

Abstract
We introduce microfluidics technologies as a key foundational technology for synthetic biology experimentation. Recent advances in the field of microfluidics are reviewed and the potential of such a technological platform to support the rapid development of synthetic biology solutions is discussed.
Keywords: microfluidic, technologies, engineering principles
1. Introduction
Over the last 50 years, discoveries in molecular biology, genomics and proteomics have helped to identify many key cellular components and processes. Concurrently, enabling technologies have been developed to allow manipulation and monitoring of biological systems. These include recombinant DNA technology, DNA synthesis and high-throughput screening technologies. Synthetic biology is a newly emerging discipline that exploits the recent advances in molecular and cellular biology to design, build and manufacture new biological systems and devices. Its uniqueness lies in the application of engineering principles to build biology from biology.
Synthetic biology is expected to have a great impact on our ability to produce bioenergy, biomaterials and novel therapeutics. It will also improve our capacity to use the sensing capabilities of natural systems, and more generally, should provide a seamless interface for biological engineers to interact with the human body or the environment. Several aspects of this nascent field have been previously discussed in some excellent review articles (Hasty et al. 2002; Kaern et al. 2003; McDaniel & Weiss 2005; Sismour & Benner 2005; Andrianantoandro et al. 2006; Heinemann & Panke 2006; Boyle & Silver 2009).
At a fundamental level synthetic biology is based on well-characterized and functionally predictable DNA ‘bioparts’, analogous to the manner in which modern computers depend on reliable electronic components to function. These bioparts can be assembled into newly designed systems using an engineering framework of modelling and simulation. For example, in bioenergy applications bacteria may be redesigned to produce large amounts of biofuels or hydrogen. Other applications include synthesizing complex drug molecules in bacterial and yeast cells or creating new biosensors to detect toxins or hospital-based infections. To realize the full potential of synthetic biology, it is essential that new technologies are established to allow both the robust characterization of bioparts and the generation of new bioparts with new functions.
1.1. Synthetic biology driving concepts
One of the main driving concepts in synthetic biology is the establishment of a conceptual framework based on the engineering design principles of standardization, modularity, abstraction and modelling. Modern engineering disciplines have developed robust methodologies and processes to cope with the increasing complexity of engineered systems. These key principles promote interoperability between systems, re-usability of existing components, and the management of complex solutions using software-based techniques. One aim of synthetic biology is to promote the application of these principles in the design and re-design of biological systems (Endy 2005).
1.2. Synthetic biology workflow
To illustrate the engineering approach to synthetic biology, the classical ‘engineering development cycle’ can be used to facilitate future synthetic biology projects (Kitney et al. 2007). The traditional engineering cycle has been developed in order to efficiently manage multi-step complex engineering projects. It is based on five well-defined steps, namely specification, design, modelling, fabrication and quality control (figure 1). The crucial task of the engineer is to identify, understand and interpret the constraints on a design in order to produce a successful solution. By understanding these constraints, engineers derive specifications for the limits within which a viable system could be produced and operated.
Figure 1.

Synthetic biology workflow.
Once defined, the specifications give the minimal standard expected for the system under development. From specifications different designs might need to be evaluated prior to full-scale production. Predictions on the performance of interim designs in relation to their specification is an essential part of this process. Confidence in the given designs usually comes from prior knowledge of how specific parts behave and interact with each other, as well as from detailed modelling and in silico testing of proposed designs. Only when a design performs to its specification will fabrication be initiated. When a system is constructed, it is essential to assess its performance that forms part of a quality control step. If the system and design fail to comply, other iterations of the engineering cycle might be necessary to achieve an appropriate design solution. Although this cycle is well established in engineering, there are few rigorous examples of applying the same approach in synthetic biology.
One of the major challenges in synthetic biology is in the reliability and robustness of new biological designs implemented in living organisms. However, constraining synthetic biology designs in an engineering-based workflow allows proper quantitative assessments of the design process. Although the application of these engineering approaches in biological engineering will be challenging, it is essential if synthetic biology is to fully develop into an application-driven science.
1.3. Foundational technologies for synthetic biology
In order to support this vision of synthetic biology, it will be critical to develop enabling technologies to support each step of the engineering cycle (figure 1). For example, to support the ‘design’ and ‘modelling’ steps, computer-aided design (CAD) tools to deal with the complexity of the task will be needed. Such tools were first developed in the fields of electrical and mechanical engineering, and have now reached a level of maturity that allows full in silico development. In synthetic biology, such CAD tools would ideally be linked to registries of existing and well-characterized bioparts (Arkin 2008). They would also provide modelling capabilities to estimate the static and dynamical properties of the synthetic biological system under study before its construction. A recent review surveys the ongoing efforts to provide synthetic biology CAD tools (Goler et al. 2008).
