

Abstract
In the last decade, significant progress has been made in expanding the scope and depth of publicly available immunological databases and online analysis resources, which have become an integral part of the repertoire of tools available to the scientific community for basic and applied research. Herein, we present a general overview of different resources and databases currently available. Because of our association with the Immune Epitope Database and Analysis Resource, this resource is reviewed in more detail. Our review includes aspects such as the development of formal ontologies and the type and breadth of analytical tools available to predict epitopes and analyze immune epitope data. A common feature of immunological databases is the requirement to host large amounts of data extracted from disparate sources. Accordingly, we discuss and review processes to curate the immunological literature, as well as examples of how the curated data can be used to generate a meta-analysis of the epitope knowledge currently available for diseases of worldwide concern, such as influenza and malaria. Finally, we review the impact of immunological databases, by analyzing their usage and citations, and by categorizing the type of citations. Taken together, the results highlight the growing impact and utility of immunological databases for the scientific community.
Keywords: Database, Epitope, Epitope prediction tools, T cell, Antibody
Introduction
In recent years, immunological databases and analysis resources (DBARs) have become a common tool, widely and increasingly utilized by the biological and immunological research communities. DBARs are utilized by scientists working in academia, biotechnology companies, and large pharma alike, to aid in the design and evaluation of new vaccines, diagnostics, and immunotherapeutics. The basic scientist utilizes them to aid the design and interpretation of experiments probing the nature of host pathogen interactions, autoimmune diseases, cancer, transplantation, and allergies. In addition, bioinformatics scientists utilize immunological databases as a source of data to explore, refine, and develop new tools and algorithms. Finally, it should be underscored that the development of immunological databases has played an important role in the design of formal data ontologies, and their integration within the broad, mainly grass roots efforts to develop a global ontology of biological events and investigations.
For the purpose of this review, we briefly summarize databases and data analysis resources of potential immunological interest and then focus in detail on two main categories of DBARs—databases hosting primary data and experimental details relating to immune epitopes and analysis resources that host tools to analyze such data and/or to predict epitopes or epitope characteristics in unknown antigenic systems. Because of our role in the development of the Immune Epitope Database and Analysis Resource (IEDB), this resource is reviewed in more detail as both a prototype and test case.
The task of compiling a listing of all online resources of potential immunological interest is in and of itself not an easy one, perhaps a testament to the tremendous growth and richness of the field. Herein, a list of over 40 different DBARs (Table 1) has been assembled by compiling resources known to us and immunological databases published in the 2009 Nucleic Acids Research Immunological Database List (www.oxfordjournals.org/nar/database/cat/14). The list has been broadly classified into ten different categories, relating to the scope of each particular DBAR. Table 1 lists each DBAR, its scope, the principal investigator(s), and the year each DBAR was established. In some cases, multiple DBARs with similar scopes were consolidated, such as DBARs with common principal investigators (i.e., Rhagava, Brusic, and Flower), and the various National Institute of Allergy and Infectious Diseases (NIAID) Bioinformatics Resource Centers (Aurrecoechea et al. 2007; Squires et al. 2008; Greene et al. 2007a, b; McNeil et al. 2007; Brinkac et al. 2009; Snyder et al. 2007; Lawson et al. 2007; Greene et al. 2007a, b).
Table 1.
Database and analysis resources of immunological interest
| Database/resource | Scope | Principal investigator(s) | Year est. | Link |
|---|---|---|---|---|
| Immune Epitope Database and Analysis Resource (IEDB) | Epitope | Sette and Peters | 2005 | www.iedb.org |
| SYFPEITHI | Epitope | Rammensee and Stevanovic | 1999 | www.syfpeithi.de |
| JenPep, AntiJen | Epitope | Flower | 2002 | www.jenner.ac.uk/Antijen/ |
| HIV Molecular Immunology Database (Los Alamos) | Epitope | Korber | 1995 | www.hiv.lanl.gov |
| MHCPEP, FIMM | Epitope | Brusic | 1994 | a |
| NetMHC, NetCTL1.0 | Epitope | Lund | 2003 | www.cbs.dtu.dk/ |
| HLA-Ligand | Epitope | Hildebrand | 2003 | a |
| Epitome | Epitope | Schlessinger | 2006 | http://cubic.bioc.columbia.edu/services/Epitome/ |
| EPIMHC/Rankpep | Epitope | Reche | 2005 | http://bio.dfci.harvard.edu/Tools/ |
| SUPERFICIAL | Epitope | Preissner | 2005 | http://bioinformatics.charite.de/superficial/ |
| MAPPP | Epitope | Kaufmann | 2003 | www.mpiib-berlin.mpg.de/MAPPP/ |
| EPIPREDICT | Epitope | Wiesmüller | 2001 | www.epipredict.de/ |
| BCIpep, ProPred, MHCBN, ABCPred, BcePred, HaptenDB | Epitope/haptens | Saha, Bhasin, & Raghava | 2001 | www.imtech.res.in/bic/ |
| SuperHapten | Haptens | Guenther & Preissner | 2007 | http://bioinformatics.charite.de/superhapten/ |
| HPTAA | Human genes and diseases | Wang | 2006 | www.hptaa.org/ |
| IL2Rgbase | Human genes and diseases | Puck | 1996 | http://research.nhgri.nih.gov/scid/ |
| NetChop | Human proteasomes | Brunak | 2002 | www.cbs.dtu.dk/services/NetChop/ |
| PAProc | Human proteasomes | Nussbaum | 2001 | www.paproc.de/ |
| dbMHC | Immunological | Helmberg & Feolo | 2000 | www.ncbi.nlm.nih.gov/gv/mhc/ |
| IPD (ESTDAB, HPA, KIR, MHC) | Immunological | Robinson, Waller, & Marsh | 2006 | www.ebi.ac.uk/ipd/ |
| IMGT (LIGM-DB, MHC-DB, PRIMER-DB, GENE-DB, 3Dstructure-DB) | Immunological | Lefranc | 1989 | www.imgt.org/ |
| VBASE2 | Immunological | Retter | 2005 | www.vbase2.org/ |
| InnateDB | Immunological | Lynn | 2008 | http://innatedb.ca/ |
| Systemsimmunology.org | Immunological | Collaborative program | 2007 | www.systemsimmunology.org/ |
| ImmPort | Immunological | Scheuermann and Karp | 2005 | www.immport.org/ |
| RCSB PDB | Macromolecular structures | Berman, Quesada, and Bourne | 1998 | www.pdb.org |
| MHC–Peptide Interaction Database | Metabolic and signaling pathways | Ranganathan | 2003 | http://surya.bic.nus.edu.sg/mpidt/ |
| TmaDB | Micorarray and gene expression | Sharma-Oates | 2005 | www.bioinformatics.leeds.ac.uk/tmadb/ |
| GPX-macrophage | Micorarray and gene expression | Grimes | 2005 | http://gpxmea.gti.ed.ac.uk/ |
| Interferon Stimulated Gene Database | Micorarray and gene expression | Williams | 2001 | www.lerner.ccf.org/labs/williams/xchip-html.cgi |
| NIAID Bioinformatics Resource Centers | Microbes and infectious diseases | Greene, Collins, Roos, Stevens, Sobral, Scheuermann, White, Lefkowitz | 2006 | www.pathogenportal.org/ |
| MUGEN Mouse Database | Vertebrate genomes | Kollias | 2003 | www.mugen-noe.org/ |
aDatabase/resource is no longer available online
Databases hosting immune epitope data
With respect to databases hosting primary data and experimental details relating to immune epitopes, several different resources should be considered. Some resources such AntiJen (Toseland et al. 2005), FIMM (Schönbach et al. 2005), and HLA-ligand (Sathiamurthy et al. 2003) are not currently maintained and/or the data contained within them were migrated to newer versions and websites. With regard to the scope of the data curated, there is considerable overlap between some of the main databases. However, some clear distinctions can be made. For example, the SYFPEITHI database (Rammensee et al. 1999) currently contains the most comprehensive collection of naturally processed and cancer-derived epitopes. The HIV Molecular Immunology Database (Los Alamos) contains the most comprehensive and highly curated collection of HIV/SIV derived epitopes (Korber et al. 2007). Finally, while the Immune Epitope Database and Analysis Resource (Peters et al. 2005) does not currently curate cancer- and HIV-derived epitopes, it does contain the most comprehensive and highly curated epitope collection relating to infectious diseases, microbes (excluding HIV), allergens, and autoimmunity. It is expected that all transplantation epitopes will become available in the IEDB within the next year.
