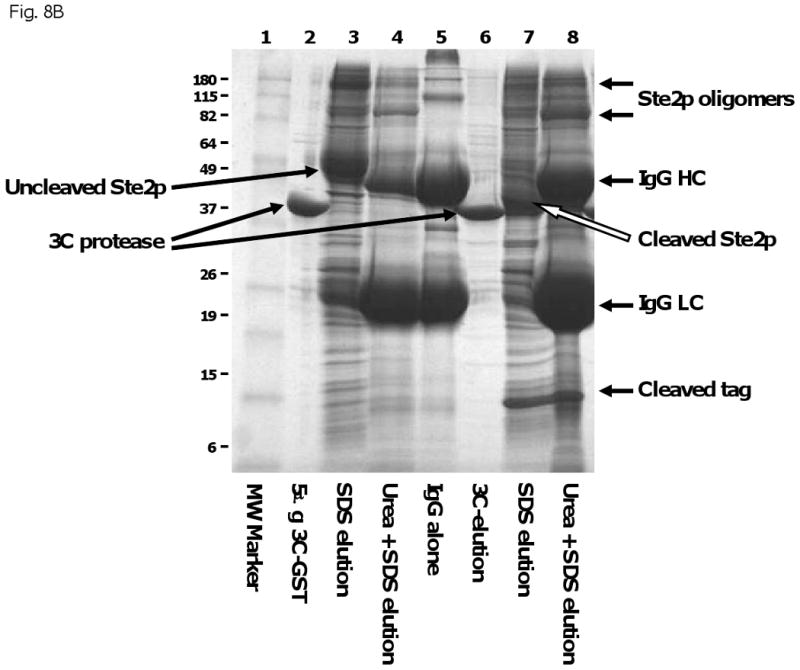
A) Inefficient cleavage from affinity matrix. Bor1p was expressed with a ZZ-His6 tag from the MORF vector BG1766 [12]. The membrane fraction from 300 O.D. 600 × mls was solubilized in 1% DDM, then bound to IgG-Sepharose and eluted by cleavage overnight with the indicated amounts of GST-tagged 3C protease in the presence of 0.01% DDM. The eluted material was solubilized in SDS loading buffer, then the equivalent of 300 O.D.600 × ml from each culture was separated by SDS polyacrylamide electrophoresis followed by staining with Coomassie blue. Lane 2 contains IgG-Sepharose to which no protein had been added, treated with SDS-urea loading buffer containing 2 mM Pefabloc SC. Lane 3 contains Bor1p eluted from IgG-Sepharose using SDS-urea loading buffer rather than 3C protease. Lanes 4 and 9 contain the indicated amounts of purified GST-tagged 3C protease. Lanes 8 and 13 show material that remained on the column after overnight elution with 3C protease but that could subsequently be eluted by treatment with SDS-urea loading buffer. B) Inefficient elution of cleaved protein from IgG. A mutated version of STE2 containing the N25Q and N32Q to prevent glycosylation [29] was expressed with a ZZ-His10 tag from vector pSGP40. The membrane fraction containing 400 μg total membrane protein was solubilized in 1% DDM, applied to 40 μl IgG-Sepharose, washed in 0.1% DDM, then eluted by overnight treatment with GST-tagged 3C protease (lane 6). Following elution with 3C, the material remaining associated with the IgG-Sepharose was eluted by sequential treatment with 5% SDS (lane 7), then SDS-urea loading buffer (lane 8). For reference, the same amount of membrane protein was bound to IgG-Sepharose and eluted directly with 5% SDS (lane 3), followed by SDS-urea buffer (lane 4). Lane 1 contains molecular weight markers, as indicated. Lane 2 contains 5 μg of purified GST-tagged 3C protease. Lane 5 shows a mock elution in which IgG-Sepharose to which no protein had been added was treated with SDS-urea loading buffer.
Detergent concentrations during purification and concentration
Crystallization of proteins generally requires the use of high protein concentrations that approach the solubility limit. This is most generally achieved through the use of ultrafiltration membranes to concentrate samples as a final stage following purification. On the other hand, it is believed by many investigators that crystallization of membrane proteins is optimally achieved at relatively low concentrations of detergent that provide minimal masking of potential protein-protein contacts and minimize inter-micellar interactions involving protein free micelles [47, 48]. Some membrane proteins are also rendered inactive by high detergent concentrations [49]. In addition to these considerations, it is desirable to maintain a well-controlled concentration of detergents to allow for reproducible concentrations for crystallization.
In considering the amount of detergent present in preparations of TMPs, it is important to distinguish between detergent that is directly associated with protein and free detergent in solution, which may be present either as monomer or, if the concentration exceeds the cmc, as micelles. As pointed out by Wiener [48], at protein concentrations typically used for crystallization, the amount of free detergent may be a very small fraction of the total detergent, since most of the detergent will be bound to the protein.
A problem arises from the fact that some detergents that are commonly used for membrane protein crystallization form micelles with sizes that are comparable to the size of the purified protein-detergent complexes. For example, in the case of Bor1p, based on its sequence (including amino acids left after cleavage of the tag), the protein has a molecular mass of 65,900 Da and binds approximately an equal mass of detergent (see below), resulting in a predicted mass of ∼130,000 Da for the protein-detergent complex containing monomeric protein. However, we find it necessary to use an ultrafiltration membrane with a cut-off of 50 kDa to prevent unacceptable losses of protein. By comparison, micelles of DDM have a molecular mass in the range 40,000-75,000 Da and those of C12E8 have a molecular mass in the range 50,000-65,000 Da, according to information provided by Anatrace Inc. Although monomers of detergent can pass through these ultrafiltration membranes, for detergents with low cmcs, and therefore low concentrations of monomers, the rate of loss of monomers is too slow to allow significant passage of detergent mass through the membrane. In addition, a large fraction of the detergent in a preparation of purified TMP exists in the form of protein-detergent complexes that are too large to pass through the membranes. Thus, concentration of purified protein with either 50kDa or 100 kDa cutoff membranes will result in concentration of detergent to high, often ill-defined, concentrations. This problem is compounded for smaller protein targets and by the rather broad molecular weight cut-offs of currently available ultrafiltration membranes. Some of these problems could, eventually, be circumvented by new types of ultrafiltration filters [50].