When it comes to the fabrication/construction and quality assessment of newly designed synthetic biology systems, many challenges still need to be addressed. Current synthetic biology projects require improved technologies for inexpensive DNA synthesis of medium-to-large size DNA constructs, including ultimately whole genomes (Baker et al. 2006; Czar et al. 2009). It will also be critical to find better technologies to assess in a systematic and robust way the responses or performances of biological designs in living organisms and non-living biochemical extracts. Currently, these steps are slow, costly and rarely automated or standardized. As an example, the first biopart characterization provided by Canton and colleagues took nearly 3 years to complete (Canton et al. 2008). It is a remarkable contribution that sets the scene for what the synthetic biology community should produce. Nonetheless, it only describes some properties of a homoserine lactone inducible promoter. Moreover, all measurements involved fluorescent protein expression, a conventional plate reader and a flow cytometer. Clearly, the task of characterizing biological systems is enormous and better technologies are required to make this process faster and more reliable. It will not be unusual for synthetic biology projects to go through many iterations of the engineering design cycle before achieving an optimal solution. It will therefore be imperative to develop technologies and processes that allow a near seamless integration between the different steps within the workflow.
One key part of the synthetic biology project pipeline is fabrication of the biological design, from DNA assembly to implementation within the chosen chassis. Once assembled, a quality control step will characterize the performance of the implemented biological design. These two aspects will ultimately enable the success of rational engineered synthetic biology projects. The application of microfluidics technologies specifically in these two areas could lead to such technologies becoming foundational in the field of synthetic biology, much like DNA synthesis and assembly. In the following sections, microfluidics technologies are introduced in the light of the technical challenges in synthetic biology design and implementation.
2. Microfluidics: a brief introduction
In simple terms, microfluidics describes the investigation of analytical systems that manipulate, process and control small volumes of fluids (typically on the picolitre to nanolitre scale). Development of microfluidic technologies has been stimulated by a variety of fundamental features that supplement system miniaturization. These features include the ability to process and handle small volumes of fluid, enhanced analytical performance (in terms of speed, efficiency and control) when compared with macroscale methods, low unit cost and perhaps most importantly the ability to access a large number of individual experiments per unit time.
Microfluidic devices employ functional components such as channels, filters, separation columns, electrodes and reactors, whose characteristic dimensions are most conveniently measured in micrometres. Importantly, they can be manufactured using a diversity of fabrication techniques originally developed in the microelectronics and semiconductor-processing industries. Indeed, since most features (such as channel widths and depths) are relatively large (>1 µm), fabrication is typically straightforward and can be achieved using well-established methodologies (Beebe et al. 2002). However, more recently soft lithographic techniques have become increasingly popular for rapid prototyping of elastomeric devices using materials such as polydimethylsiloxane (Quake & Scherer 2000).
A material effect of reactor miniaturization is that fluid properties become increasingly controlled by viscous forces rather than inertial forces. This relationship can be understood through use of the dimensionless Reynolds number (Re),
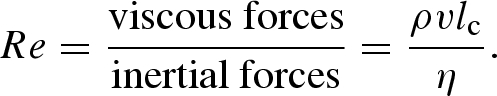 |
Here ρ is the density of the fluid, η the viscosity of the fluid, v the characteristic velocity and lc the characteristic length. Because small length scales are characteristic of microfluidic systems, associated Reynolds numbers are also small (Re is usually less than 1), indicating that flow is laminar (Stone & Kim 2001). For example, in blood capillaries, Reynolds numbers are typically <101–2. This contrasts with macroscale conduits (Re > 103) in which flow regimes are almost always turbulent. Significantly, in laminar flow regimes, fluid follows streamlines that are constant with time and do not overlap. As a result, they are highly repeatable and controllable. Complete reviews of the physics of microfluidic flows can be found in Beebe et al. (2002) and Squires & Quake (2005).
Since the length scales associated with microfluidic structures are small, diffusion times can be extremely short. Accordingly, diffusion aids mass transport for flows in microdevices. Unfortunately, for slow microfluidic flows diffusion alone is usually not sufficient to fully mix fluids. In this situation, rapid mixing with low reagent consumption can be achieved using chaotic advection. Put simply, chaotic advection enhances mixing in laminar-flow systems by continuously ‘stretching’ and ‘refolding’ concentrated solute volumes (Song et al. 2006). Numerous microfluidic mixers designed to be effective in the laminar flow regime have been reported in the literature. Further discussion of both passive and active micromixers can be found elsewhere (Bessoth et al. 1999; Campbell & Grzybowski 2004; Nguyen & Wu 2005).
For reactive systems, a key issue faced when using continuous flow microfluidics is the parabolic nature of pressure-driven laminar flows that leads to hydrodynamic dispersion (Krishnadasan et al. 2004). This means the progression of chemical reactions is influenced and complicated by the local features of the velocity distribution in the channel (Stone & Kim 2001). Optimal microfluidic geometries that minimize solute dispersion have been explored extensively and are reviewed elsewhere (Dutta et al. 2006). In addition, the high surface area to volume ratios characteristic of microscale environments, although beneficial in terms of thermal management, dictate that a significant proportion of the contained fluid will be in intimate contact with the microchannel surface during transit and thus adsorption of macromolecules is a significant issue.
