

Abstract
Background
Sideroblastic anemias are heterogeneous congenital and acquired bone marrow disorders characterized by pathologic iron deposits in mitochondria of erythroid precursors. Among the congenital sideroblastic anemias (CSAs), the most common form is X-linked sideroblastic anemia, due to mutations in 5-aminolevulinate synthase (ALAS2). A novel autosomal recessive CSA, caused by mutations in the erythroid specific mitochondrial transporter SLC25A38, was recently defined. Other known etiologies include mutations in genes encoding the thiamine transporter (SLC19A2), the RNA-modifying enzyme pseudouridine synthase 1 (PUS1), a mitochondrial ATP-binding cassette transporter (ABCB7), glutaredoxin 5 (GLRX5), as well as mitochondrial DNA deletions. Despite these known diverse causes, in a substantial portion of CSA cases a presumed genetic defect remains unknown.
Procedure
In the context of the recent discovery of SLC25A38 as a major novel cause, we systematically analyzed a large cohort of previously unreported CSA patients. Sixty CSA probands (28 females, 32 males) were examined for ALAS2, SLC25A38, PUS1, GLRX5, and ABCB7 mutations. SLC19A2 and mitochondrial DNA were only analyzed if characteristic syndromic features were apparent.
Results
Twelve probands had biallelic mutations in SLC25A38. Seven ALAS2 mutations were detected in eight sporadic CSA cases, two being novel. We also identified a novel homozygous null PUS1 mutation and novel mitochondrial DNA deletions in two patients with Pearson syndrome. No mutationswere encountered in GLRX5, ABCB7, or SLC19A2.
Conclusions
The remaining undefined probands (43%) can be grouped according to gender, family and clinical characteristics, suggesting novel X-linked and autosomal recessive forms of CSA.
Keywords: sideroblastic anemia, iron, ringed sideroblasts, microcytic anemia
INTRODUCTION
The defining feature of a sideroblastic anemia is the presence of bone marrow ring sideroblasts: erythroid precursors with pathologic mitochondrial iron deposits. Most commonly, and in adults, these cells are found in association with myelodysplastic syndromes, in which their pathogenesis is obscure. Sideroblastic anemia also occurs after exposure to certain drugs or alcohol and with copper deficiency. On the other hand, at least seven forms of congenital sideroblastic anemia (CSA) are defined at the molecular level, each of which has provided some insight into cellular pathways associated with disordered mitochondrial iron metabolism (Table I) [1,2].
Table I.
Genetically Defined Forms of Congenital Sideroblastic Anemia (CSA)
| CSA Form | Gene | Anemia form | No. of reported mutations | No. of reported probands |
|---|---|---|---|---|
| X-linked sideroblastic anemia (XLSA) | Erythroid 5-aminolevulinate synthase (ALAS2) | microcytic | 48 | ~76 |
| Autosomal recessive CSA | Mitochondrial transporter (SLC25A38) | microcytic | 11 | 15 |
| Glutaredoxin 5 deficiency | Glutaredoxin 5 (GLRX5) | microcytic | 1 | 1 |
| X-linked sideroblastic anemia and ataxia (XLSA/A) | ATP-binding cassette transporter (ABCB7) | microcytic | 3 | 3 |
| Mitochondrial myopathy, lactic acidosis and sideroblastic anemia (MLASA) | Pseudouridine synthase 1 (PUS1) | normocytic | 2 | 4 |
| Thiamine-responsive megaloblastic anemia, with diabetes and deafness (TRMA) | High-affinity thiamine transporter (SLC19A2) | macrocytic/megaloblastic | 28 | ~50 |
| Pearson marrow-pancreas syndrome | Mitochondrial DNA deletions, duplications, rearrangements | macrocytic | ~40 | >40 |
X-linked sideroblastic anemia (XLSA, OMIM# 300751) is considered the most common CSA. Cooley first described it in 1945 [3]. Decades later, linkage of the disorder to the 5-aminolevulinate synthase 2 (ALAS2) locus (Xp11.21) was established. ALAS2, also known as erythroid ALAS, catalyzes the first and rate-limiting step in the heme biosynthetic pathway in erythroid cells. To date 48 distinct disease-causing mutations in ALAS2 have been identified in nearly 80 unrelated individuals or families. All of the mutations affect the catalytic domain of the ALAS2 enzyme (encoded by exons 5–11) [4]. One putative mutation in the ALAS2 promoter was reported in two families [5], but its significance is uncertain as the variant occurs in some normal populations [6]. XLSA is characterized by microcytic-hypochromic anemia, often with two discrete populations of red cells; one microcytic and the other normocytic (erythrocyte dimorphism); systemic iron overload as a consequence of the chronic ineffective erythropoiesis; and, frequently, a response to supplementation with pyridoxine, the precursor of pyridoxal 5′-phosphate (PLP), which is the essential cofactor of ALAS. As in all CSA forms, the precise mechanism of the mitochondrial iron accumulation is unclear.