To maintain a low and controlled detergent concentration in samples prepared for crystallization, we have adopted a strategy of solubilization of membranes at relatively high detergent concentrations, reduction of detergent concentrations during affinity chromatography, further reduction of detergent concentrations during preparative size exclusion chromatography, and the use of repeated cycles of concentration and dilution to achieve high concentrations of protein while minimizing free detergent in the final stages of purification. The use of high detergent concentrations in initial stages of purification is required to achieve effective solubilization of membrane preparations. Repeated washing steps at low detergent concentrations applied to proteins bound to affinity matrices provide a rapid and effective way of removing initial high detergent concentrations. This is also a step at which it is convenient to exchange the partially purified protein from one detergent that may provide effective solubilization, to another detergent that may prove more effective for crystallization.
It is possible to concentrate purified protein-detergent complexes prior to crystallization without accumulating unacceptably high concentrations of detergent by diluting the protein preparations to the point where the free detergent concentration is at or below the cmc. This increases the proportion of detergent monomers in solution, thus increasing the proportion of free detergent that can pass through an ultrafiltration membrane. If the sample can be diluted to the point where the free detergent concentration drops below the cmc, essentially all the free detergent can be removed. The extent to which the preparation can be diluted is limited by lowest detergent concentration that can be achieved without aggregation or precipitation of the protein. However, some proteins appear to be able to tolerate detergent concentrations lower than the cmc, at least for limited lengths of time. Figure 9 shows an example of a concentration step in which a Bor1p-DDM complex was subjected to multiple rounds of dilution and concentration. The first two rounds did not actually bring the DDM concentration below the cmc (∼0.006%) and resulted in removal of only small amounts of detergent. However, subsequent rounds achieved the desired dilution, allowing removal of significant amounts of free detergent, eventually leaving a population of detergent molecules consisting mainly of detergent that is tightly bound to protein and can not readily be removed by further dilution steps.
Fig. 9.

Decrease in levels of DDM in the concentrator concentrate during repeated dilutions and re-concentration of Bor1p. A purified preparation of Bor1p was subjected to repeated cycles of 10-fold dilution with size exclusion buffer containing 0.001% DDM, followed by re-concentration using an Amicon Ultra (50 kDa cut-off) to the original volume. Detergent concentrations were determined by colorimetric assay [27]. The total amount of detergent is shown for a sample in which the volume changes at different steps of concentration. The final concentrations of protein and detergent were 10 mg/ml and 1.8 % w/v in a final volume of 300 μl.
Size exclusion chromatography coupled with UV, light scattering and refractive index detection for characterization of purified proteins
We have used triple-detection size-exclusion chromatography for characterization of the oligomeric state and detergent-binding properties of purified membrane proteins. When calibrated using proteins of known molecular masses and extinction coefficients, TDSEC allows determination of protein molecular mass independent of the hydrodynamic properties of a given protein species [51-53]. It also allows determination of the amount of protein-bound detergent and lipid, as well as detection of lipid and detergent micelles that are not associated with protein [54].
Typical chromatograms from the TDSEC system are shown in Fig. 10. Features of the chromatograms generally include: 1) a light scattering peak or peaks corresponding to the void volume of the column. This peak is unlikely to contain protein since it is observed following injections of detergent-containing buffers without protein (but not following injections of buffer without detergent) and since it typically does not contain any significant components that are detectable by UV absorbance. It is unlikely to contain a significant mass of detergent, since no significant peak of refractive index is detected at this elution volume. Instead, it is apparently is comprised of small amounts of either large detergent aggregates or large particles released during switching of the injection valve in the presence of detergents. The peak is not diminished by pre-filtration of injected samples. 2) One or more protein peaks. These contain protein (detectable by UV absorbance), bound lipid, and detergent. The mass contributions of these different components can be distinguished using the multiple detection systems [51-53]. 3) One or more micelle peaks. These are characterized by significant increases in light scattering and refractive index, but little or no UV absorbance. However, some detergents exhibit detectable absorbances at 280 nm. In some cases, multiple putative micelle peaks are detected. These may reflect different elution times of mixed detergent/lipid micelles compared to pure lipid micelles. The existence of multiple distinct populations indicates that there may be stable (on the time scale of minutes) populations that contain different defined amounts of lipids. In some cases, if the loaded sample contains less detergent than the running buffer or if the sample is loaded under conditions where the cmc is higher than for the running buffer, negative deflections of light scattering and refractive index traces are detected corresponding to depletion of detergent or solutes from the mobile phase due to different mobilities of these components [55]. For accurate determination of protein molecular masses, it is important to establish that the micelle peaks do not significantly overlap with the protein-containing peaks [56]. 4) One or more peaks that elute at or beyond the included volume of the column. This generally results from discrepancies between the concentrations of detergent monomers, glycerol, or salts in comparing the loaded sample with the running buffer. Some small molecule inhibitors used in protein purifications may elute beyond the included volume due to interactions with the column matrix.
Fig. 10.