As noted, microfluidic systems have received much attention in recent years because of the promise of significantly improved analytical performance when compared with conventional instrument formats. In the proceeding sections the latest advances in the design and development of microfluidic systems for synthetic biology applications will be described. In particular, devices supporting the fabrication step of the synthetic biology development cycle—synthesis and sequencing of DNA and cell-based and cell-free protein expression—are highlighted.
2.1. Biological synthesis in microfluidic devices
Because of the unique environments provided by microfluidic networks, a variety of synthetic processes can be performed in both continuous flow and batch formats. Increased efficiencies of mixing and separation combined with high rates of thermal and mass transfer make microfluidic reactors ideal for processing complex reactions, improving reaction selectivities, reducing reaction times and generating high-quality products. Indeed, DNA amplification, combinatorial and high-throughput small molecule chemistries, immunoassays, and tissue and cell culturing are all areas that have benefited from advances in microfluidic chip technology (Khandurina & Guttman 2002; Andersson & van den Berg 2003; Bange et al. 2005; Zhang et al. 2006).
In the current context Kong and co-workers reported initial steps towards gene synthesis in a microfluidic device. In these studies the parallel synthesis of 1 kb long genes from minute concentrations of oligonucleotides was demonstrated. Specifically, both synthesis and amplification are performed concurrently for a range of genes and gene segments, including a full-length green fluorescent protein (GFP) construct (993 bp). Four parallel nanolitre-volume chambers are loaded with polymerase construction and amplification (PCA) mixtures with a primary valve controlling all the input lines and a secondary valve addressing all the output lines. On an underlying layer, a series of water lines are filled to minimize evaporation during thermocycling. Significantly, synthesis is achieved using extremely small concentrations of each oligonucleotide. In addition, the reduced reactor volumes minimize the quantity of reagent needed and hence lower the overall cost per reaction. What is perhaps most exciting in terms of potential application in synthetic biology is the possibility that such reactors (for high-quality DNA template synthesis) could be integrated with additional microfluidic modules for in vitro protein expression (Kong et al. 2007).
2.2. Sequencing-by-synthesis in microfluidic devices
In 2004, Kartalov and Quake reported the construction of a microfluidic chip capable of reading up to 4 bp using a sequencing-by-synthesis paradigm (Kartalov & Quake 2004). The technique involves exposing a primed DNA template to known standard nucleotide, its fluorescently tagged analogue and a DNA polymerase. If the tagged nucleotide is the complement of the template base at the end of the primer, the polymerase will extend the primer with that nucleotide. After a wash step, the fluorescence signal reveals if the nucleotide is correct. If it is not, the other nucleotides, their fluorescently tagged analogue and DNA polymerase are sequentially delivered to the template and the resulting fluorescence signal is measured to allow base identification. The microfluidic system (consisting of multiple microfluidic channels and valves) offers significant improvements to analytical throughput owing to fast reagent diffusion. Indeed, the duration of a sequencing run using the microfluidic design is comparable to, or better than, the best current technologies. In addition, a microfluidic approach offers parallel processing of DNA templates and consumption of small volumes of reagents, thereby reducing the cost of individual measurements owing to the economy of scale. While this demonstration of the sequencing-by-synthesis technique is the first of its kind in a microfluidic device, the small read length of 3–4 bp, which is determined by signal-to-noise limitations, is a clear restriction in integrated lab-on-a-chip applications. Kartalov and Quake indicate that improvements to both the surface chemistry and polymerase will enable significantly longer read lengths. In fact, the company Helicos Biosciences Corporation was formed from the sequencing-by-synthesis foundational technology and has reported read length capabilities greater than 25 bp from individual templates. Furthermore, Pacific Biosciences utilizes a single molecule real time (SMRT) chip consisting of thousands of zero-mode waveguides that provide extremely small detection volumes (cylindrical holes approx. 100 nm in diameter) to employ sequencing-by-synthesis. The method utilizes nucleotides with a fluorescent dye attached to the phosphate chain rather than to the base; the phosphate chain is cleaved when the nucleotide is incorporated into the strand and diffuses rapidly out of the detection volume, leaving a low background in the detection volume. These modifications enable read lengths of tens of thousands of nucleotides (Eid et al. 2009).