We sought to perform a comparative analysis of the data housed in each DBAR in terms of references curated, number and types of epitopes and assays, number of antigens and proteins from which the epitopes are derived, and host organisms in which the immune response directed against the epitope originated. This analysis was challenging because, in some cases, the resources are no longer available online, while, in other cases, the data are not available, as the databases can only be searched for specific records, and global searches are not feasible.
With these caveats in mind, the results of our analysis were compiled following thorough examination and querying of each DBAR or extracted from metrics published by the DBARs themselves and are listed in Table 2. Although the bases for the comparisons are IEDB data parameters, a concerted effort was made to retrieve equivalent metrics from the other DBARs. However, the exact definitions of each parameter may not necessarily be consistent among the various DBARs.
Table 2.
Data content in epitope DBARs
| Epitope database/resource | References curated | T cell epitopes | B cell epitopes | T cell response assays | B cell response assays | MHC peptide binding assays | MHC ligand elution assays | Epitope source organisms | Host organisms |
|---|---|---|---|---|---|---|---|---|---|
| Immune Epitope Database and Analysis Resource (IEDB) | 7,840 | 43,258 | 38,552 | 113,232 | 81,084 | 157,021 | 961 | 2,117 | 49 |
| AntiJen | U | 2,960 | U | 3,994 | U | 15,557 | U | U | U |
| SYFPEITHI | 1,134 | 7,782 | N/A | N/A | N/A | N/A | U | U | 15 |
| HIV Molecular Immunology Database (Los Alamos) | 2,538 | U | U | 7,084 | 1,751 | U | U | 1 | 17 |
| MHCBN | 1,519 | 4,907 | N/A | U | N/A | 24,739 | N/A | U | 7 |
| Epitome | U | N/A | 10,180 | N/A | N/A | N/A | N/A | U | U |
| EPIMHC | U | 4,867 | N/A | N/A | N/A | 8,201 | N/A | 19 | 9 |
MHCPEP/FIMM and HLA-ligand are not shown as these resources are no longer available online
N/A not applicable, U unavailable
As shown in Table 2, in terms of curated references, the HIV Molecular Immunology Database hosts data derived from about 2,500 references and the IEDB from about 7,000. Given that the focuses of these two databases are non-overlapping, these two DBARs combined are the most comprehensive in terms of curated references. However, it should be pointed out that neither the IEDB nor the HIV Molecular Immunology Database currently curate cancer references. Other databases, such as MHCBN (Lata et al. 2009), EPIMHC (Reche et al. 2005), and SYFPEITHI can be used to fill this gap.
Perhaps, as a result of its broad focus and comprehensive approach to curation, the IEDB seems to be the most comprehensive repository in terms of number of epitopes and specific assays curated. Exceptions to this are found in the realms of MHC ligand elution assays and information on peptides interacting with TAP. The former are abundantly represented in the SYFPEITHI resource. While actual numbers were not available to us, we estimate that the number of records relating to this type of assay present in SYFPEITHI vastly outnumbers those present in the IEDB. The MHCBN database provides a search interface that enables the user to query for TAP-associated peptides in human, mouse, or rat hosts and provides the results in terms of binding affinity.
In conclusion, each of the DBARs examined has a clear focus in terms of the scope of the data it houses. In the following section, we describe in more detail the IEDB, in whose design and implementation our group has been involved.
The development of a formal ontology for the IEDB
The IEDB is unique within DBARs hosting primary data in two respects. First, the IEDB was designed with an experiment-centric view. Rather than hosting lists of epitopes and associated characteristics, the IEDB data structure is based on curation of the actual experimental data associated with a given potential epitope structure. For this reason, the experimental details relating to the organism that represents the source of the epitope and the details relating to the host whose immune system recognized the epitopes are both captured. Likewise, the experimental circumstances surrounding the immunization, the assay system, and the ultimate readout utilized are also captured.
Second, the experimental data are captured in a searchable format, thus allowing the user to select the type of host, experimental procedures, taxonomic domains, or immunological outcome of interest. This circumvents the need for somewhat arbitrary stipulations of how to define an epitope and allows searches to be flexible and adapted to specific questions (Vita et al. 2008).
Soon after work commenced on the IEDB, it became apparent that development of a formal ontology was necessary to accurately represent experimental detail in a relational database, encompassing for each captured experiment as many as 300 different data fields. Formal ontologies, as described in detail elsewhere (Schulze-Kremer 2002; Bard and Rhee 2004) are a formal representation of the different entities encountered in a given domain and their relation to each other. Development of a formal ontology for the IEDB became instrumental in ensuring the uniform and consistent curation of the data, so that different curators could consistently represent different papers and experiments. Ultimately, a formal ontology allowed the representation of complex processes in a computer-readable format and made it possible to integrate the knowledge contained in different databases.
The first version of the IEDB ontology was developed before any information had been curated and was used to guide the design of the database itself (Sathiamurthy et al. 2005). With the database implemented and data being curated, a more formal and comprehensive ontology was developed. This was done in parallel with the initiation of a collaborative project, the Ontology of Biomedical Investigations (OBI), which aims to represent entities necessary to describe investigations in general, such as assays, reagents, and data (Lord et al. 2009).
Thus, the information captured in the IEDB can be described in the same terms as in other resources that also utilize OBI. The specific terms necessary to describe epitopes and their recognition were captured in the ONTology of Immune Epitopes (ONTIE; Greenbaum et al. 2009a, b). Having the IEDB data represented using terms rigorously defined in a formal ontology has facilitated the ability to perform data consistency checks, formulate highly expressive queries, and has enhanced the potential for seamless interoperability with other data resources (Peters and Sette 2007).