Elution profiles of purified preparations of 20 μg Bor1p using analytical size exclusion chromatography (TDSEC) on a Shodex KW-803 silica column. The protein used for each panel had been initially solubilized in 1% DDM. A) Static light scattering traces for the protein purified in DDM before and after 4 cycles of ∼10-fold dilution and reconcentration (in buffer containing 0.001% DDM) using a Millipore Amicon regenerated cellulose 50 kDa concentrators. The protein had initially been concentrated 9-fold after elution from the preparative size exclusion column in 0.05% DDM. B) TDSEC of Bor1p following cycles of dilution and reconcentration in DDM containing no added lipid. The protein had been exchanged into 0.1% DDM during affinity chromatography, then to 0.02% DDM during preparative size exclusion chromatography. C) TDSEC for Bor1p in C12E8 with lipid (phosphatidylserine (C18:0,C18:1), 0.1 mg/ml in a solution containing 6 mg/ml protein) added after the final cycles of protein dilution and re-concentration. The protein had been exchanged into 0.05% C12E8 during affinity chromatography, then to 0.03% C12E8 during preparative size exclusion chromatography. D) TDSEC of Bor1p in C12E8 with lipid (phosphatidylserine (C18:0,C18:1), 0.1 mg/ml; (at ∼0.6 mg/ml protein) added prior to the final cycles of dilution and re-concentration. The protein had been exchanged into 0.05% C12E8 during affinity chromatography, then to 0.03% C12E8 during preparative size exclusion chromatography.
We used TDSEC to determine the molecular mass of the protein and of the protein/detergent/lipid complex and the amount of non-protein (detergent, lipid, or carbohydrate) moieties bound to protein purified under different conditions. Chromatograms in Figures 10A and 10B and summarized in Table 2 compare the properties of Bor1p in DDM and C12E8. In each case, the protein was initially solubilized in 1% DDM, however, in one case, the protein was maintained in 0.1% DDM whereas in the other case the protein was transferred to 0.05% C12E8. The protein-only molecular masses determined from the TDSEC measurements indicate that Bor1p is a monomer in DDM and a dimer in C12E8. Consistent with this, the protein elutes from the analytical size exclusion column considerably faster in C12E8 than in DDM, indicating that the hydrodynamic radius of the protein-detergent complex is larger in C12E8. In C12E8 there is approximate agreement between the molecular mass of the complex determined from TDSEC and from mobility on the size exclusion column. However, the protein molecular mass in DDM calculated based on TDSEC is somewhat higher than the calculated monomer molecular mass of the protein and the mass of the protein-detergent complex calculated form TDSEC is higher than that determined based on the size exclusion mobility. This could indicate that the major protein peak actually consists of an incompletely resolved mixture of monomer and dimer. Both the TDSEC analysis and colorimetric assay of DDM concentrations indicate that Bor1p binds slightly more than its own weight in these detergents (Table 2).
Table 2.
Molecular masses of Bor1p protein and protein-detergent complex purified in C12E8 or DDM. Based on the protein sequence, the calculated molecular mass for Bor1p (monomer) including residues of the tag that remain after 3C cleavage is 65.9 kDa.
| Bor1p in DDM | Bor1p in C12E8 (Lipid added after concentration) |
Bor1p in C12E8 (Lipid added prior to concentration) |
|
|---|---|---|---|
| MW Bor1p (TDSEC)a | 80 ± 5 kDab | 150 ± 20 kDa c | 40 ± 20 kDa |
| MW of complex (TDSEC)d | 180 ± 10 kDa | 310 ± 30 kDa | 320 ± 30 kDa |
| MW of complex (SEC mobility)e | 120 ± 5 kDa | 470 ± 30 kDa | 690 ± 60 kDa |
| Detergent/Protein (g/g) (TDSEC)f | 1.2 ± 0.2 | 1.1 ± 0.2 | NDg |
| Detergent/Protein (g/g) (colorimetric)h | 1.1 | NDi | NDi |
Calculated using the triple detector method [52]. Values are indicated as mean ± standard deviation.
n = 2.
n = 25
Calculated using the triple detector method [52]using a value of dn/dc for detergent of 0.143 for DDM and 0.109 for C12E8. In each case, the value of dn/dc for lipid, if present was assumed to be the same as that for detergent.
Calculated based on the mobility of the protein containing peak, compared with the mobilities of the standard proteins (see Materials and Methods), n = 3.
Calculated as the ratio of the protein and lipid components, each determined by the triple detector TDSEC method.
Not determined due to overlap of protein and free lipid/detergent micelle peaks.
Determined by colorimetric assay of fractions from TDSEC chromatography. The amount of detergent in non-peak containing fractions was subtracted from the detergent concentration of the protein-containing peak. The protein concentration was determined based on staining on SDS-PAGE in comparison to known concentrations of purified 3C protease.
Not determined because the colorimetric assay for detergent is based on detection of sugar moieties and, thus is not applicable to C12E8.
During detergent extraction of membrane proteins from the lipid bilayer, bound lipids may be removed, potentially destabilizing the protein. Thus natural or synthetic lipids are sometimes added during purification to improve protein stability and function and in some cases improving crystal diffraction [57-59]. In Fig. 10C, we show the effects of adding back lipid to Bor1p prior to the final cycles of concentration and dilution used for detergent removal from the purified protein. The addition of lipid resulted in a drastic shift of the mobility of the protein-detergent complex on the size exclusion column indicative of a large increase in the size of the complex. The addition of lipid also led to a decrease in the protein-only molecular mass calculated by TDSEC, indicative of a decrease in protein oligomerization. The underlying reason for these increases in complex size are not known, but apparently are related to the process of concentrating protein in the presence of lipids, since such shifts were not seen when the same concentrations of lipids were added to protein after the concentration step (Fig. 10B). Removal of detergent, resulting in loss of mixed lipid/detergent micelles, may lead to accumulation of lipid vesicles or aggregates that associate with the protein.