Mutations in the erythroid specific mitochondrial transporter SLC25A38 have been discovered as the cause of an autosomal recessive, nonsyndromic form of CSA [7]. Affected patients present with severe microcytic anemia and systemic iron overload in early childhood. It is hypothesized that SLC25A38 facilitates ALA production by importing glycine into mitochondria, and/or by exchanging glycine for ALA across the mitochondrial inner membrane.
Severe sideroblastic anemia associated with a homozygous splice site mutation in glutaredoxin 5 (GLRX5), an integral component of the assembly machinery of iron-sulfur clusters within the mitochondria, has been reported in one patient [8].
Apart from these genetically defined forms of CSA, four syndromic types are recognized. XLSA with ataxia (XLSA/A, OMIM# 301310) was first described in 1985 in two kindreds [9]. Affected individuals presented with mild microcytic anemia and neurologic deficits of delayed motor development, non- or slowly-progressive ataxia and severe cerebellar hypoplasia or atrophy. To date, three distinct missense mutations in the ABCB7 gene have been described in these patients [10–12]. ABCB7 resides on the X chromosome and encodes a mitochondrial transporter protein, which is involved in the biogenesis of cytosolic iron sulfur clusters [11].
Mitochondrial myopathy, lactic acidosis and sideroblastic anemia (MLASA, OMIM # 600462) is characterized by muscle weakness, normocytic anemia and lactic academia with onset in childhood [13]. Among four families with this syndrome two distinct mutations in pseudouridine synthase (PUS-1) are reported [14–16]. Defective pseudouridylation of tRNAs is thought to lead to faulty translation of mitochondrial genes and altered mitochondrial metabolism [14,16,17].
The syndrome of thiamine responsive megaloblastic anemia (TRMA, OMIM# 249270), which is associated with diabetes and deafness, has the unusual CSA phenotype of megaloblastic erythroid maturation with ring sideroblasts. Mutations in the high-affinity thiamine transporter SLC19A2 underlie this syndrome. A clinical response to pharmacologic doses of thiamine is a hallmark of the disorder [18–20]. Impairment of the thiamine dependent generation of succinyl CoA was suggested to result in the ring sideroblast abnormality [21].
The Pearson marrow-pancreas syndrome (PMPS, OMIM # 557000) is associated with large-scale mitochondrial DNA (mtDNA) deletions, rearrangements or duplications. The degree of the heteroplasmic distribution of mtDNA defects is thought to correlate with the clinical course [22–24]. The clinical presentation includes macrocytic anemia, metabolic acidosis and exocrine pancreatic insufficiency within the first six months of life, the anemia often being the most prominent feature. The marrow characteristically shows non-megaloblastic maturation with vacuolization of erythroid and myeloid precursors and ring sideroblasts [25].
The molecular characterization of these forms of CSA has accrued largely from studies of individual families or cases as reported in the literature. However, a significant percentage of patients with CSA appear to remain genetically undefined. Here, we present a systematic molecular genetic analysis of an original cohort of 83 probands with CSA, which was assembled over an eighteen-year period. Twenty-three probands in this cohort were previously found to have ALAS2 mutations [4]. In this study, the remaining 60 probands (28 female, 32 male) were formally examined for mutations in known candidate genes.
PATIENTS AND METHODS
Patients
Most of the patients were referred to S.S.B. for analysis of the ALAS2 gene or for clinical consultation. CSA was ascertained by bone marrow morphology, red cell parameters and anemia of life-long or many years duration. CSA and CSA syndromes were defined as described in the introduction as well as in the original reports [3,7–9,13,19,25]. Informed consent was obtained from participants in the study, as approved by the Institutional Review Board of the University of Oklahoma Health Sciences Center and the Children’s Hospital Boston Committee on Clinical Investigation.
Isolation of DNA
Genomic DNA was isolated from peripheral blood or bone marrow, using the QIAamp DNA Blood Maxi Kit (Qiagen, Valencia, CA) or the Puregene DNA Purification Kit (Gentra Systems, Inc., Minneapolis, MN).