2.3. Cell-based gene expression in microfluidic devices
Thompson et al. (2004) have recently described a microfluidic-based living cell array (LCA) to perform high-throughput molecular stimulation and continuous monitoring of expression events in individual live cells. The device utilizes a dilution module consisting of a microfluidic gradient generator that creates eight outlet concentrations from two inlet concentrations that feed into eight separate chambers in the downstream cell culture module (figure 2b). Cells are introduced and allowed to attach in the cell culture module. Subsequently, a gene expression inducer is diluted in the dilution module and then delivered to the individual cell culture chambers. Gene expression dynamics were then extracted using time-lapsed phase contrast and fluorescence imaging. The LCA method is of particular interest since a variety of conditions can be screened simultaneously and thus affords efficient exploration of a wide parameter space for molecular stimulation of cells. The automated microfluidic LCA platform has been used as a real-time gene expression array to monitor coordinated temporal expression activity of multiple pathways (King et al. 2007) and to monitor expression activity with temporal input stimuli (King et al. 2008).
Figure 2.

(a) Schematic of the living cell array (LCA) device. (b) The microfluidic network design of the LCA and images of dilution module and cell culture chamber (Thompson et al. 2004).
In addition, Balagadde et al. (2005) demonstrated programmed population control of Escherichia coli cells with a microchemostat enabling long-term culture. This microfluidic bioreactor was used to monitor the growth dynamics of cell populations using a synthetic circuit that regulates the cell density by negative feedback based on quorum sensing. The circuit is induced with isopropyl-β-thiogalactopyranoside (IPTG), and subsequently the cell density is detected by a signalling molecule (acyl-homerserine lactone or AHL), which modulates the expression of the killer gene (lacZα-ccdB) that controls the cell death rate. The microchemostat consists of 16 nl fluidic loops with valves that meter medium in, waste out and recover cells. The oscillatory cell growth induced by the synthetic circuit is more stable in the microchemostat than in normal macroscale culture formats, and direct visualization of cells by optical microscopy can be used to obtain gene expression dynamics over a range of conditions and at high throughput (Balagadde et al. 2005).
2.4. Cell-free protein expression in microfluidic devices
Very recently, Khnouf et al. (2009) carried out cell-free expression of luciferase in a passively pumped microfluidic device consisting of an array of 192 microchannels connected by pairs of wells. The device is designed to be compatible with a standard 384-well microplate format (figure 3a). To initiate the assay, a large drop of the protein expression solution is placed in the outlet well and a smaller drop of nutrient solution is placed in the inlet well. The difference in the surface tension of the two drops drives the fluid from the inlet to the outlet where luciferase expression takes place (figure 3b,c). Importantly, the passive pumping mechanism allows continuous replenishment of nutrients and enhances expression levels over five times. For the investigated operating conditions, the amount of luciferase expression is found to be dependent on the volume, the feeding frequency and delivery rate of nutrient solution and independent of the volume of expression solution. The simple 192-channel design offers simultaneous expression of protein under different conditions, gives two orders of magnitude savings in reagent consumption compared with commercial expression instrumentation and is compatible with commercial reagent dispensers and microplate readers. Significantly, these continuous flow microfluidic devices represent a first step towards the design and development of modules for synthetic biology applications. Nevertheless, optimization and integration of these types of designs will be required to further the goals of synthetic biology.
Figure 3.

(a) Image of the passive pumping microfluidic device for cell-free protein expression. The design is compatible with the 384-well microplate format. (b) A large droplet is placed on the outlet and a smaller droplet is placed on the inlet. Passive pumping from the inlet to the outlet occurs because of the difference in surface tension between the droplets. (c) Protein expression consisting of DNA translation and protein translation occurs in the outlet droplet (Khnouf et al. 2009).
3. Segmented flow microfluidics
An emergent area of microfluidic research is the generation and utilization of segmented (or multiphase) flows (Song et al. 2006; Huebner et al. 2008). Here, flow instabilities within microchannel networks are used to spontaneously form droplets when multiple laminar streams of aqueous reagents are injected into an immiscible carrier fluid. These aqueous droplets are dispersed in the continuous oil phase and define femtolitre to nanolitre volumes of aqueous reagents. Importantly, the droplets do not touch the microchannel surfaces and can be stabilized against coalescence using surfactant molecules localized at the oil–water interface. The ability to confine reagents inside these small volume droplets eliminates hydrodynamic dispersion, offers rapid and facile mixing (Song et al. 2003; Song & Ismagilov 2003; Tice et al. 2003, 2004; Bringer et al. 2004; Liau et al. 2005) and provides for a well-defined and isolated reaction environment (Joanicot & Ajdari 2005). Such droplets have controllable compositions, and residence (or reaction) times are directly related to their position in the microfluidic channel after formation, i.e. for a uniform velocity u, every position d along the channel corresponds to a time point t, where t = u/d (Song et al. 2003).