Analysis resources: prediction of T cell epitopes
In parallel to databases hosting primary data, a number of online resources provide tools that facilitate predictions of T cell epitopes on the basis of MHC class I and class II binding, their propensity to being transported by TAP, or their generation by proteosomal processing (for class I restricted epitopes only). In terms of predicting MHC binding, the simplest approach is based on motifs describing primary and secondary anchors associated with epitopes or ligands for specific allelic molecules. Several different resources provide motif listings (see SYFPEITHI, Center for Biological Sequence Analysis, the HIV Molecular Immunology Database), but it is widely recognized that predictions based on motifs alone are associated with poor performance because too many potential leads are identified and many epitopes lack canonical motifs (Ruppert et al. 1993). Accordingly, more sophisticated predictive tools have been developed, such as quantitative matrices, artificial neural networks, and support vector machines.
In discussing these types of analytical tools in the context of the various analysis resources, two separate issues can be identified. First, the evaluation of the accuracy and sensitivity of the tools provided by the various resources and second, the breadth of MHC class I and class II molecules for which such predictions are available.
A rigorous evaluation of the performance of the various tools was lacking until recently when side-by-side evaluations of various tools were presented for both MHC class I and class II molecules (Peters et al. 2006; Lin et al. 2008; Wang et al. 2008). In those evaluations, care was taken to ensure that all methods were benchmarked on large and rigorously curated datasets and that the methods were not evaluated using the same datasets utilized for training the algorithms. Rigorous measures of the accuracy, sensitivity, and true predictive value of the algorithms were defined and consistently applied. When such across-the-board, plain-level field evaluations were performed, it was found that, in general, different methods provide relatively similar levels of performance. Within the different methods evaluated, however, non-linear methods such as those using artificial neural networks and consensus tended to provide the best overall performance compared with linear ones (e.g., scoring matrices).
The main determinant of the performance of a specific algorithm, in actuality, appeared to be the amount of data available to train and evaluate the predictions. In that respect, it is predicted that the performance of MHC binding predictions will continue to improve as the quantity of experimental data available to the bioinformatics community continues to steadily increase. As expected, the overall performance of MHC class I predictions was significantly better than their class II counterpart. No systematic evaluations of the value of proteosomal cleavage and TAP transport predictions have been published, but our empirical experience suggests that, although of theoretical relevance, these predictions generally provide little, if any, improvement in performance over predictions of MHC binding alone.
Table 3 provides a summary of the scope of T cell prediction tools available in epitope-related DBARs. The IEDB provides the largest number of predictors, reflective of the fact that multiple predictive methods are offered while also allowing the user to generate consensus predictions, which have been shown to be most effective (Mallios 2003; Wang et al. 2008; Zhang et al. 2009). In terms of breadth of algorithms available, the HIV Molecular Immunology Database provides the most extensive library of MHC class I predictors. However, the only method utilized for prediction is HLA binding motif, which has been shown to be less accurate and sensitive than other methods, such as neural networks and Stabilized Matrix Method (Peters and Sette 2005; Lundegaard et al. 2008). Most other resources are comparable in the breadth (number) of MHC class II allelic predictors. In terms of hosts for which predictors are available, in general, human and murine MHC are the most frequently found. Predictions for other hosts are also offered such as non-human primates (chimpanzee, macaque), rat, and cow.
Table 3.
Tools content in epitope DBARs
| Epitope database/resource | T cell epitope prediction tools (predictors)a | MHC class I prediction methods | MHC class I prediction alleles | MHC class II prediction methods | MHC class II prediction alleles | B cell tools available | Analysis tools available |
|---|---|---|---|---|---|---|---|
| Immune Epitope Database and Analysis Resource (IEDB) | 993 | 6 | 82 | 5 | 67 | 3 | 4 |
| SYFPEITHI | 154 | 1 | 37 | 1 | 6 | N/A | N/A |
| HIV Molecular Immunology Database (Los Alamos) | 205 | 1 | 165 | 1 | 40 | 1 | 12 |
| ProPred, ABCPred, BcePred | 51 | N/A | N/A | 1 | 51 | 8 | N/A |
| Rankpep | 199 | 1 | 86 | 1 | 61 | N/A | N/A |
| MAPPP | 264 | 2 | 44 | N/A | N/A | N/A | N/A |
aThese figures represent the total number of all permutations of prediction methods, prediction alleles, and prediction peptide lengths available
N/A not applicable, U unavailable
In summary, a variety of different analysis resources provide tools that can be utilized to predict class I and class II restricted epitopes, by a number of different methods and for a number of different alleles. However, several areas appear to be worth considering for future developments, and they include improving the performance of class II predictive tools, expanding the breadth of class II alleles for which predictive tools are available and also increasing coverage of host species beyond mice and humans.
The prediction of B cell epitopes
In contrast to the progress made in the realm of MHC binding prediction tools, the prediction of B cell epitopes has, thus far, proven a more challenging task. The performance of various B cell prediction tools was evaluated by Blythe and Flower (2005) and also scrutinized in a specific focus panel (Greenbaum et al. 2007). A key difference from T cell epitope prediction tools is that the specificity associated with MHC molecules is not present, and as such, the prediction methods are developed to be generally applicable irrespective of genetic polymorphisms and species of the immune responses host.
Most algorithms utilized are based on the assumption that epitopes recognized by antibody responses are exposed on protein surfaces and/or are enriched in the content of specific amino acid residues. Accordingly, various combinations of structural predictions, molecular modeling, hydrophilicity, and solvent exposure scales are utilized. At best, the various methods are associated with area under the curve (AUC) values around 0.7 (with 0.5 being the AUC value of random predictions and up to 0.99 and 0.89 for state-of-the-art MHC binding predictions for certain class I and class II alleles, respectively).
Several DBARs [ABCPred and BcePred (Saha and Raghava 2007), IEDB (Ponomarenko et al. 2008)] host state-of-the-art B cell epitope prediction tools. BcePred provides prediction of linear B cell epitopes utilizing the traditional approach based on physicochemical properties, a strategy which has in the past been shown to be only marginally stronger than random (Blythe and Flower 2005). ABCpred, on the other hand, utilizes a more progressive recurrent neural network approach, which, when evaluated on protein sequences not used in the development of its algorithm, was shown to produce relatively better predictive performance (Saha and Raghava 2006). Newer approaches are also being developed, especially in the context of a recent initiative from the NIAID that awarded large-scale B cell epitope discovery contracts with the recommendation to utilize the data generated to improve B cell epitope prediction methods.
Analysis tools: sequence conservation, population coverage, and epitope visualization
The two preceding sections describe bioinformatics tools aimed at the prediction of T and B cell epitopes. In addition, various other types of tools are available to the scientific community. These tools can be collectively designated as analytical tools, as they are designed to assist in understanding the data associated with various epitopes rather than prediction of new ones. Examples of these tools are sequence conservation tools, population coverage tools, and epitope visualization tools.
The HIV Molecular Immunology Database provides a number of analysis tools designed to aid researchers in applying epitope knowledge to vaccine design. For example, the Hepitope tool tests for HLA alleles that are enriched in a set of individuals that react with a set of known reactive peptides. The Epicover tool, which computes how well a potential vaccine cocktail (antigen set) covers potential user-specified epitopes, can also be harnessed for vaccine development (Thurmond et al. 2008). An alignment tool called Epilign is also available and allows the user to align epitopes or functional domains to HIV1, HIV2, or SIV.