Mutation analysis
DNA was amplified by PCR using the conditions and primers as listed in Supplementary Table I. All exon primers were designed to include at least 30 bp of flanking intronic sequence and were synthesized by Integrated DNA Technologies (Coralville, IA). PCR products were examined by gel electrophoresis and purified by either treating the reaction mixture with Exonuclease I (New England Biolabs, Ipswich, MA) and shrimp alkaline phosphatase (Roche Diagnostics, Indianapolis, IN), or by using the QIAquick Gel Extraction Kit (Qiagen, Valencia, CA). Both DNA strands were sequenced using fluorescent dye-termination chemistry (Children’s Hospital Boston Molecular Genetics Core Facility). Sequences were analyzed with the program Sequencher 4.8 DNA sequence assembly software (Gene Codes, Ann Arbor, MI). Mutations were confirmed by repeat amplification and sequencing.
X–chromosome inactivation assay
The Human Androgen Receptor Assay (HUMARA) of X chromosome inactivation was performed as previously described [26–28].
Mitochondrial genome deletion mapping and assessment of heteroplasmy
Mitochondrial genome deletions were screened by performing long-range PCR of total bone marrow DNA using the Expand Long Template PCR system (Roche, Indianapolis, IN) according to manufacturer’s instructions and primers as listed in Supplementary Table II. Samples indicating a deleted species were further characterized with restriction enzyme mapping of purified amplicons. Minimal intervals containing the deletion junctions were PCR amplified and sequenced. Nucleotide positions of the deletions were determined from the revised Cambridge Reference Sequence of the human mitochondrial DNA in GenBank as RefSeq AC_000021.2 gi: 115315570, and sequence analysis was performed using data from www.mitomap.org and www.mitowheel.org. Heteroplasmy was quantified by real-time PCR (with primers as in Supplementary Table II) and comparing the amplification of the deletion junction to a non-deleted segment of the mitochondrial genome.
RESULTS
Mutation analysis
ALAS2 mutations
In all 60 probands, ALAS2 was characterized by sequencing the coding region, intron-exon boundaries, proximal promoter and intron 8, which contains an erythroid-specific enhancer [29]. Seven distinct missense mutations were detected amongeight singleton patients; all are in the catalytic domain (exons 5–11) of the enzyme (Table II). No mutations were found in the promoter or intron 8 of ALAS2. Twoof the ALAS2 mutations are novel: V301A in a male patient, and R517G in a female patient with an unfortunate skewed X chromosome inactivationpattern in peripheral blood leukocytes as assessed by the humanandrogen receptor assay (HUMARA). The patients with the F165L and R170C mutations were responsive to pyridoxine, consistent with the mutations affecting enzyme stability and cofactor binding, respectively [30].
Table II.
Mutations Identified in the Cohort of 60 Patients with Congenital Sideroblastic Anemia
| Patient | Gender | Gene | Mutation | Exon | Codon change | Severity of anemiaa |
|---|---|---|---|---|---|---|
| 1 | M | ALAS2 | 547C>A | 5 | F165L | moderate |
| 2 | M | ALAS2 | 560C>T | 5 | R170C | severe |
| 3 | M | ALAS2 | 954T>Cb | 7 | V301Ab | moderate |
| 4 | M | ALAS2 | 1283C>T | 9 | R411C | moderate |
| 5 | M | ALAS2 | 1407G>A | 9 | R452H | mild |
| 6 | M | ALAS2 | 1407G>A | 9 | R452H | mild |
| 7 | Fc | ALAS2 | 1601C>Gb | 10 | R517Gb | moderate |
| 8 | M | ALAS2 | 1611C>T | 10 | P520L | moderate |
| 9 | F | PUS1 | 885C>T | 3 | R144W | severe |
| 10 | M | PUS1 | 917C>Tb | 4 | Q154Xb | moderate |
| 11 | F | mtDNA | 2501 bp mtDNA delb | - | - | severe |
| 12 | M | mtDNA | 4836 bp mtDNA delb | - | - | severe |
| 13–24d | 5M, 7F | SLC25A38 | Multipleb | 4–7 | Multiple | severe |
Mild, Hb >10 g/dL; moderate, Hb 7–10 g/dL; severe Hb <7 g/dL;
Novel mutation;
Has marked skewing of X-chromosome inactivation;
The details of these cases are presented in Reference 8
A novel mutation in PUS-1
The sequence of PUS-1 was determined in all probands lacking an ALAS2 mutation. We identified one novel homozygous null mutation in a familial case. The mutation predicts a nonsense mutation at glutamine 154 (Q154X) that would lead to a protein lacking the 266 C-terminal amino acids. The second mutation, which was reported independently previously [14], predicts the codon change R144W; this mutation appears to be a recurrent allele in individuals of Persian Jewish descent occurring as a result of a founder effect.