3.1. Bulk in vitro compartmentalization
In vitro compartmentalization (IVC) of reactions in bulk water-in-oil emulsions has been identified as a promising method for massively parallel processing (Griffiths & Tawfik 2006). In principle, such an approach allows access to the huge combinatorial parameter spaces required for screening, selecting and developing both natural and ‘artificial’ biological and macromolecular systems by directed evolution (Kelly et al. 2007). For example, aqueous solutions containing a gene library could be emulsified with a homogenizer in an oil–surfactant mixture within a matter of minutes to produce a water-in-oil emulsion containing in excess of 1010 droplets per millilitre. Such a combinatorial approach would greatly benefit synthetic biology, providing a powerful paradigm in the characterization of biological systems. Each droplet would constitute an independent experiment where the inputs of the synthetic biology system would be stimulated in a specific way. A large collection of droplets could potentially provide a rich source of data to assess the behaviour of the synthetic biology system over a wide range of conditions.
Although IVC can decrease volumes by as much as 109 compared with conventional microtitre plate screening methods (Kelly et al. 2007), control of emulsion characteristics in bulk systems is difficult since the formation of droplets via bulk methods generates a large distribution of microdroplet sizes. For example, Tawfik and Griffiths have reported size distributions in excess of 100 per cent in diameter when forming aqueous microdroplets in oil (Tawfik & Griffiths 1998). This polydispersity can, however, be minimized using a homogenizer and higher stirring frequencies.
Emulsion polymerase chain reaction (ePCR) in bulk systems has been demonstrated and utilized as a way of amplifying DNA fragments (Nakano et al. 2003) for two novel sequencing techniques using IVC. The first is multiplex polony sequencing developed by Church and co-workers where PCR colonies (or polonies) consisting of short genomic fragments are compartmentalized and amplified by ePCR into beads. Subsequently, the microbeads are anchored in an array and the sequence is determined one base at a time by ligation of fluorescent oligonucleotides (Shendure et al. 2005). This technology has been transferred to commercial systems. The Polonator G.007, by Dover Systems, gives a 13 bp read per DNA tag in the array and the SOLiD™ 3 System from Applied Biosystems gives a slightly longer read length of 50 bp per DNA tag in the array.
A second important sequencing technique is picotitre plate pyrosequencing (Margulies et al. 2005). Here, ePCR is first employed to amplify DNA fragments on microbeads and then followed by a pyrosequencing step. Pyrosequencing is analagous to sequencing-by-synthesis (see §2.2) but instead relies on detection of pyrophosphate release on nucleotide incorporation (Ronaghi et al. 1996). This technology has been developed into a commercial product, the Genome Sequencer FLX System from 454 Life Sciences, and can achieve read lengths of 400 bases.
Extensive research has been carried out using IVC since the original pioneering work in the late 1990s. For a more complete description of the range of high-throughput, cell-free technologies utilizing IVC, the reader is referred to the reviews elsewhere (Griffiths & Tawfik 2006; Kelly et al. 2007). One limitation associated with IVC in bulk emulsions is that the delivery of additional reagents via droplet fusion for more complex multi-step processes is difficult, or often impossible (Kelly et al. 2007). Microfluidic IVC technology, as described in detail in the proceeding sections, offers precise formation, control and manipulation of droplets, which may enable the realization of these complex applications.
3.2. Microfluidic in vitro compartmentalization
Production of microdroplets within microfluidic systems offers important advantages over bulk methods for creating water-in-oil emulsions for IVC. As previously noted, highly monodisperse droplet populations ranging in size from 20 to 1000 µm can be formed by shearing one fluid phase with the other, and varying flow rates of both the continuous and the dispersed phases (Anna et al. 2001; Xu et al. 2001; Nisisako et al. 2002; Garstecki et al. 2006). Indeed, such microdroplet systems have been used for numerous applications including the production of microparticles by polymerization (Xu et al. 2001), the synthesis of nanoparticles (Khan et al. 2004; Shestopalov et al. 2004), the encapsulation of cells or subcellular organelles (El-Ali et al. 2005; He et al. 2005), screening of protein crystallization (Zheng et al. 2003), kinetic measurements (Song & Ismagilov 2003), chemical amplification using synthetic reaction networks (Gerdts et al. 2004) and DNA analysis (Burns et al. 1998). More detailed reviews summarizing the applications of microdroplet-based systems in chemistry and biology can be found elsewhere (Song et al. 2006; Huebner et al. 2008). In terms of the needs of synthetic biology, such controlled droplet formation is critical in guaranteeing reproducible and stable experimental conditions during characterization.
3.2.1. Microdroplet control and dynamics
The ability to control and manipulate individual droplets is of critical importance when performing complex chemical or biological analyses on chip. Numerous passive microfluidic channel geometries have been demonstrated to control droplet splitting (Song et al. 2003; Link et al. 2004; Tan et al. 2004), droplet fusion (Song et al. 2003; Tan et al. 2004; Bremond et al. 2008; Niu et al. 2008) and droplet sorting (Tan et al. 2004). Active approaches for droplet manipulation have also been demonstrated and applied to valving, sorting, fusing and splitting (Priest et al. 2006; Ahn et al. 2006a,b; Baroud et al. 2007). Some of these structures are summarized in figure 4.