TDSEC provides a convenient method for evaluating the extent of detergent removal from proteins, based on the relative areas of the protein-detergent peak and non-protein containing detergent/lipid micelle peaks. This is particularly important in cases where there is not a facile method for determining detergent concentration. Fig. 10A is a comparison of TDSEC chromatograms before and after detergent reduction by dilution and concentration as described above that demonstrates effective detergent removal based on the loss of the micelle peak, based on the relative decrease in area of the detergent micelle after detergent reduction.
Reduction of excess detergent concentrations does not seem to alter the mobility or detergent content of the protein-containing peaks, as would be expected if the solutions contained two distinct populations of detergent, one of these comprised free micelles and the other comprised of detergent tightly bound to protein. As reported for detergent-binding studies of bacteriorhodopsin by small-angle neutron scattering and sedimentation equilibration by analytical ultracentrifugation, changes in free detergent concentrations apparently have little effect on the number of detergent molecules binding the membrane protein (Santionicola, et.al., 2008). However, at low detergent concentrations near or below the cmc, the protein may aggregate, resulting in increased high molecular weight oligomers detected by TDSEC, or in a loss in protein signal accompanied by an increase in system backpressure, since the size exclusion column can act as a filter to remove insoluble aggregates.
A limitation of the system we have used for TDSEC, that may also be generally relevant to other systems, is the instability of baseline traces from the detectors when columns are equilibrated with buffers of high viscosity such as those containing high levels of glycerol. High viscosity, combined with the temperature sensitivity of the viscosity, may lead to fluctuations in output traces resulting from pressure fluctuations in the flow cells of the detectors. Performing the analysis at room temperature allows the system to be run at glycerol levels up to 15%. The presence of high glycerol concentrations can also result affect the appearance of micelles peaks in TDSEC, since changing the glycerol concentration of detergent-containing solutions can also change detergent cmcs.
Discussion
We describe here a set of procedures that constitute a pipeline for targeting, expression, and purification of transmembrane proteins from the yeast Saccharomyces cerevisiae. These procedures allow rapid cloning of membrane protein targets into multiple vectors that provide expression under control of the yeast GAL1 or ADH2 promoters and result in fusion of the targets to various combinations of tags at their C-termini. Transmembrane proteins expressed from these vectors can be purified at levels yielding as much as 2 mg per liter of yeast culture. While such yields are lower than can be achieved using prokaryotic expression systems, the yeast-derived transmembrane proteins targets we have expressed can be expected to contain native eukaryotic post-translational modifications and to be associated with native yeast lipids. Furthermore, we find that the yeast-expressed proteins can generally be solubilized with mild detergents [13], in contrast to the general insolubility of eukaryotic membrane proteins expressed in bacteria, particularly the tendency for such proteins to be found in inclusion bodies.
Use of the well-characterized yeast genome as the source for transmembrane protein targets allows selection of proteins that are likely to be capable of expression at high levels. Three previous studies have determined expression levels of endogenous transmembrane proteins in yeast. These include: 1) an analysis of expression levels of integrated copies of GFP fusions [30]; 2) analysis of expression levels of membrane proteins expressed from genes cloned into multicopy plasmids under control of their native promoters [31]; and 3) analysis of expression levels of tagged proteins expressed from genes cloned into multicopy plasmids under control of a constitutively active promoter [12]. To achieve optimum expression levels, we focused on genes that were highly expressed under galactose control. Not all genes that were highly expressed as chromosomal integrants or under control of the constitutive promoter were among the highest expressing when expressed under galactose control. The extent of annotation of the genome of Saccharomyces cerevisiae facilitated the selection of target genes with known functions that are not likely to be components of large complexes. Functional analysis of purified proteins remains an important part of characterization and definition of conditions suitable for crystallization that is beyond the scope of the studies presented here.
While the procedures we have developed allow production of transmembrane proteins on a scale sufficient for crystallization trials, there remains room for improvement; even modest increases in protein yield would be of considerable utility. Some of the current limitations on production that may prove to be most readily overcome are: 1) Cell density in cultures. Our current protocols attain a cell density of 30-35 grams/liter (wet weight of cells). However, protocols for culturing baker's yeast to densities yielding hundreds of grams of cell wet weight per liter have been reported [60]. 2) Promoter strength and induction. We have achieved an increase of approximately two-fold in yield (on a per liter basis) of several different purified proteins through use of the ADH2 promoter compared to a galactose-inducible promoter. Use of the ADH2 promoter also provides simplification of induction conditions, since it is activated by depletion of glucose with no need to add galactose. Expression under ADH2 control also allows use of yeast expression hosts with mutations in the galactose induction pathway. (Such mutations are found in many commonly used strains, including the protease deficient strain BJ2168, used in this study.) Procedures for optimization of ADH2 induction have not been optimized and no general comparative analysis of the best promoter for membrane protein expression in yeast is currently available. 3) Plasmid loss. Plasmid loss during cell culture appears to constitute a significant restriction on levels of TMP expression. Such losses are expected when cultures are grown in rich media that provides the highest cell densities. One approach to overcoming this problem may be multi-copy chromosomal integration of target genes as described by Wittrup and co-workers [61]. Another approach is the engineering of essential genes into expression vectors to allow growth on rich media. However, as noted by others [40], we find that plasmid loss is an ongoing process that occurs even in selective media, resulting in failure of the plasmid-deficient cells to grow. 4) Efficiency of cell lysis. Although we have established protocols that provide usable efficiencies of yeast cell lysis while minimizing exposure of lysates to high-shear environments, a considerable fraction of expressed membrane proteins remains in low speed pellet fractions following cell disruption. This could reflect protein in unlysed cells, or protein that forms large aggregates or is trapped in large organelles at lysis. Such protein could possibly be recoverable. 5) Membrane protein solubilization. While some TMP targets are nearly completely solubilized by the detergents used in this project, some proteins could not be completely solubilized by even the most effective detergents that were tried. Thus, finding an optimum combination of buffer conditions, additives, and detergent types could lead to increases in yield. However, there remains a possibility that protein fractions that are difficult to solubilize may be denatured or non-native, so that increased recovery would actually be undesirable.