Mitochondrial DNA deletions in two patients
We detected and mapped large heteroplasmic mitochondrial genome deletions in two patients, thus confirming the diagnosis of Pearson syndrome. Patient 11 was found to have a novel deletion of 2501 bp inclusive of nucleotides 10949–13449 and flanked by a 5 bp repeat (CAACC), resulting in truncation of the mitochondrial genes ND4 and ND5 and three tRNA genes (LCUN, SAGY, and H). Quantitative PCR measurements of DNA from peripheral blood and bone marrow revealed that the deleted mitochondrial DNA species represented approximately 60–80% of total mitochondrial DNA. Patient 12 was found to have a novel deletion of 4836 bp inclusive of nucleotides 8517–13352 without a flanking repeat, resulting in deletion or truncation of genes ATP8, ATP6, COIII, ND3, ND4L, ND4, and ND5, and five tRNA genes (G, R, LCUN, SAGY, and H). Quantitative PCR analysis of DNA from peripheral blood of this patient showed that the deleted mitochondrial DNA species constituted approximately 7% of mitochondrial DNA.
SLC25A38, GLRX5 and ABCB7 mutation analyses, and cases still undefined
During the course of this project, mutations in SLC25A38 were discovered as a cause of CSA by the Atlantic Medical Genetics and Genomics Initiative and three of us (MDF, DRC, SSB) [7]. As described further by Guernsey et al, SLC25A38 was sequenced in all probands from our cohort lacking ALAS2 mutations and eleven distinct biallelic mutations were identified in 12 probands. Sequencing of the GLRX5 and ABCB7 genes revealed only known polymorphisms. In the remaining 36 probands (43% of the cohort), which include seven familial cases, a disease-causingmutation was not found.
Clinical features
Patients with identified mutations
The age range at diagnosis of patients with XLSA was between six months and forty-nine years. All but one patient had microcytic anemia. Patient 7 (Table II) has macrocytic anemia with an increased red cell distribution width (RDW) and marked erythrocyte dimorphism, as previously reported in female patients with XLSA [4]. In two patients, the anemia was pyridoxine responsive (Patient 1 and 2). None of the probands have required chronic blood transfusions. The patients with MLASA presented at ages eleven years (Patient 9) and nine months (Patient 10). Both have normocytic anemia and patient 9 requires regular blood transfusions. Their myopathy is progressive. Both cases with mitochondrial DNA deletions presented at birth with severe macrocytic anemia. They require chronic blood transfusions and have developed severe iron overload. Patient 11 also has diabetes and hypothyroidism. The disabling mutations in SLC25A38 lead to severe microcytic-hypochromic anemia and systemic iron overload very early in life. Most of them require blood transfusions.
Genetically undefined patients with CSA
The 36 probands without disease-causing mutations we tentatively sub-categorize according to clinical characteristics (Table III). Group I has a pyridoxine responsive phenotype highly reminiscent of XLSA due to ALAS2 mutations. Group II, has an otherwise similar, but pyridoxine non-responsive phenotype and males predominate. Group III is distinguished by a normocytic or macrocytic anemia occurring only in individuals with two X chromosomes (9 females and one XXY male). Patients in group IV presented in infancy or during childhood with severe microcytic anemia, and are predominantly transfusion dependent. The phenotype of this group is similar to that of the patients who were found to have mutations in SLC25A38, suggesting additional recessive causes of CSA.
Table III.
Proposed Grouping of Genetically Undefined Probands/Kindreds in the Cohort of 60 Patients with Congenital Sideroblastic Anemia According to Clinical Features
| Group | No. of probands | Multiplex kindreds | Severity of anemiaa | Microcytic anemia | Range of ages at diagnosis | Response to pyridoxine | Females/Males | Key features |
|---|---|---|---|---|---|---|---|---|
| I | 7 | - | Moderate to severe | Yes | 2–68 | Yes | 3/4 | Phenotype similar to XLSA, anemia pyridoxine responsive |
| II | 6 | 4 | Mild to moderate | Yes | 13–50 | No | 1/5 | Phenotype similar to XLSA, anemia pyridoxine unresponsive |
| III | 10 | 1 | Mild to severe | No | Infancy–39 | No | 9/1b | Not microcytic anemia, presence of two X- chromosomes |
| IV | 13 | 2 | Moderate to severe | Yes | Infancy - childhood | No | 5/8 | Transfusion dependence early in life; apparent autosomal recessive, or de novo, defect |
as defined in Table II;
XXY male; XLSA, X-linked sideroblastic anemia
DISCUSSION
In this analysis of a large collection of index cases with CSA, as summarized in Fig. 1, causative mutations in previously identified CSA associated genes_were identified in 57 percent. While the cohort is undoubtedly biased toward individuals in whom an ALAS2 mutation was a clinical consideration, our findings confirm the general impression that XLSA represents the most common form of CSA that can be ascertained with current mutation detection strategies. ALAS2 missense mutations constituted 37 percent of the entire cohort of 83 probands (Fig. 1). The two novel ALAS2 mutations, V301A and R517G, described here further expand the spectrum of distinct mutations recognized in XLSA. One of these (R517G), occurring in a woman with adult onset sideroblastic anemia associated with unfortunate skewed X-chromosome inactivation in peripheral blood cells, underscores the not uncommon expression of this disorder in females as a consequence of age-related, non-random X inactivation in hematopoietic cells [4].