Figure 4.

(a) Passive droplet break-up at T-junction. The flow resistance of the two side arms controls the size of the daughter droplets. Image from Link et al. (2004). (b) Flows of small, medium and large droplets in the passive merging chamber. Image from Niu et al. (2008). (c) (i) Complete and (ii) incomplete passive sorting by channel resistance. Image from Tan et al. (2004). (d) (i) With the laser off, droplets split into equal drops at bifurcation and (ii) with the laser on, asymmetric daughter droplets are formed. Image from Baroud et al. (2007). (e) Droplets of different sizes are coalesced by an electric field. Image from Ahn et al. (2006b). (f) By applying an electric field, droplets are redirected into the collect channel (see d). Image from Ahn et al. (2006a). (g) Using multiple electrodes on left and right, the droplets can be bi-directionally manipulated. Image from Ahn et al. (2006a).
3.2.2. The physics of multiphase flow
There are many competing physical phenomena governing multiphase flows (for both gases and liquids). However, for small-scale liquid–liquid flows (representative of microdroplets) viscous and interfacial forces generally dominate. The dimensionless group that relates these two forces is the capillary number,
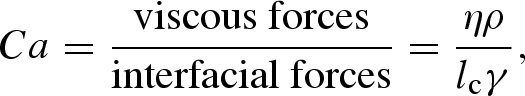 |
where η is the viscosity of the continuous phase, lc the characteristic length and γ the interfacial tension. For flows of liquid–liquid systems with high γ (and thus low Ca), the fluids segment into droplets readily over a wide range of flow conditions, while flows stratify when there are no interfacial forces between the liquids (i.e. high Ca; Shui et al. 2007). Hence from the definition of Ca, flow patterns can be modified by modulating the interfacial forces between the fluids through the addition of surfactants, adjustment of the continuous fluid viscosity, a change in the fluid velocity or alterations in the microfluidic geometry. For more involved descriptions of flow characteristics and dimensionless groups for generalized multiphase flows, the reader is referred to other comprehensive reviews (Baroud & Willaime 2004; Gunther & Jensen 2006; Shui et al. 2007).
In the proceeding sections of this article, novel droplet-based microfluidic systems demonstrating polymerase chain reaction (PCR) and cell-based and cell-free protein expression are described.
3.2.3. PCR in microdroplets
PCR is unquestionably one of the most important tools in modern biology, with applications ranging from forensics to diagnostics, cloning and sequencing (deMello 2003). Although simple to implement, PCR in macroscale thermal cyclers is slow and inefficient because of large thermal masses. Transferral of PCR to microfluidic formats has been shown to be efficient in reducing the cost of fabrication and consumption of biological samples, but also time of DNA amplification. Recently, Kiss and co-workers and Schaerli and co-workers have reported microfluidic devices for performing PCR in microdroplets. Kiss et al. (2008) encapsulate the PCR reagents into picolitre sized droplets that then travel along long serpentine channels spatially arranged such that the droplets pass through alternating denaturation and annealing zones for efficient amplification. They demonstrate the amplification of a 245 bp DNA template in 35 min at dilute concentrations, as low as one template in 167 droplets (0.003 pg µl−1). Similarly, Schaerli et al. (2009) have developed a microfluidic device where DNA amplification is achieved by moving droplets radially across a circular device and through different temperature zones (hot at the centre and cool at the periphery). A schematic of the fluidic network is shown in figure 5. The authors reported successful amplification of an 85 bp template at four starting concentrations, including amplification of a single copy of DNA. Encapsulation of reagents into droplets offers significant advantage over continuous flow microfluidic PCR (Kopp et al. 1998) since template and polymerase adsorption to channel walls is avoided. The authors also show amplification of a 505 bp DNA fragment, which in principle is long enough to code for small proteins. Further optimization of the thermal cycling and residence times is expected to allow for efficient amplification of even longer DNA templates. A droplet-based PCR device would have many advantages as a tool in synthetic biology. For example, it could be an essential building block for on-chip DNA fragment assembly, using ligation-independent cloning techniques (Yehezkel et al. 2008). It could also be an essential part of a directed evolution framework where error-prone PCR could be used to generate diversity (Yehezkel et al. 2008).
Figure 5.

Radial droplet-based PCR device. Oil enters at inlet A and two aqueous fluids enter at inlets B1 and B2 and form droplets at the T-junction C. The droplets pass through the hot zone at D where denaturation takes place and primer annealing and template extension occur in the cooler zone E. After 34 cycles, the droplets exit at F (Schaerli et al. 2009).