The levels of protein production that we have achieved, when expressed on a mg/liter basis, are much lower than those reported for expression of membrane proteins in the methylotrophic yeast Pichia pastoris [49]. However, since Pichia can be cultured to densities of several hundred grams per liter whereas our typical cultures of baker's yeast grow to about 30-35 grams per liter, the amounts of target TMP produced per cell weight in the two types of yeast are comparable. This means that the use of Pichia for expression does not provide any advantage over baker's yeast in terms of the extent of purification required to achieve near homogeneity, as long as sufficient volumes of yeast cells can be cultured. On the other hand, there are numerous advantages to the use of Saccharomyces cerevisiae membrane protein expression, including: 1) The high degree of genetic and proteomic characterization of the genome of Saccharomyces cerevisiae compared to Pichia. Genomes of baker's yeast and related species have been repeatedly sequenced and are readily available in an extensively annotated public database [33]. The full sequence of Pichia pastoris is not publicly available as of this writing. The importance of Saccharomyces cerevisiae as a widely used, tractable, model organism has resulted in extensive application of technologies for characterizing protein-protein interactions, subcellular locations of proteins, expression patterns of proteins, and identification of protein functions in this species. Little of this information is available for Pichia. 2) Ease of cloning and transforming. Plasmid vectors capable of autonomously replicating in yeast cells are generally used for high expression of proteins in Saccharomyces cerevisiae. In contrast, the most commonly used vectors for Pichia are generally integrated into the chromosomes during transformation. Obtaining Pichia transformants that incorporate multiple copies of integrated genes requires achieving high transformation efficiencies in order to select for clones with enhanced antibiotic resistance. 3) Ease of culture. The expression systems we describe here use cultures grown in flasks or fermentors, aerated by ambient oxygen, with expression induced either by depletion of glucose or by transfer to galactose. High density growth of Pichia generally requires use of a fermentor under rigorously controlled conditions to allow initial growth in a carbon source such as glycerol followed by transfer to methanol, often requiring aeration with pure oxygen. 4) Ease of cell lysis. Breakage of the cell wall of baker's yeast is generally easier than for Pichia, requiring less vigorous agitation or lower pressures for treatments of cell pellets. 5) Ease of genetic manipulation and availability of modified strains. A wide variety of strains of Saccharomyces cerevisiae is readily available, including protease deficient strains [41], strains showing enhanced expression under galactose control [62], and strains optimized for incorporation of selenomethionine for x-ray analysis [15]. Only a limited set of protease deficient strains are available in Pichia, and these sometimes associated with slow growth rates and reduced levels of protein expression.
Inefficient affinity purification remains a major problem for some membrane protein targets. In general, binding and recovery from IgG-Sepharose are more efficient that from IMAC resins. Some target proteins we have tested do not efficiently bind to either IMAC or IgG affinity matrices (results not shown). In addition, the affinity tags fused to most targets were inefficiently cleaved compared with what is seen with soluble proteins in similar vectors, and some proteins failed to elute from affinity matrices despite having their tags proteolytically removed. The basis for these effects may, in part, reside in the presence of bound detergent. Detergent is expected to interact primarily with hydrophobic regions of proteins, which should not include the relatively hydrophilic tags. In many cases, the sites of tagging are separated by more than a hundred amino acids from the predicted transmembrane regions. The apparent blocking of binding to affinity matrices and of proteolytic cleavage may be the result of close proximity in three-dimensional space between the locations of the tags and the transmembrane regions or may result from hydrophilic interactions between detergent headgroups and proteins. Blockage of proteolysis of a soluble protein by bound non-ionic detergent has been observed previously[63].
Different detergents can differentially affect the conformation and oligomeric states of proteins. As shown in Fig. 10 and Table 2, the protein molecular mass of the protein detergent complex containing Bor1p is somewhat greater than the predicted monomer molecular mass of 65,900 Da when the protein is maintained in DDM, but is approximately equal to the predicted dimer mass when the protein is transferred to C12E8. Although Bor1p exhibits some sequence similarity to the AE1 anion exchanger [28, which exists as a dimer {Casey, 1991 #76], and has previously been reported to have a tendency to form dimers [28], the functional oligomeric state of Bor1p is not known. While the ability of a protein to elute as a symmetrical monomer peak by size exclusion chromatography is often presented as a desirable hallmark of a TMP suitable for crystallization, it may be that in some cases, a detergent such as DDM may disrupt normal interactions leading to oligomerization, as has been reported for rhodopsin [64]. Bor1p appears to bind approximately the same mass of detergent per mass of protein in its monomeric and oligomeric state (Table 2).
The structure and physical chemistry of protein-detergent complexes in solution and the mechanisms by which such complexes interact to form crystals remain poorly understood aspects of membrane protein structural biology. The TDSEC experiments presented in Fig. 10, as well as others reported previously [56], confirm that detergent is present in solutions containing purified TMPs both as protein-detergent complexes and as free detergent. The free detergent component is comprised of both detergent monomers and micelles consisting of pure detergent or mixtures of detergent and lipids. The ability to reproducibly separate free from bound detergent by size exclusion chromatography indicates that the complexes do not exchange appreciably on the time scale of an approximately 20-minute chromatography run. Consistent with previous characterizations of protein-detergent complexes, the membrane proteins that we have purified bind more than their own mass in detergent. Thus, in samples containing protein at the high concentrations useful for crystallization, protein-bound detergent is present at a significant excess over both the final free detergent concentrations and the concentration of detergent monomers. The stable nature of the protein-detergent complexes we have purified is also supported by the observation that the amount of detergent bound to purified TMPs (as determined by TDSEC) does not show any significant dependence on total detergent concentration in the solution (results not shown.)