The IEDB also hosts several analytical tools. The epitope conservancy tool, for example, enables the user to specify the sequence of a set of epitopes of interest, and the tool can return the degree to which each epitope is conserved in a set of related protein sequences of interest, also specified by the user (Bui et al. 2007a, b). Within the IEDB, a tool also allows users to compute the population coverage projected for a given T cell epitope(s) based on its known HLA restriction or binding characteristics and on the frequency of HLA molecules in different ethnic groups (Bui et al. 2006).
Another class of analytical tools can be collectively designated as epitope visualization tools. These tools range from tools that allow visualizing the location of an epitope or a series of residues within a given 3D structure, to genome browsers that map and visualize the epitope location within different ORFs and their respective location within genomic information (Beaver et al. 2007). MHCBN offers a peptide mapping tool that displays the location of known MHC binders, TAP binders, and T cell epitopes available in MHCBN database on the protein sequence provided by the user.
The curation of immune epitope data
It is becoming more and more apparent that curation of large amounts of biological data is a requisite to the establishment of large data depositories in general. A key element of large-scale curation is the development of objective criteria for curation, which is dependent on development of ontologies, as described above. Another key element is process automation, which is, in turn, dependent upon ontology and objective process development.
These issues apply to the development of biological databases in general and to immunological DBARs in particular. As stated above and described in more detail elsewhere (Vita et al. 2008), the curation of experimental data at the level of detail and granularity required by the IEDB ontology requires the establishment of a rigorous yet objective process to ensure consistency and compatibility with partial automation.
The IEDB process of curating relevant scientific published literature starts with automated PubMed queries that are executed at 3-month intervals. These queries are designed to be broad in nature, in order to capture as many potentially relevant papers as possible. Specifically, of the over 18 million papers listed in the PubMed resource, we have to date identified approximately 145,000 as being potentially relevant. The abstracts of these potentially relevant references are then scanned by automated text classifiers (Wang et al. 2007) and also further inspected by senior immunologists, to select the truly relevant references to continue in the curation process per se.
A total of roughly 24,000 references have been identified at this stage and divided into major reference classes (infectious disease, HIV, autoimmunity, allergy, transplantation, cancer, and “others”). Within each class, each reference is then placed in one of several categories. For example, the autoimmunity class is further categorized into diabetes, multiple sclerosis, rheumatoid arthritis, lupus, etc. Within each category, subcategories are used to more accurately categorize the references. For example, the diabetes category includes subcategories of insulin and GAD. The classes, categories and subcategories, are used to prioritize and organize the curation flow. They have also provided interesting insights relating to global disease morbidity and mortality data. We have found that, in most cases, diseases associated with high morbidity and mortality have been the most studied, while some areas such as dengue, Schistosoma, HSV-2, Bordetella pertussis and Chlamydia trachomatis were associated with far less extensive coverage. These types of analyses may provide a justification for focusing research towards relatively less well-studied yet critical disease areas (Davies et al. 2009).
Following categorization, the references are curated by a staff of doctoral-level curators. Quality control is provided by computer-based validation and by a system of peer review of curated records (Vita et al. 2006). Currently, the curation of microbes and allergen epitopes is essentially complete and up-to-date, while curation of autoimmune and transplant epitopes is ongoing.
Meta-analysis of influenza immune epitope data
A corollary of the availability of large amounts of data in specialized data repositories is that the data itself can be mined to investigate trends that might not be revealed by examining the data included in a given study because of small sample size (Liberati et al. 2009). Meta-analysis of immunological data is particularly effective in revealing, in a given field of research, pathogen, or disease system, which areas have been targeted most extensively by research and which areas conversely represent knowledge gaps. Immune epitope data meta-analyses for a given disease or pathogen facilitate the use of the data, engage community experts, and can lead to formulation of novel hypotheses. Typically, a meta-analysis is based on the inventory of current knowledge of T cell and antibody epitopes, host organisms, disease states, conservancy, and other relevant variables.
In the case of influenza, analysis of the immunologic data available in the literature as of the end of 2006 (Bui et al. 2007a, b) provided a comprehensive catalog of influenza epitopes, thus establishing a resource for investigators wishing to utilize them in basic studies, or in the evaluation of different vaccination strategies or vaccine constructs. The analysis, however, also revealed several gaps existing at that point in time. Relatively few epitopes were defined in birds and non-human primates, and there was a striking paucity of well-defined antibody epitopes, especially in humans. Few epitopes were characterized for their protective potential. Overall, a limited number of epitopes were reported for avian influenza strains and subtypes. Finally, other than HA and NP proteins, there were relatively few epitopes reported for the other influenza proteins. It should be noted that several of these knowledge gaps have since been significantly bridged by researchers from many different groups (Ekiert et al. 2009; Sun et al. 2009; Yu et al. 2008).
An updated analysis of influenza epitope data with special emphasis on swine-origin H1N1 (Greenbaum et al. 2009a, b) examined the sequence of reported epitopes, which, by definition, represent the pool of preexisting immunity in the general human population. As expected, the majority of antibody epitopes were not conserved in the novel swine-origin influenza (S-OIV), supporting the notion that widespread vaccination with an S-OIV-specific vaccine is required to prevent infection in the general populace. However, the majority of the epitopes recognized by CD8+ T cells were completely invariant. Based on these results, it was hypothesized and then experimentally demonstrated that some T cell immunity is preexisting in the general population against S-OIV and of magnitude similar to that preexisting against seasonal H1N1 influenza.
Meta-analysis of immune epitope data of additional pathogens
Additional epitope meta-analyses relating to the knowledge associated with tuberculosis, botulinum/anthrax toxins, malaria, and poxviruses have been produced (Bui et al. 2007a, b; Blythe et al. 2007; Zarebski et al. 2008; Vaughan et al. 2009; Moutaftsi 2010).
While the Mycobacterium tuberculosis genome contains approximately 4,000 potentially expressed proteins, epitopes have only been identified from approximately 150 of them (∼4%). Furthermore, 23 of these proteins contain ten epitopes or more. These 23 proteins account for more than 71% of the total epitopes identified. It is possible that immune responses are highly focused on very few antigens, and the immune system is oblivious to the vast majority of the coding ORFs. More likely, many antigens have not been characterized and investigated, and a genome-wide approach to epitope/antigen identification would reveal many additional antigens. Another noteworthy finding was that, while epitopes have been described for various disease states, such as clinically active versus latent tuberculosis, exposed, but not converted, and BCG vaccinated studies comparing these different patient populations side-by-side in a systematic fashion have been scarce.
A similar analysis revealed a wealth of plasmodial epitope data available for the scientific community (Vaughan et al. 2009), including a total of 1,566 unique epitopes consisting of 892 T cell (mostly CD4+) and 896 B cell. Strikingly, as in the case of mycobacteria, epitopes were derived from relatively few antigens. While antigens from all life cycle stages were represented, most epitopes were derived from antigens expressed at the parasite surface during liver and asexual blood stages. In all, epitope data were available for only 46 plasmodial antigens, and more than 95% of the malaria genome was not represented. As in the case of M. tuberculosis, high throughput epitope/antigen identification might reveal many new promising antigens.
Indeed, analysis of poxviruses and vaccinia virus highlighted that a large number of antigens spanning virtually every ORF can be targeted by immune responses in complex pathogens with a genome composed of more than 200 different ORFs (Moutaftsi et al. 2007; Sette et al. 2008). In conclusion, meta-analysis of epitope data is a novel avenue to provide the scientific community with a “forest” rather than “tree” level view of the content and granularity of the scientific literature related to specific disease indications.