Figure 1.
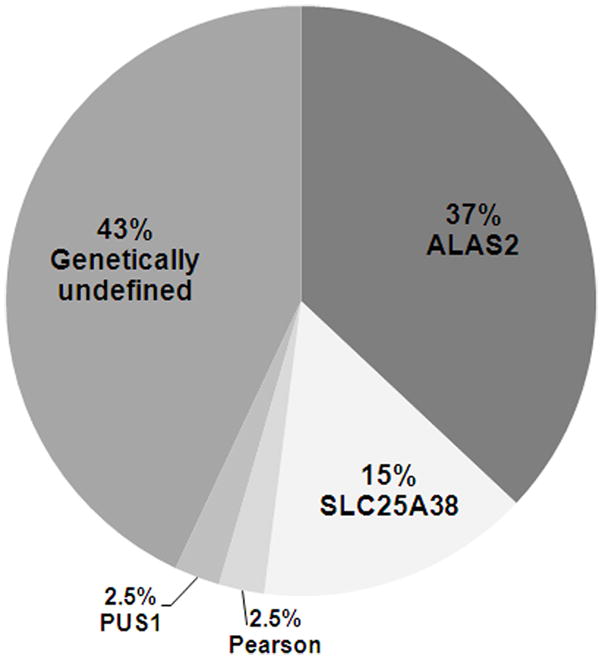
Categories of Identified mutations in the cohort of patients with congenital sideroblastic anemia. The relative percentages of the disease causing mutations in the entire cohort of 83 initial probands (23 previously found to be ALAS2 defects) are indicated. The distribution probably reflects a degree of referral bias, and so may not be readily generalized.
As reported separately [7], the discovery that molecular defects in the mitochondrial carrier family gene SLC25A38 are associated with nonsyndromic, autosomal recessive CSA led to the identification of multiple biallelic mutations in this gene in three distinct families and nine additional unrelated individuals from our cohort. Mutations in SL25A38 appear to be a relatively common cause of CSA, accounting for 15 percent of probands.
Our results indicate that GLRX5 deficiency is indeed very uncommon, as, despite its clinical overlap with other nonsyndromic causes of CSA, we did not identify a single additional mutation. Likewise, although our cohort is almost certainly biased against patients with syndromic features that would suggest MLASA, XLSA/A, TRMA or Pearson syndrome, as illustrated by the finding of PUS1 mutations and mtDNA deletions, the value of a systematic approach to mutation detection is underscored, particularly as associated features may be subtle and go unrecognized at presentation. The ABCB7 variants of XLSA/A may be underrepresented as the mild anemia in the context of a clinically most apparent ataxia phenotype may hinder its recognition. The clinically distinctive features of TRMA, that is only variably associated with bone marrow ring sideroblasts, would likewise cause these individuals to be referred for SLC19A2 sequencing without consideration of a non-syndromic cause of the anemia. The 43% of the cohort who are genetically undefined constitute diverse phenotypes as described under Results and in Table III. In many cases the clinical features would suggest novel X-linked or autosomal recessive causes of CSA yet to be discovered.
The evidence that sideroblastic anemia is an inherited trait in Group III is less convincing than in the other groups, as it tends to occur later in life and there is only one multiplex family. Interestingly, one of the few recurring cytogenetic abnormalities in patients with the acquired myelodysplastic syndrome, refractory anemia with ringed sideroblasts (RARS), is an isodicentric chromosome Xq13.1 (idicXq13.1), which occurs only in females [31]. Thus, although highly unlikely, it is possible that Group III has a cytogenetically cryptic acquired Xq13.1 rearrangement. Alternatively, it is possible that germline X-linked lethal mutations at this or another X-chromosome locus could result in a similar congenital phenotype [32].