Taken together, the approaches we describe allow selection, expression, purification, and characterization of yeast TMP targets for structure determination. These procedures are based on homologous expression of these proteins in yeast cells. The overall yields, concentrations, and purities of the purified proteins are useful for x-ray diffraction analysis. An important aspect of the approach is precise control and monitoring of detergent concentrations and protein oligomeric states, aspects that we anticipate will be useful for exploring conditions for crystallization and for achieving understanding of the underlying process of crystallization of protein-detergent complexes.
Acknowledgments
We thank Drs. Elizabeth Mathew and Shabnam Alam for technical assistance in preparing expression vectors. This work was supported by the Center for High Throughput Structural Biology of the NIGMS Protein Structure Initiative (U54GM074899).
Footnotes
Abbreviations used: transmembrane proteins, TMPs; protein-detergent complex (PDC); ligation-independent cloning, LIC; dodecyl-β-D-maltopyranoside, DDM, dodecyloctaethylene, C12E8, Fos-choline®-16 (FC-16); 4-(2-Aminoethyl)-), benzenesulfonyl fluoride, hydrochloride, AEBSF; phenylmethanesulphonyl fluoride, PMSF; immobilized metal affinity chromatography, IMAC; triple detection size exclusion chromatography, TDSEC; open reading frames, ORFs; critical micelle concentration, cmc
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.White SH. Membrane Proteins of Known Structure. http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html.
- 2.Sanders CR, Sonnichsen F. Solution NMR of membrane proteins: practice and challenges. Magn Reson Chem. 2006;44(Spec No):S24–40. doi: 10.1002/mrc.1816. [DOI] [PubMed] [Google Scholar]
- 3.Hiller S, Wagner G. The role of solution NMR in the structure determinations of VDAC-1 and other membrane proteins. Curr Opin Struct Biol. 2009;19:396–401. doi: 10.1016/j.sbi.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Midgett CR, Madden DR. Breaking the bottleneck: eukaryotic membrane protein expression for high-resolution structural studies. J Struct Biol. 2007;160:265–274. doi: 10.1016/j.jsb.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 5.Harries WE, Akhavan D, Miercke LJ, Khademi S, Stroud RM. The channel architecture of aquaporin 0 at a 2.2-A resolution. Proc Natl Acad Sci U S A. 2004;101:14045–14050. doi: 10.1073/pnas.0405274101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 7.Toyoshima C, Nakasako M, Nomura H, Ogawa H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution. Nature. 2000;405:647–655. doi: 10.1038/35015017. [DOI] [PubMed] [Google Scholar]
- 8.Ferguson AD, McKeever BM, Xu S, Wisniewski D, Miller DK, Yamin TT, Spencer RH, Chu L, Ujjainwalla F, Cunningham BR, Evans JF, Becker JW. Crystal structure of inhibitor-bound human 5-lipoxygenase-activating protein. Science. 2007;317:510–512. doi: 10.1126/science.1144346. [DOI] [PubMed] [Google Scholar]
- 9.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gelperin DM, White MA, Wilkinson ML, Kon Y, Kung LA, Wise KJ, Lopez-Hoyo N, Jiang L, Piccirillo S, Yu H, Gerstein M, Dumont ME, Phizicky EM, Snyder M, Grayhack EJ. Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes Dev. 2005;19:2816–2826. doi: 10.1101/gad.1362105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.White MA, Clark KM, Grayhack EJ, Dumont ME. Characteristics affecting expression and solubilization of yeast membrane proteins. J Mol Biol. 2007;365:621–636. doi: 10.1016/j.jmb.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alexandrov A, Martzen MR, Phizicky EM. Two proteins that form a complex are required for 7-methylguanosine modification of yeast tRNA. RNA. 2002;8:1253–1266. doi: 10.1017/s1355838202024019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malkowski MG, Quartley E, Friedman AE, Babulski J, Kon Y, Wolfley J, Said M, Luft JR, Phizicky EM, DeTitta GT, Grayhack EJ. Blocking S-adenosylmethionine synthesis in yeast allows selenomethionine incorporation and multiwavelength anomalous dispersion phasing. Proc Natl Acad Sci U S A. 2007;104:6678–6683. doi: 10.1073/pnas.0610337104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sherman F. Getting started with yeast. Methods Enzymol. 2002;350:3–41. doi: 10.1016/s0076-6879(02)50954-x. [DOI] [PubMed] [Google Scholar]
- 17.Mohanty AK, Wiener MC. Membrane protein expression and production: effects of polyhistidine tag length and position. Protein Expr Purif. 2004;33:311–325. doi: 10.1016/j.pep.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 18.Alexandrov A, Dutta K, Pascal SM. MBP fusion protein with a viral protease cleavage site: one-step cleavage/purification of insoluble proteins. Biotechniques. 2001;30:1194–1198. doi: 10.2144/01306bm01. [DOI] [PubMed] [Google Scholar]
- 19.Lindskog S, Nyman PO. Metal-Binding Properties of Human Erythrocyte Carbonic Anhydrases. Biochim Biophys Acta. 1964;85:462–474. doi: 10.1016/0926-6569(64)90310-4. [DOI] [PubMed] [Google Scholar]