The impact of immunological databases
As described in the previous sections, a number of different DBARs are available to the scientific community. In this and following sections, we explore in more detail the specific impact that these resources have had on the scientific community. We first sought to estimate the impact of each DBAR in terms of the number of PubMed citations of primary publications describing each DBAR. To this end, we assembled a list of publications associated with each DBAR. First, we performed PubMed queries, adopting a uniform and unbiased approach that would facilitate interdatabase comparisons and designed to retrieve publications relevant to each DBAR. Accordingly, each query consisted of the name(s) of the principal investigator(s), in addition to the word “database” followed by a wild-card character. The resulting publication list was then manually inspected to exclude those publications that were obviously irrelevant. The error rate of this approach was analyzed by using the IEDB as a test case. This query strategy produced 35 publications. Cross-checking against our master list, the query successfully retrieved 22 of the 36 (61%) IEDB-related references.
Table 4 lists the number of publications obtained for each DBAR following this procedure, both in absolute terms and in terms of publications/year. It was found that a total of eight DBARs were associated with at least ten different publications, with yearly rates in the 0.67 to 4.5 publications range. Within this group were several different epitope-based DBARs, including SYFPEITHI, AntiJen, BCIpep, and, the HIV Molecular Immunology Database.
Table 4.
Publication and citation metrics for immunologically relevant DBARs
| Database/resource | Publications | Publications/year | Citations | Citations/year |
|---|---|---|---|---|
| Immune Epitope Database and Analysis Resource (IEDB) | 22 | 4.40 | 564 | 112.80 |
| SYFPEITHI | 11 | 1.00 | 1,254 | 114.00 |
| JenPep, AntiJen | 10 | 1.25 | 342 | 42.75 |
| HIV Molecular Immunology Database (Los Alamos) | 10 | 0.67 | 1,091 | 72.73 |
| MHCPEP, FIMM | 9 | 0.56 | 373 | 23.31 |
| NetMHC, NetCTL1.0 | 4 | 0.57 | 94 | 13.43 |
| HLA-Ligand | 2 | 0.29 | 53 | 7.57 |
| Epitome | 1 | 0.25 | 24 | 6.00 |
| EPIMHC | 1 | 0.20 | 18 | 3.60 |
| SUPERFICIAL | 1 | 0.20 | 4 | 0.80 |
| MAPPP | 1 | 0.14 | 50 | 7.14 |
| EPIPREDICT | 1 | 0.11 | 19 | 2.11 |
| BCIpep, ProPred, MHCBN, ABCPred, BcePred, HaptenDB | 12 | 1.50 | 481 | 60.13 |
| SuperHapten | 1 | 0.33 | 5 | 1.67 |
| HPTAA | 1 | 0.25 | 3 | 0.75 |
| IL2Rgbase | 1 | 0.07 | 72 | 5.14 |
| NetChop | 2 | 0.25 | 170 | 21.25 |
| PAProc | 2 | 0.22 | 135 | 15.00 |
| dbMHC | 7 | 0.70 | 2,321 | 232.10 |
| IPD (ESTDAB, HPA, KIR, MHC) | 3 | 0.75 | 72 | 18.00 |
| IMGT (LIGM-DB, MHC-DB, PRIMER-DB, GENE-DB, 3Dstructure-DB ) | 77 | 3.67 | 3,286 | 156.48 |
| VBASE2 | 1 | 0.20 | 14 | 2.80 |
| RCSB PDB | 54 | 4.50 | 19,589 | 1632.42 |
| MHC–Peptide Interaction Database | 2 | 0.29 | 25 | 3.57 |
| TmaDB | 1 | 0.20 | 13 | 2.60 |
| GPX-Macrophage | 1 | 0.20 | 1 | 0.20 |
| Interferon Stimulated Gene Database | 1 | 0.11 | 304 | 33.78 |
| NIAID Bioinformatics Resource Centers | 17 | 4.25 | 197 | 49.25 |
| MUGEN Mouse Database | 1 | 0.14 | 3 | 0.43 |
The number of primary publications for each DBAR was obtained using PubMed queries, and the citations made to these primary publications were quantified using the ISI Web of Knowledge and Google Scholar. Multiple citations from a single paper were tabulated separately. Duplicate citations from the ISI Web of Knowledge and Google Scholar were excluded from the total number, except in cases where there were over 200 citations for a given DBAR publication that made finding duplicates manually intensive. In such cases, the resource with the highest number of citations for a given DBAR publication was used
The number of citations made to these primary publications was also quantified by the ISI Web of Knowledge and Google Scholar. This analysis revealed that immunological and epitope-related databases have a very significant impact. Taken together the 13 main epitope-related DBARs receive on average 466 citations per year, roughly equivalent to one third of the annual citations generated by the RCSB PDB resource, which has been in operation since 1998, has a much broader scope containing over 60,000 structures of all biological molecules and as such is of relevance for immunological and non-immunological applications alike.
Probing the nature of citations to assess database usage
The preceding sections illustrate the breadth of DBARs available, and how these resources are widely utilized and cited in the scientific literature. We were interested in probing the usage of the DBARs in more detail and specifically ascertaining how the online resources are used. While it is not immediately straightforward to establish the specific use associated with each user and visit, analysis of the papers referencing a DBAR can more readily provide insights in this respect.
To this end, the IEDB citations were further evaluated by subdividing them by year and category (general IEDB, analysis resource, and curation/meta-analyses). It was found that citations of general IEDB publications grew at the fastest rate and doubled each of the first 3 years, subsequently reaching a plateau. By contrast, citations of publications relating to the analysis resource as well as the meta-analyses grew at slower rates, but now represent the dominant source of IEDB citations. Citations of the meta-analysis started later than the other two categories (the first meta-analysis was published in 2007), but also appears to grow at a similar rate (data not shown).
Next, each manuscript citing at least one IEDB publication was manually reviewed. We found that those citations fall into several broad categories, such as retrieval of specific T or B cell datasets (20% and 6%, respectively) and utilization of specific tools (24%). A number of references utilized the IEDB to further ontology development and/or integration (8%) or to develop and improve predictive or analytical tools (23%). The distribution of each of the citation categories is shown in Fig. 1.
Fig. 1.
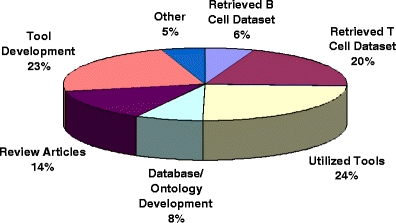
IEDB citation categorization by nature of the citation made
Thus, this analysis suggests that the usage of immunological database and analysis resources is roughly balanced (IEDB dataset retrieval constitutes 26% of citations, while tool usage accounts for 24%). Furthermore, the results indicate that over 80% of all citations are attributable to practical applications of DBARs, either in terms of tool/dataset use or further development of new tools and applications.
Conclusions and discussion
The last decade has witnessed unprecedented growth in the number of publicly available immunological databases and analysis resources (Bourne 2005). Though these resources can be of considerable value to scientists working in myriad settings, the explosive rate of their proliferation presents challenges to those scientists to maintain a clear appreciation of the resources at their disposal. We thus undertook this review to investigate and present the content of various immunological DBARs, the scope of their predictive and analytical capabilities, and their overall impact on the scientific community with the ultimate goal of informing the readers and potentially guiding them to the resource(s) that may be of greatest utility for their research interests.