All of the patients eventually found to have mutations in SLC25A38 [7] resemble Group IV. The two families and the 13 individuals in Group IV may well have unconventional mutations in this gene. However, the absence of any CSA patients with only one aberrant SLC25A38 allele detected with the sequencing strategy used would suggest that such unconventional mutations are unusual and that there are additional recessive causes of CSA yet to be discovered.
Beyond the referral bias, there are several other limitations to this study. The greatest caveat is the fact that, with the exception of ALAS2 in which we analyzed the proximal promoter and an intronic enhancer, we sequenced only exons and intron/exon boundaries. In doing so, we would likely miss genomic rearrangements, transcriptional control elements, and other unusual mutations that alter gene expression. Furthermore, our mtDNA analysis strategy will not detect small deletions, certain rearrangements, and all point mutations. Lastly, by restricting the analysis of mtDNA and SLC19A2 to those cases in which there was at least a suggestion of a syndromic phenotype we would not uncover disease phenotypes outside of the spectrum conventionally associated with these mutations.
Notably, the known defective proteins in the various CSAs all affect mitochondrial metabolism as depicted in the sketch in Figure 2. Deficient protoporphyrin biosynthesis due to impaired ALAS2 and mitochondrial transporter SLC25A38 reduces the utilization of iron that is imported into the mitochondrion at an apparent normal rate. Proteins GLRX5 and ABCB7 participate in the generation of iron-sulfur clusters [11,33], which directly regulate cellular iron homeostasis through their role in the function of the cytosolic iron-sulfur protein IRP1 [34], and when defective would influence cellular iron uptake and ferritin translation as well as the translation of ALAS2. The other defects, residing in PUS1, the high-affinity thiamine transporter of TRMA and mtDNA, are postulated to affect iron metabolism in a secondary manner. Thus, the common denominator among the molecularly defined forms of CSA is an evident link between the affected proteins and mitochondrial iron metabolism. The precise mechanisms leading to the severe mitochondrial iron accumulation in the ring sideroblast remain to be elucidated.
Figure 2.

Sketch of a mitochondrion depicting the sites of the proteins that are defective in the various congenital sideroblastic anemias. Gly – glycine; ALA – 5-aminolevulinic acid; SLC25A38 – mitochondrial carrier protein SLC25A38; ALAS2 – 5-aminolevulinate synthase 2; PLP – pyridoxal 5′-phosphate; SLC19A2 – high-affinity thiamine transporter SLC19A2; mtDNA – mitochondrial DNA; tRNA – transfer RNA; PUS1 – pseudouridine synthase 1; Fe-S – iron-sulfur; GLRX5 – glutaredoxin 5; ABCB7 – ATP-binding cassette transporter ABCB7; MFRN 1 – mitoferrin 1.
In conclusion, this study provides the largest and most comprehensive analysis of the genetic causes of CSA reported to date. It emphasizes the frequency of ALAS2 and SLC25A38 mutations, equally accompanied by a lack of as yet identifiable underlying defects in a large proportion of cases. In the latter, autosomal recessive as well as X-linked defects appear likely and may be revealed in known CSA causing genes with other approaches of mutational analysis. Identification of genetic defects not only permits precise diagnosis, but also allows for accurate family screening, more appropriate genetic counseling and a better estimation of prognosis. Continued study of affected patients can be expected to lead to new insights into the causes of unexplained CSA and the poorly understood area of mitochondrial and cellular iron metabolism in the erythroid cell.
Supplementary Material
Acknowledgments
We express our gratitude to the patients, their families and referring physicians. We thank Mathew Heeney for help with consenting some of the patients, Philip Wise and Eric Wasson for technical assistance, and Mark Samuels and his colleagues at the Atlantic Medical Genetics and Genomics Initiative for discussions concerning SLC25A38 in advance of publication. This work was supported in part by NIH grant R01 DK080011 (M.D.F); The U.S. Department of Veterans Affairs, University of Oklahoma Health Sciences Center Provost’s Fund and The Oklahoma Center for Advancement of Science and Technology (S.S.B.); NIH grant K08 HL089150 (S.A.), and NIH grant K24 HL004184 (E.J.N.).
Footnotes
Conflict of Interest
The authors have none to declare.