- 20.Donovan JW. The Spectrophotometric Titration of the Sulfhydryl and Phenolic Groups of Aldolase. Biochemistry. 1964;3:67–74. doi: 10.1021/bi00889a012. [DOI] [PubMed] [Google Scholar]
- 21.Buhner M, Sund H. Yeast alcohol dehydrogenase: SH groups, disulfide groups, quaternary structure, and reactivation by reductive cleavage of disulfide groups. Eur J Biochem. 1969;11:73–79. doi: 10.1111/j.1432-1033.1969.tb00741.x. [DOI] [PubMed] [Google Scholar]
- 22.Foster JF, Yang Jen Tsi. On the Mode of Interaction of Surface Active Cations with Ovalbumin and Bovine Serum Albumin. J Am Chem Soc. 1954;76:1015–1019. [Google Scholar]
- 23.Cohn EJ, Hughes WL, Weare JH. Preparation and Properties of Serum and Plasma Proteins. XIII. Crystallization of Serum Albumins from Ethanol-Water Mixtures. J Am Chem Soc. 1947;69:1753–1761. doi: 10.1021/ja01199a051. [DOI] [PubMed] [Google Scholar]
- 24.Koechlin BA. Preparation and Properties of Serum and Plasma Proteins. XXVIII. The β1-Metal-combining Protein of Human Plasma. J Am Chem Soc. 1952;74:2649–2653. [Google Scholar]
- 25.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 26.Masuko T, Minami A, Iwasaki N, Majima T, Nishimura S, Lee YC. Carbohydrate analysis by a phenol-sulfuric acid method in microplate format. Anal Biochem. 2005;339:69–72. doi: 10.1016/j.ab.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 27.Urbani A, Warne T. A colorimetric determination for glycosidic and bile salt-based detergents: applications in membrane protein research. Anal Biochem. 2005;336:117–124. doi: 10.1016/j.ab.2004.09.040. [DOI] [PubMed] [Google Scholar]
- 28.Zhao R, Reithmeier RA. Expression and characterization of the anion transporter homologue YNL275w in Saccharomyces cerevisiae. Am J Physiol Cell Physiol. 2001;281:C33–45. doi: 10.1152/ajpcell.2001.281.1.C33. [DOI] [PubMed] [Google Scholar]
- 29.Mentesana PE, Konopka JB. Mutational analysis of the role of N-glycosylation in alpha-factor receptor function. Biochemistry. 2001;40:9685–9694. doi: 10.1021/bi0108507. [DOI] [PubMed] [Google Scholar]
- 30.Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 31.Osterberg M, Kim H, Warringer J, Melen K, Blomberg A, von Heijne G. Phenotypic effects of membrane protein overexpression in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2006;103:11148–11153. doi: 10.1073/pnas.0604078103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tusnady GE, Simon I. The HMMTOP transmembrane topology prediction server. Bioinformatics. 2001;17:849–850. doi: 10.1093/bioinformatics/17.9.849. [DOI] [PubMed] [Google Scholar]
- 33.Saccharomyces Genome Database. http://www.yeastgenome.org/
- 34.Alexandrov A, Vignali M, LaCount DJ, Quartley E, de Vries C, De Rosa D, Babulski J, Mitchell SF, Schoenfeld LW, Fields S, Hol WG, Dumont ME, Phizicky EM, Grayhack EJ. A facile method for high-throughput co-expression of protein pairs. Mol Cell Proteomics. 2004;3:934–938. doi: 10.1074/mcp.T400008-MCP200. [DOI] [PubMed] [Google Scholar]
- 35.Aslanidis C, de Jong PJ. Ligation-independent cloning of PCR products (LIC-PCR) Nucleic Acids Res. 1990;18:6069–6074. doi: 10.1093/nar/18.20.6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dieckman L, Gu M, Stols L, Donnelly MI, Collart FR. High throughput methods for gene cloning and expression. Protein Expr Purif. 2002;25:1–7. doi: 10.1006/prep.2001.1602. [DOI] [PubMed] [Google Scholar]
- 37.Lee KM, DaSilva NA. Evaluation of the Saccharomyces cerevisiae ADH2 promoter for protein synthesis. Yeast. 2005;22:431–440. doi: 10.1002/yea.1221. [DOI] [PubMed] [Google Scholar]
- 38.Price VL, Taylor WE, Clevenger W, Worthington M, Young ET. Expression of heterologous proteins in Saccharomyces cerevisiae using the ADH2 promoter. Methods Enzymol. 1990;185:308–318. doi: 10.1016/0076-6879(90)85027-l. [DOI] [PubMed] [Google Scholar]
- 39.Park EH, Shin YM, Lim YY, Kwon TH, Kim DH, Yang MS. Expression of glucose oxidase by using recombinant yeast. J Biotechnol. 2000;81:35–44. doi: 10.1016/s0168-1656(00)00266-2. [DOI] [PubMed] [Google Scholar]
- 40.O'Kennedy RD, Patching JW. Effects of medium composition and nutrient limitation on loss of the recombinant plasmid pLG669-z and beta-galactosidase expression by Saccharomyces cerevisiae. J Ind Microbiol Biotechnol. 1997;18:319–325. doi: 10.1038/sj.jim.2900387. [DOI] [PubMed] [Google Scholar]
- 41.Jones EW. Vacuolar proteases and proteolytic artifacts in Saccharomyces cerevisiae. Methods Enzymol. 2002;351:127–150. doi: 10.1016/s0076-6879(02)51844-9. [DOI] [PubMed] [Google Scholar]
- 42.Membrane Protein Databank. http://www.mpdb.ul.ie/
- 43.Quartley E, Alexandrov A, Mikucki M, Buckner FS, Hol WG, DeTitta GT, Phizicky EM, Grayhack EJ. Heterologous expression of L. major proteins in S. cerevisiae: a test of solubility, purity, and gene recoding. J Struct Funct Genomics. 2009;10:233–247. doi: 10.1007/s10969-009-9068-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mohanty AK, Simmons CR, Wiener MC. Inhibition of tobacco etch virus protease activity by detergents. Protein Expr Purif. 2003;27:109–114. doi: 10.1016/s1046-5928(02)00589-2. [DOI] [PubMed] [Google Scholar]