After compiling a list of resources of potential immunological interest, we systematically examined those DBARs hosting experimental data relating to immune epitopes. Our survey revealed that in terms of data content, each DBAR examined tends to have a clear strength in certain data subsets or disease areas and can, therefore, perhaps better cater to the needs of scientists seeking those particular data. A noteworthy trend among DBARs is the growing integration of formal data ontologies (Noy et al. 2009; Yip 2009). Such standardization has already proven to facilitate interdatabase connections and data sharing, as evidenced by links between resources of diverse focuses. A prime example of this is found in the IEDB, where users can follow links from epitope data to relevant data in BioHealthBase and EuPathDB, which, in turn, also place links from their respective websites to the IEDB. Therefore, it is envisioned that data will become increasingly accessible and integrated with other data resources in the near future.
We further considered the DBARs that provide access to predictive and analytical tools for immune epitope data. Our intent was not specifically to comparatively examine the performance of these tools, as such analyses have been published elsewhere (Mallios 2003; Blythe and Flower 2005; Peters et al. 2005; Peters et al. 2006; Saha and Raghava 2006; Lin et al. 2008; Lundegaard et al. 2008; Wang et al. 2008; Zhang et al. 2009). However, several conclusions about the current state of predictive tools did emerge from this examination. Specifically, our survey highlights clear shortcomings in the predictive tools available. Namely, MHC class II and B cell epitope predictive tools merit improvement, both in terms of predictive performance and, for MHC class II, in terms of coverage of species and alleles currently available. We anticipate progress in these realms will follow the emergence of larger experimental datasets that will become publicly available in the near future.
We also explored the impact of immunological databases by examining their impact on the scientific community, as well as their strength as a means to clearly and concisely represent empirical data in a centralized resource. To this end, we undertook an effort to systematically quantify the impact of immunological DBARs by collecting metrics on their publication and citation rates. The high cumulative citation rate of the epitope-related DBARs is a clear indicator of the degree to which these resources permeate the scientific community and help guide research. A closer examination of the nature of these citations, using the IEDB as an example DBAR, revealed that these citations are mostly attributable to practical applications of the IEDB and represents further evidence of the direct impacts of DBARs.
Another indicator of DBAR impact is their utility for performing systematic meta-analyses (Kaczorowski 2009). To illustrate this point, we presented several examples of meta-analyses that have been performed to date based on the data available in the IEDB. With these examples, we hope to both raise the reader's awareness of their existence and to promote further meta-analyses as a means of driving and guiding continued applications of empirical data.
In this review, we have highlighted both the present utility of the diverse collection of immunological databases and analysis resources, while also exposing areas that require further development. In the final analysis, it is clear that, while immunological DBARs are presently widely utilized by the scientific community, in many respects, the field is still in its early stages, and continued development and refinement are necessary. Hence, it is reasonable to anticipate that the future years will see a diminishing lag between the emergence of robust experimental data and the ability of the scientific community to efficiently access and interpret such data.
Acknowledgments
We gratefully acknowledge the IEDB team and funding by the National Institutes of Health contract HHSH266200400006C.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Aurrecoechea C, Heiges M, Wang H, Wang Z, Fischer S, Rhodes P, Miller J, Kraemer E, Stoeckert CJ, Jr, Roos DS, Kissinger JC. ApiDB: integrated resources for the apicomplexan bioinformatics resource center. Nucleic Acids Res. 2007;35(Database issue):D427–D430. doi: 10.1093/nar/gkl880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard JB, Rhee SY. Ontologies in biology: design, applications and future challenges. Nat Rev Genet. 2004;5(3):213–222. doi: 10.1038/nrg1295. [DOI] [PubMed] [Google Scholar]
- Beaver JE, Bourne PE, Ponomarenko JV. EpitopeViewer: a Java application for the visualization and analysis of immune epitopes in the Immune Epitope Database and Analysis Resource (IEDB) Immunome Res. 2007;3:3. doi: 10.1186/1745-7580-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blythe MJ, Flower DR. Benchmarking B cell epitope prediction: underperformance of existing methods. Protein Sci. 2005;14(1):246–248. doi: 10.1110/ps.041059505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blythe MJ, Zhang Q, Vaughan K, de Castro R, Jr SN, Bui HH, Lewinsohn DM, Ernst JD, Peters B, Sette A. An analysis of the epitope knowledge related to mycobacteria. Immunome Res. 2007;3:10. doi: 10.1186/1745-7580-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne P. Will a biological database be different from a biological journal? PLoS Comput Biol. 2005;1(3):179–181. doi: 10.1371/journal.pcbi.0010034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkac LM, Davidsen T, Beck E, Ganapathy A, Caler E, Dodson RJ, Durkin AS, Harkins DM, Lorenzi H, Madupu R, Sebastian Y, Shrivastava S, Thiagarajan M, Orvis J, Sundaram JP, Crabtree J, Galens K, Zhao Y, Inman JM, Montgomery R, Schobel S, Galinsky K, Tanenbaum DM, Resnick A, Zafar N, White O, Sutton G. Pathema: a clade-specific bioinformatics resource center for pathogen research. Nucleic Acids Res. 2009;38(Database issue):D408–D414. doi: 10.1093/nar/gkp850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui HH, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinformatics. 2006;7:153. doi: 10.1186/1471-2105-7-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui HH, Peters B, Assarsson E, Mbawuike I, Sette A. Ab and T cell epitopes of influenza A virus, knowledge and opportunities. Proc Natl Acad Sci U S A. 2007;104(1):246–251. doi: 10.1073/pnas.0609330104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui HH, Sidney J, Li W, Fusseder N, Sette A. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinformatics. 2007;8:361. doi: 10.1186/1471-2105-8-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies V, Vaughan K, Damle R, Peters B, Sette A. Classification of the universe of immune epitope literature: representation and knowledge gaps. PLoS One. 2009;4(9):e6948. doi: 10.1371/journal.pone.0006948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekiert DC, Bhabha G, Elsliger MA, Friesen RH, Jongeneelen M, Throsby M, Goudsmit J, Wilson IA. Antibody recognition of a highly conserved influenza virus epitope. Science. 2009;324(5924):246–251. doi: 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum JA, Andersen PH, Blythe M, Bui HH, Cachau RE, Crowe J, Davies M, Kolaskar AS, Lund O, Morrison S, Mumey B, Ofran Y, Pellequer JL, Pinilla C, Ponomarenko JV, Raghava GP, van Regenmortel MH, Roggen EL, Sette A, Schlessinger A, Sollner J, Zand M, Peters B. Towards a consensus on datasets and evaluation metrics for developing B-cell epitope prediction tools. J Mol Recognit. 2007;20(2):75–82. doi: 10.1002/jmr.815. [DOI] [PubMed] [Google Scholar]