3.2.4. Cell-based protein expression in microdroplets
Huebner et al. (2007) have performed quantitative analysis of the expression of yellow fluorescent protein in E. coli cells encapsulated within microdroplets and simultaneously measured drop size, fluorescence yield and cell occupancy. A confocal laser-induced fluorescence detection system (figure 6b) is capable of resolving single fluorophore events at frequencies greater than 100 kHz. On-line fluorescence readout both low and high cell occupancies is given in figure 6c,d, respectively. These time-resolved fluorescence measurements of encapsulated E. coli cells expressing the yellow fluorescent mutant ‘Venus’ under flow in microdroplets compare well with conventional absorbance measurements, suggesting that droplet-based processing mimics expression and growth in bulk. The ability to conduct cell-based expression in droplets is key for synthetic biology. It provides an opportunity to develop and characterize synthetic biology systems in vivo. For example, by taking advantage of the combinatorial power provided by droplet-based microfluidics, many different hosts, or chassis, could be tested simultaneously. Depending on experimental conditions, single cell analysis could also be performed in droplets. When it comes to system optimization, an in vivo evolution framework could be implemented by carefully controlling the selective pressure applied on the droplet-based culture.
Figure 6.

(a) Image of aqueous droplet flow in the microdevice at a T-junction. Inlets A and B are aqueous solutions and C is a water-immiscible oil solution. (b) Schematic of the laser induced fluorescence optical set-up. (c,d) Optical readout of 0.5 s traces recorded under low (cell suspension: 0.3 µl min−1; media: 2.7 µl min−1) and high (cell suspension: 2.1 µl min−1; media: 0.9 µl min−1) cell loading conditions. Arch-shaped signal (see insets) corresponds to the weak fluorescent media. Vertical spikes correspond to signal of the expressed fluorescent protein (Huebner et al. 2007).
3.2.5. Cell-free protein expression in microdroplets
Dittrich et al. (2005) demonstrated cell-free protein expression in water-in-oil emulsion drops formed in a microfluidic device. A schematic diagram of their design concept for in vitro evolution of proteins in compartmentalized flow in microfluidic channels is given in figure 7. The compounds for cell-free expression (GFP and RNA polymerase) in one inlet stream and the templates from the gene library (amino acids, nucleotides and energy equivalents) in the other inlet stream are brought together when they reach two oil streams at a flow-focusing junction. Microdroplets are formed and the contents are able to rapidly mix inside the droplet. The droplets are stored in the end reservoir at 37°C for 50 min and flowed back into the microchannel for on-line detection. Laser-induced fluorescence by epifluorescence confocal microscopy is used for transient high-sensitivity analysis of the contents of the emulsions, which were estimated to be 60 nM GFP on average.
Figure 7.

Schematic of the in vitro evolution of proteins in microfluidic channels (Dittrich et al. 2005).
Building on the work of Dittrich et al. (2005), Courtois et al. (2008) created an integrated device for measuring cell-free protein expression in microdroplets. Droplets are formed at a flow-focusing junction, pass through a winding channel to rapidly mix the contents by chaotic advection, and are finally stored in a reservoir with V-shaped entrances and exits to slow entering droplets gradually to prevent fusion and to avoid droplets trapped in corners as they leave the reservoir. Solutions obtained from a commercial in vitro transcription and translation (IVTT) kit were mixed by bringing together two streams at equal flow rates at the point of droplet formation, one containing E. coli lysate and amino acids and the other containing the remainder of the kit (ribosomes, tRNA, translation factors and ribonucleotides) and the DNA template (pIVEX-GFP with a T7 promoter). The concentration of DNA template was diluted substantially (to 0.1, 0.3 and 0.5 pM) to ensure that on an average there is less than one template molecule per droplet; at these concentrations the majority of droplets contain no template (80% at 0.5 pM), a small amount contains more than one template (2% at 0.5 pM), and the remaining contain a single template (18% at 0.5 pM; Courtois et al. 2008). After 6.5 h of incubation at room temperature, the expression is measured by laser-induced fluorescence detection at the outlet to the reservoir where droplets are allowed to flow out. It was found that the number of molecules of GFP produced in droplets containing one template was between 12 000 and 30 000, depending on the pre-emulsion pIVEX-GFP concentration, which is much higher than protein expression in droplets with multiple template copies (4000–8000 protein molecules) using the same microdroplet configuration. The demonstration of protein expression using a single template in a droplet shows promise for using microdroplet technology platforms in synthetic biology. Cell-free expression systems can be foreseen as a potential chassis for synthetic biology systems. Therefore a droplet-based version would provide an ideal high-throughput platform to develop and characterize synthetic biology systems in vitro.
4. Detection
There is a science to be developed concerned with the characterization of re-useable and modular bioparts (Arkin 2008; Canton et al. 2008). For synthetic biology to reach its full potential, it must provide bioparts with enough information attached so that a predictable behaviour can be determined and the parts can be re-used without expert knowledge. The best way of characterizing bioparts is open to debate in the community (Lucks et al. 2008), and is likely to remain open for years to come. Ultimately, to build robust and reliable biological systems, it is critical to measure device performance for a wide array of conditions and thus have available an array of detection techniques to assess biological performance. As evidenced by the proof-of-principle continuous and microdroplet-based microfluidic devices described in the previous sections, fluorescence detection is the most common method utilized because of its demonstrated measurement sensitivity in small detection volumes. Conventional laser-induced fluorescence (LIF) is often used and is capable of high sensitivity with detection limits in the picomolar range (Schwarz & Hauser 2001).