- 45.Lee BK, Jung KS, Son C, Kim H, VerBerkmoes NC, Arshava B, Naider F, Becker JM. Affinity purification and characterization of a G-protein coupled receptor, Saccharomyces cerevisiae Ste2p. Protein Expr Purif. 2007;56:62–71. doi: 10.1016/j.pep.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shi C, Shin YO, Hanson J, Cass B, Loewen MC, Durocher Y. Purification and characterization of a recombinant G-protein-coupled receptor, Saccharomyces cerevisiae Ste2p, transiently expressed in HEK293 EBNA1 cells. Biochemistry. 2005;44:15705–15714. doi: 10.1021/bi051292p. [DOI] [PubMed] [Google Scholar]
- 47.Screpanti E, Padan E, Rimon A, Michel H, Hunte C. Crucial steps in the structure determination of the Na+/H+ antiporter NhaA in its native conformation. J Mol Biol. 2006;362:192–202. doi: 10.1016/j.jmb.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 48.Wiener MC. A pedestrian guide to membrane protein crystallization. Methods. 2004;34:364–372. doi: 10.1016/j.ymeth.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 49.Lerner-Marmarosh N, Gimi K, Urbatsch IL, Gros P, Senior AE. Large scale purification of detergent-soluble P-glycoprotein from Pichia pastoris cells and characterization of nucleotide binding properties of wild-type, Walker A, and Walker B mutant proteins. J Biol Chem. 1999;274:34711–34718. doi: 10.1074/jbc.274.49.34711. [DOI] [PubMed] [Google Scholar]
- 50.Mehta A, Zydney AL. Effect of membrane charge on flow and protein transport during ultrafiltration. Biotechnol Prog. 2006;22:484–492. doi: 10.1021/bp050324x. [DOI] [PubMed] [Google Scholar]
- 51.Folta-Stogniew E. Oligomeric states of proteins determined by size-exclusion chromatography coupled with light scattering, absorbance, and refractive index detectors. Methods Mol Biol. 2006;328:97–112. doi: 10.1385/1-59745-026-X:97. [DOI] [PubMed] [Google Scholar]
- 52.Hayashi Y, Matsui H, Takagi T. Membrane protein molecular weight determined by low-angle laser light-scattering photometry coupled with high-performance gel chromatography. Methods Enzymol. 1989;172:514–528. doi: 10.1016/s0076-6879(89)72031-0. [DOI] [PubMed] [Google Scholar]
- 53.Wen J, Arakawa T, Philo JS. Size-exclusion chromatography with on-line light-scattering, absorbance, and refractive index detectors for studying proteins and their interactions. Anal Biochem. 1996;240:155–166. doi: 10.1006/abio.1996.0345. [DOI] [PubMed] [Google Scholar]
- 54.Strop P, Brunger AT. Refractive index-based determination of detergent concentration and its application to the study of membrane proteins. Protein Sci. 2005;14:2207–2211. doi: 10.1110/ps.051543805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Felinger A, Pasti L, Dondi F, van Hulst M, Schoenmakers PJ, Martin M. Stochastic theory of size exclusion chromatography: peak shape analysis on single columns. Anal Chem. 2005;77:3138–3148. doi: 10.1021/ac050042b. [DOI] [PubMed] [Google Scholar]
- 56.Slotboom DJ, Duurkens RH, Olieman K, Erkens GB. Static light scattering to characterize membrane proteins in detergent solution. Methods. 2008;46:73–82. doi: 10.1016/j.ymeth.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 57.Guan L, Smirnova IN, Verner G, Nagamori S, Kaback HR. Manipulating phospholipids for crystallization of a membrane transport protein. Proc Natl Acad Sci U S A. 2006;103:1723–1726. doi: 10.1073/pnas.0510922103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 59.Zhang H, Kurisu G, Smith JL, Cramer WA. A defined protein-detergent-lipid complex for crystallization of integral membrane proteins: The cytochrome b6f complex of oxygenic photosynthesis. Proc Natl Acad Sci U S A. 2003;100:5160–5163. doi: 10.1073/pnas.0931431100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alberghina L, Porro D, Martegani E, Ranzi BM. Efficient production of recombinant DNA proteins in Saccharomyces cerevisiae by controlled high-cell-density fermentation. Biotechnol Appl Biochem. 1991;14:82–92. [PubMed] [Google Scholar]
- 61.Parekh RN, Shaw MR, Wittrup KD. An integrating vector for tunable, high copy, stable integration into the dispersed Ty delta sites of Saccharomyces cerevisiae. Biotechnol Prog. 1996;12:16–21. doi: 10.1021/bp9500627. [DOI] [PubMed] [Google Scholar]
- 62.Mylin LM, Hofmann KJ, Schultz LD, Hopper JE. Regulated GAL4 expression cassette providing controllable and high-level output from high-copy galactose promoters in yeast. Methods Enzymol. 1990;185:297–308. doi: 10.1016/0076-6879(90)85026-k. [DOI] [PubMed] [Google Scholar]
- 63.Dumont ME, Richards FM. Insertion of apocytochrome c into lipid vesicles. J Biol Chem. 1984;259:4147–4156. [PubMed] [Google Scholar]
- 64.Jastrzebska B, Maeda T, Zhu L, Fotiadis D, Filipek S, Engel A, Stenkamp RE, Palczewski K. Functional characterization of rhodopsin monomers and dimers in detergents. J Biol Chem. 2004;279:54663–54675. doi: 10.1074/jbc.M408691200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]