References
- 1.Bottomley SS. Congenital sideroblastic anemias. Curr Hematol Rep. 2006;5(1):41–49. [PubMed] [Google Scholar]
- 2.Fleming MD. The genetics of inherited sideroblastic anemias. Semin Hematol. 2002;39(4):270–281. doi: 10.1053/shem.2002.35637. [DOI] [PubMed] [Google Scholar]
- 3.Cooley TB. A severe type of hereditary anemia with elliptocytosis. Interesting sequence of splenectomy. Am J Med Sci. 1945;209:561–568. [Google Scholar]
- 4.Bottomley SS. Sideroblastic Anemias. In: John P, Greer JF, Rodgers George M, Paraskevas Frixos, Glader Bertil, editors. Wintrobe’s Clinical Hematology. 12. Lippincott Williams & Wilkins; 2008. pp. 835–855. [Google Scholar]
- 5.Bekri S, May A, Cotter PD, et al. A promoter mutation in the erythroid-specific 5-aminolevulinate synthase (ALAS2) gene causes X-linked sideroblastic anemia. Blood. 2003;102(2):698–704. doi: 10.1182/blood-2002-06-1623. [DOI] [PubMed] [Google Scholar]
- 6.May A, Barton C, Masters G, et al. Severe sideroblastic anemia in an ALAS2 compound heterozygote for −206, a common polymorphism, and a novel mutation in exon 11 (Lys535del) linked to lack of haemoglobinisation in vitro and ineffective erythropoiesis in vivo. ASH. 2005;106:988a. [Google Scholar]
- 7.Guernsey DL, Jiang H, Campagna DR, et al. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nature genetics. 2009;41(6):651–653. doi: 10.1038/ng.359. [DOI] [PubMed] [Google Scholar]
- 8.Camaschella C, Campanella A, De Falco L, et al. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood. 2007;110(4):1353–1358. doi: 10.1182/blood-2007-02-072520. [DOI] [PubMed] [Google Scholar]
- 9.Pagon RA, Bird TD, Detter JC, et al. Hereditary sideroblastic anaemia and ataxia: an X linked recessive disorder. J Med Genet. 1985;22(4):267–273. doi: 10.1136/jmg.22.4.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allikmets R, Raskind WH, Hutchinson A, et al. Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A) Hum Mol Genet. 1999;8(5):743–749. doi: 10.1093/hmg/8.5.743. [DOI] [PubMed] [Google Scholar]
- 11.Bekri S, Kispal G, Lange H, et al. Human ABC7 transporter: gene structure and mutation causing X-linked sideroblastic anemia with ataxia with disruption of cytosolic iron-sulfur protein maturation. Blood. 2000;96(9):3256–3264. [PubMed] [Google Scholar]
- 12.Maguire A, Hellier K, Hammans S, et al. X-linked cerebellar ataxia and sideroblastic anaemia associated with a missense mutation in the ABC7 gene predicting V411L. Br J Haematol. 2001;115(4):910–917. doi: 10.1046/j.1365-2141.2001.03015.x. [DOI] [PubMed] [Google Scholar]
- 13.Casas KA, Fischel-Ghodsian N. Mitochondrial myopathy and sideroblastic anemia. Am J Med Genet A. 2004;125A(2):201–204. doi: 10.1002/ajmg.a.20368. [DOI] [PubMed] [Google Scholar]
- 14.Bykhovskaya Y, Casas K, Mengesha E, et al. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA) Am J Hum Genet. 2004;74(6):1303–1308. doi: 10.1086/421530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernandez-Vizarra E, Berardinelli A, Valente L, et al. Nonsense mutation in pseudouridylate synthase 1 (PUS1) in two brothers affected by myopathy, lactic acidosis and sideroblastic anaemia (MLASA) J Med Genet. 2007;44(3):173–180. doi: 10.1136/jmg.2006.045252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeharia A, Fischel-Ghodsian N, Casas K, et al. Mitochondrial myopathy, sideroblastic anemia, and lactic acidosis: an autosomal recessive syndrome in Persian Jews caused by a mutation in the PUS1 gene. J Child Neurol. 2005;20(5):449–452. doi: 10.1177/08830738050200051301. [DOI] [PubMed] [Google Scholar]
- 17.Patton JR, Bykhovskaya Y, Mengesha E, et al. Mitochondrial myopathy and sideroblastic anemia (MLASA): missense mutation in the pseudouridine synthase 1 (PUS1) gene is associated with the loss of tRNA pseudouridylation. J Biol Chem. 2005;280(20):19823–19828. doi: 10.1074/jbc.M500216200. [DOI] [PubMed] [Google Scholar]