- Greenbaum JA, Vita R, Zarebski L, Emami H, Sette A, Ruttenburg A, Peters B (2009a) ONTology of Immune Epitopes (ONTIE) Representing the Immune Epitope Database in OWL. The 12th Annual Bio-Ontologies Meeting, ISMB 2009, pages 45–48
- Greenbaum JA, Kotturi MF, Kim Y, Oseroff C, Vaughan K, Salimi N, Vita R, Ponomarenko J, Scheuermann RH, Sette A, Peters B. Pre-existing immunity against swine-origin H1N1 influenza viruses in the general human population. Proc Natl Acad Sci USA. 2009;106:20365–20370. doi: 10.1073/pnas.0911580106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene JM, Plunkett G, 3rd, Burland V, Glasner J, Cabot E, Anderson B, Neeno-Eckwall E, Qiu Y, Mau B, Rusch M, Liss P, Hampton T, Pot D, Shaker M, Shaull L, Shetty P, Shi C, Whitmore J, Wong M, Zaremba S, Blattner FR, Perna NT. A new asset for pathogen informatics—the Enteropathogen Resource Integration Center (ERIC), an NIAID Bioinformatics Resource Center for Biodefense and Emerging/Re-emerging Infectious Disease. Adv Exp Med Biol. 2007;603:28–42. doi: 10.1007/978-0-387-72124-8_4. [DOI] [PubMed] [Google Scholar]
- Greene JM, Collins F, Lefkowitz EJ, Roos D, Scheuermann RH, Sobral B, Stevens R, White O, Di Francesco V. National Institute of Allergy and Infectious Diseases bioinformatics resource centers: new assets for pathogen informatics. Infect Immun. 2007;75(7):3212–3219. doi: 10.1128/IAI.00105-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczorowski J. Standing on the shoulders of giants: introduction to systematic reviews and meta-analyses. Can Fam Physician. 2009;55(11):1155–1156. [PMC free article] [PubMed] [Google Scholar]
- Korber BTM, Brander C, Haynes BF, Koup R, Moore JP, Walker BD, Watkins DI. HIV molecular immunology 2006/2007. Los Alamos, New Mexico: Los Alamos National Laboratory, Theoretical Biology and Biophysics; 2007. [Google Scholar]
- Lata S, Bhasin M, Raghava GP. MHCBN 4.0: a database of MHC/TAP binding peptides and T-cell epitopes. BMC Res Notes. 2009;2:61. doi: 10.1186/1756-0500-2-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson D, Arensburger P, Atkinson P, Besansky NJ, Bruggner RV, Butler R, Campbell KS, Christophides GK, Christley S, Dialynas E, Emmert D, Hammond M, Hill CA, Kennedy RC, Lobo NF, MacCallum MR, Madey G, Megy K, Redmond S, Russo S, Severson DW, Stinson EO, Topalis P, Zdobnov EM, Birney E, Gelbart WM, Kafatos FC, Louis C, Collins FH. VectorBase: a home for invertebrate vectors of human pathogens. Nucleic Acids Res. 2007;35(Database issue):D503–D505. doi: 10.1093/nar/gkl960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gøtzsche PC, Ioannidis JP, Clarke M, Devereaux PJ, Kleijnen J, Moher D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. J Clin Epidemiol. 2009;62(10):e1–e34. doi: 10.1016/j.jclinepi.2009.06.006. [DOI] [PubMed] [Google Scholar]
- Lin HH, Ray S, Tongchusak S, Reinherz EL, Brusic V. Evaluation of MHC class I peptide binding prediction servers: applications for vaccine research. BMC Immunol. 2008;9:8. doi: 10.1186/1471-2172-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord P, ShahN, Sansone S-A, Stephens S, Soldatova L (2009) The OBI Consortium. Modeling biomedical experimental processes with OBI. The 12th Annual Bio-Ontologies Meeting, ISMB 2009, pages 41+
- Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic Acids Res. 2008;36(Web Server issue):W509–W512. doi: 10.1093/nar/gkn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallios RR. A consensus strategy for combining HLA-DR binding algorithms. Hum Immunol. 2003;64(9):852–856. doi: 10.1016/S0198-8859(03)00142-3. [DOI] [PubMed] [Google Scholar]
- McNeil LK, Reich C, Aziz RK, Bartels D, Cohoon M, Disz T, Edwards RA, Gerdes S, Hwang K, Kubal M, Margaryan GR, Meyer F, Mihalo W, Olsen GJ, Olson R, Osterman A, Paarmann D, Paczian T, Parrello B, Pusch GD, Rodionov DA, Shi X, Vassieva O, Vonstein V, Zagnitko O, Xia F, Zinner J, Overbeek R, Stevens R. The National Microbial Pathogen Database Resource (NMPDR): a genomics platform based on subsystem annotation. Nucleic Acids Res. 2007;35(Database issue):D347–D353. doi: 10.1093/nar/gkl947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutaftsi M, Tscharke DC, Vaughan K, Koelle DM, Stern L, Calvo-Calle M, Ennis F, Terajima M, Sutter G, Crotty S, Drexler I, Franchini G, Yewdell JW, Head SR, Blum J, Peters B, Sette A. Uncovering the interplay between CD8, CD4 and antibody responses to complex pathogens. Future Microbiol. 2010;5(2):221–239. doi: 10.2217/fmb.09.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutaftsi M, Bui HH, Peters B, Sidney J, Salek-Ardakani S, Oseroff C, Pasquetto V, Crotty S, Croft M, Lefkowitz EJ, Grey H, Sette A. Vaccinia virus-specific CD4+ T cell responses target a set of antigens largely distinct from those targeted by CD8+ T cell responses. J Immunol. 2007;178(11):6814–6820. doi: 10.4049/jimmunol.178.11.6814. [DOI] [PubMed] [Google Scholar]
- Noy NF, Shah NH, Whetzel PL, Dai B, Dorf M, Griffith N, Jonquet C, Rubin DL, Storey MA, Chute CG, Musen MA. BioPortal: ontologies and integrated data resources at the click of a mouse. Nucleic Acids Res. 2009;37(Web Server issue):W170–W173. doi: 10.1093/nar/gkp440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters B, Sette A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics. 2005;6:132. doi: 10.1186/1471-2105-6-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters B, Sette A. Integrating epitope data into the emerging web of biomedical knowledge resources. Nat Rev Immunol. 2007;7(6):485–490. doi: 10.1038/nri2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters B, Sidney J, Bourne P, Bui HH, Buus S, Doh G, Fleri W, Kronenberg M, Kubo R, Lund O, Nemazee D, Ponomarenko JV, Sathiamurthy M, Schoenberger S, Stewart S, Surko P, Way S, Wilson S, Sette A. The immune epitope database and analysis resource: from vision to blueprint. PLoS Biol. 2005;3(3):e91. doi: 10.1371/journal.pbio.0030091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters B, Bui HH, Frankild S, Nielson M, Lundegaard C, Kostem E, Basch D, Lamberth K, Harndahl M, Fleri W, Wilson SS, Sidney J, Lund O, Buus S, Sette A. A community resource benchmarking predictions of peptide binding to MHC-I molecules. PLoS Comput Biol. 2006;2(6):e65. doi: 10.1371/journal.pcbi.0020065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarenko J, Bui HH, Li W, Fusseder N, Bourne PE, Sette A, Peters B. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics. 2008;9:514. doi: 10.1186/1471-2105-9-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanović S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50(3–4):213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- Reche PA, Zhang H, Glutting JP, Reinherz EL. EPIMHC: a curated database of MHC-binding peptides for customized computational vaccinology. Bioinformatics. 2005;21(9):2140–2141. doi: 10.1093/bioinformatics/bti269. [DOI] [PubMed] [Google Scholar]