- 18.Ricketts CJ, Minton JA, Samuel J, et al. Thiamine-responsive megaloblastic anaemia syndrome: long-term follow-up and mutation analysis of seven families. Acta Paediatr. 2006;95(1):99–104. doi: 10.1080/08035250500323715. [DOI] [PubMed] [Google Scholar]
- 19.Neufeld EJ, Fleming JC, Tartaglini E, et al. Thiamine-responsive megaloblastic anemia syndrome: a disorder of high-affinity thiamine transport. Blood Cells Mol Dis. 2001;27(1):135–138. doi: 10.1006/bcmd.2000.0356. [DOI] [PubMed] [Google Scholar]
- 20.Bergmann AK, Sahai I, Falcone JF, et al. Thiamine-Responsive Megaloblastic Anemia: Identification of Novel Compound Heterozygotes and Mutation Update. J Peds. 2009 doi: 10.1016/j.jpeds.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abboud MR, Alexander D, Najjar SS. Diabetes mellitus, thiamine-dependent megaloblastic anemia, and sensorineural deafness associated with deficient alpha-ketoglutarate dehydrogenase activity. J Pediatr. 1985;107(4):537–541. doi: 10.1016/s0022-3476(85)80011-1. [DOI] [PubMed] [Google Scholar]
- 22.Rotig A, Bourgeron T, Rustin P, et al. Phenotypic expression of mitochondrial genotypes in cultured skin fibroblasts and in Epstein-Barr virus-transformed lymphocytes in Pearson syndrome. Muscle Nerve. 1995;3:S159–164. doi: 10.1002/mus.880181431. [DOI] [PubMed] [Google Scholar]
- 23.Rotig A, Colonna M, Bonnefont JP, et al. Mitochondrial DNA deletion in Pearson’s marrow/pancreas syndrome. Lancet. 1989;1(8643):902–903. doi: 10.1016/s0140-6736(89)92897-3. [DOI] [PubMed] [Google Scholar]
- 24.Rotig A, Cormier V, Blanche S, et al. Pearson’s marrow-pancreas syndrome. A multisystem mitochondrial disorder in infancy. J Clin Invest. 1990;86(5):1601–1608. doi: 10.1172/JCI114881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pearson HA, Lobel JS, Kocoshis SA, et al. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J Pediatr. 1979;95(6):976–984. doi: 10.1016/s0022-3476(79)80286-3. [DOI] [PubMed] [Google Scholar]
- 26.Allen RC, Zoghbi HY, Moseley AB, et al. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet. 1992;51(6):1229–1239. [PMC free article] [PubMed] [Google Scholar]
- 27.Pegoraro E, Schimke RN, Arahata K, et al. Detection of new paternal dystrophin gene mutations in isolated cases of dystrophinopathy in females. Am J Hum Genet. 1994;54(6):989–1003. [PMC free article] [PubMed] [Google Scholar]
- 28.Sleddens HF, Oostra BA, Brinkmann AO, et al. Trinucleotide repeat polymorphism in the androgen receptor gene (AR) Nucleic Acids Res. 1992;20(6):1427. doi: 10.1093/nar/20.6.1427-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Surinya KH, Cox TC, May BK. Identification and characterization of a conserved erythroid-specific enhancer located in intron 8 of the human 5-aminolevulinate synthase 2 gene. J Biol Chem. 1998;273(27):16798–16809. doi: 10.1074/jbc.273.27.16798. [DOI] [PubMed] [Google Scholar]
- 30.Astner I, Schulze JO, van den Heuvel J, et al. Crystal structure of 5-aminolevulinate synthase, the first enzyme of heme biosynthesis, and its link to XLSA in humans. Embo J. 2005;24(18):3166–3177. doi: 10.1038/sj.emboj.7600792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dierlamm J, Michaux L, Criel A, et al. Isodicentric (X)(q13) in haematological malignancies: presentation of five new cases, application of fluorescence in situ hybridization (FISH) and review of the literature. British journal of haematology. 1995;91(4):885–891. doi: 10.1111/j.1365-2141.1995.tb05405.x. [DOI] [PubMed] [Google Scholar]
- 32.Pondarre C, Campagna DR, Antiochos B, et al. Abcb7, the gene responsible for X-linked sideroblastic anemia with ataxia, is essential for hematopoiesis. Blood. 2007;109(8):3567–3569. doi: 10.1182/blood-2006-04-015768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wingert RA, Galloway JL, Barut B, et al. Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis. Nature. 2005;436(7053):1035–1039. doi: 10.1038/nature03887. [DOI] [PubMed] [Google Scholar]
- 34.Rouault TA, Tong WH. Iron-sulfur cluster biogenesis and human disease. Trends Genet. 2008;24(8):398–407. doi: 10.1016/j.tig.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.