- Ruppert J, Sidney J, Celis E, Kubo RT, Grey HM, Sette A. Prominent role of secondary anchor residues in peptide binding to HLA-A2.1 molecules. Cell. 1993;74(5):929–937. doi: 10.1016/0092-8674(93)90472-3. [DOI] [PubMed] [Google Scholar]
- Saha S, Raghava GP. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins. 2006;65(1):40–48. doi: 10.1002/prot.21078. [DOI] [PubMed] [Google Scholar]
- Saha S, Raghava GP. Prediction methods for B-cell epitopes. Methods Mol Biol. 2007;409:387–394. doi: 10.1007/978-1-60327-118-9_29. [DOI] [PubMed] [Google Scholar]
- Sathiamurthy M, Hickman HD, Cavett JW, Zahoor A, Prilliman K, Metcalf S, Fernandez Vina M, Hildebrand WH. Population of the HLA ligand database. Tissue Antigens. 2003;61(1):12–19. doi: 10.1034/j.1399-0039.2003.610102.x. [DOI] [PubMed] [Google Scholar]
- Sathiamurthy M, Peters B, Bui HH, Sidney J, Mokili J, Wilson SS, Fleri W, McGuinness DL, Bourne PE, Sette A. An ontology for immune epitopes: application to the design of a broad scope database of immune reactivities. Immunome Res. 2005;1(1):2. doi: 10.1186/1745-7580-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönbach C, Koh JL, Flower DR, Brusic V. An update on the functional molecular immunology (FIMM) database. Appl Bioinformatics. 2005;4(1):25–31. doi: 10.2165/00822942-200504010-00003. [DOI] [PubMed] [Google Scholar]
- Schulze-Kremer S. Ontologies for molecular biology and bioinformatics. In Silico Biol. 2002;2(3):179–193. [PubMed] [Google Scholar]
- Sette A, Moutaftsi M, Moyron-Quiroz J, McCausland MM, Davies DH, Johnston RJ, Peters B, Rafii-El-Idrissi Benhnia M, Hoffmann J, Su HP, Singh K, Garboczi DN, Head S, Grey H, Felgner PL, Crotty S. Selective CD4+ T cell help for antibody responses to a large viral pathogen: deterministic linkage of specificities. Immunity. 2008;28(6):847–858. doi: 10.1016/j.immuni.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EE, Kampanya N, Lu J, Nordberg EK, Karur HR, Shukla M, Soneja J, Tian Y, Xue T, Yoo H, Zhang F, Dharmanolla C, Dongre NV, Gillespie JJ, Hamelius J, Hance M, Huntington KI, Jukneliene D, Koziski J, Mackasmiel L, Mane SP, Nguyen V, Purkayastha A, Shallom J, Yu G, Guo Y, Gabbard J, Hix D, Azad AF, Baker SC, Boyle SM, Khudyakov Y, Meng XJ, Rupprecht C, Vinje J, Crasta OR, Czar MJ, Dickerman A, Eckart JD, Kenyon R, Will R, Setubal JC, Sobral BW. PATRIC: the VBI PathoSystems Resource Integration Center. Nucleic Acids Res. 2007;35(Database issue):D401–D406. doi: 10.1093/nar/gkl858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squires B, Macken C, Garcia-Sastre A, Godbole S, Noronha J, Hunt V, Chang R, Larsen CN, Klem E, Biersack K, Scheuermann RH. BioHealthBase: informatics support in the elucidation of influenza virus host pathogen interactions and virulence. Nucleic Acids Res. 2008;36(Database issue):D497–D503. doi: 10.1093/nar/gkm905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Lu X, Li C, Wang M, Liu Q, Li Z, Hu X, Li J, Liu F, Li Q, Belser JA, Hancock K, Shu Y, Katz JM, Liang M, Li D. Generation, characterization and epitope mapping of two neutralizing and protective human recombinant antibodies against influenza A H5N1 viruses. PLoS One. 2009;4(5):e5476. doi: 10.1371/journal.pone.0005476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurmond J, Yoon H, Kuiken C, Yusim K, Perkins S, Theiler J, Bhattacharya T, Korber B, Fischer W. Web-based design and evaluation of T-cell vaccine candidates. Bioinformatics. 2008;24(14):1639–1640. doi: 10.1093/bioinformatics/btn251. [DOI] [PubMed] [Google Scholar]
- Toseland CP, Clayton DJ, McSparron H, Hemsley SL, Blythe MJ, Paine K, Doytchinova IA, Guan P, Hattotuwagama CK, Flower DR. AntiJen: a quantitative immunology database integrating functional, thermodynamic, kinetic, biophysical, and cellular data. Immunome Res. 2005;1(1):4. doi: 10.1186/1745-7580-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan K, Blythe M, Greenbaum J, Zhang Q, Peters B, Doolan DL, Sette A. Meta-analysis of immune epitope data for all Plasmodia: overview and applications for malarial immunobiology and vaccine-related issues. Parasite Immunol. 2009;31(2):78–97. doi: 10.1111/j.1365-3024.2008.01077.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vita R, Vaughan K, Zarebski L, Salimi N, Fleri W, Grey H, Sathiamurthy M, Mokili J, Bui HH, Bourne PE, Ponomarenko J, de Castro R, Jr CRK, Sidney J, Wilson SS, Stewart S, Way S, Peters B, Sette A. Curation of complex, context-dependent immunological data. BMC Bioinformatics. 2006;7:341. doi: 10.1186/1471-2105-7-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vita R, Peters B, Sette A. The curation guidelines of the immune epitope database and analysis resource. Cytometry A. 2008;73(11):1066–1070. doi: 10.1002/cyto.a.20585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Morgan AA, Zhang Q, Sette A, Peters B. Automating document classification for the Immune Epitope Database. BMC Bioinformatics. 2007;8:269. doi: 10.1186/1471-2105-8-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Sidney J, Dow C, Mothé B, Sette A, Peters B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol. 2008;4(4):e1000048. doi: 10.1371/journal.pcbi.1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip YL (2009) Accelerating knowledge discovery through community data sharing and integration. Yearb Med Inform 117–20 [PubMed]
- Yu X, Tsibane T, McGraw PA, House FS, Keefer CJ, Hicar MD, Tumpey TM, Pappas C, Perrone LA, Martinez O, Stevens J, Wilson IA, Aguilar PV, Altschuler EL, Basler CF, Crowe JE., Jr Neutralizing antibodies derived from the B cells of 1918 influenza pandemic survivors. Nature. 2008;455(7212):532–536. doi: 10.1038/nature07231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarebski LM, Vaughan K, Sidney J, Peters B, Grey H, Janda KD, Casadevall A, Sette A. Analysis of epitope information related to Bacillus anthracis and Clostridium botulinum. Expert Rev Vaccines. 2008;7(1):55–74. doi: 10.1586/14760584.7.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Lundegaard C, Nielsen M. Pan-specific MHC class I predictors: a benchmark of HLA class I pan-specific prediction methods. Bioinformatics. 2009;25(1):83–89. doi: 10.1093/bioinformatics/btn579. [DOI] [PMC free article] [PubMed] [Google Scholar]