Electrochemical detection (amperometric, conductivity and potentiometry) is also regarded as a promising detection method for microfluidic systems with measurement sensitivities approaching that of fluorescence detection (Wang 2002). In addition, the detector and instrumentation can be miniaturized on-chip for electrochemical methods, which is a significant improvement over the bulky off-chip instrumentation required for LIF and offers the possibility of truly self-contained devices with portable detection. To our knowledge, electrochemical detection has not yet been used in a microfluidic device towards a synthetic biology application. However, electrochemical detection used for other lab-on-a-chip systems has been previously reviewed (Wang 2002).
Finally, it should also be noted that alternative techniques have been developed for detection in microfluidic devices. These include methods based on chemiluminescence, electrochemiluminescence, infrared spectroscopy, Raman spectroscopy, absorbance spectroscopy and refractive index variation (Wang 2002; Viskari & Landers 2006).
5. Concluding remarks
Microfluidic technologies can be described broadly by the abstraction hierarchy given in figure 8. Most important to the synthetic biologist are those at the highest levels, the lab-on-a-chip layer and the protocol units layer, which enable the fabrication and quality control stages of the synthetic biology development cycle (cf. figure 1). The lower levels of the abstraction hierarchy describe the basic operations, basic components, and the physical layers. While further development of these low-level layers will be necessary for realization of true lab-on-a-chip systems for synthetic biology applications, in the future it would be ideal to have standardized microfluidic platforms and tool-kits so that the user can focus efforts on the aims of synthetic biology rather than the design and operation of the microfluidic system.
Figure 8.

Microfluidics abstraction hierarchy.
As previously described, for synthetic biology to advance to a position where it can be used for bioenergy, biomaterial or therapeutic applications, technology platforms that enable rapid characterization and fabrication of bioparts will be required. Adapting and developing existing microfluidic tools, and particularly microdroplet technologies that provide controllable, small volume compartments, provide a way toward this goal. Already microdroplet technologies are readily used for characterization of wide ranges of droplet conditions and these droplet conditions can be easily varied by tuning flow rates. Utilizing microdroplet technology, fundamental characterization for synthetic biology is already within close reach. For example, the relationship between inputs and outputs to systems (i.e. testing the effect of varying input concentrations on output expression) can be rapidly tested and mapped as transfer functions. While microdroplet systems are becoming more investigated than continuous flow microfluidic tools for testing and characterization applications, further development will be required to provide an entire suite of essential synthetic biology protocols (e.g. synthesis, sequencing, etc.).
Numerous continuous flow microfluidic and microdroplet-based devices have been described in this review. These are proof-of-principle demonstrations of microfluidic devices that conduct the types of protocols that will be necessary for fabrication and quality control in the development cycle for synthetic biology. These microfluidic platforms offer many potential benefits for advancing synthetic biology including automated, high throughput, and multiplexed operation and scalability through parallelized design. In addition, the underlying physics of microfluidic flow (i.e. laminar flow) generates flow environments that are highly controllable and reproducible. These advantages, along with small volumes requirements of reagents and samples, allow significantly reduced costs of operation and rapid testing compared with existing macroscale technologies. With further development and optimization as well as combination of these fabrication protocols, integrated lab-on-a-chip systems are achievable and are poised to advance the field of synthetic biology.
Footnotes
One contribution to a Theme Supplement ‘Synthetic biology: history, challenges and prospects’.
References
- Ahn K., Kerbage C., Hunt T. P., Westervelt R. M., Link D. R. 2006a. Dielectrophoretic manipulation of drops for high-speed microfluidic sorting devices. Appl. Phys. Lett. 88, 024104 ( 10.1063/1.2164911) [DOI] [Google Scholar]
- Ahn K., Agresti J., Chong H., Marquez M. 2006b. Electrocoalescence of drops synchronized by size-dependent flow in microfluidic channels. Appl. Phys. Lett. 88, 264105 ( 10.1063/1.2218058) [DOI] [Google Scholar]
- Andersson H., van den Berg A. 2003. Microfluidic devices for cellomics: a review. Sens. Actuators B 92, 315–325. ( 10.1016/S0925-4005(03)00266-1) [DOI] [Google Scholar]
- Andrianantoandro E., Basu S., Karig D. K., Weiss R. 2006. Synthetic biology: new engineering rules for an emerging discipline. Mol. Syst. Biol. 2, 1–14. ( 10.1038/msb4100073) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anna S. L., Bontoux N., Stone H. A. 2001. Formation of dispersions using ‘flow focusing’ in microchannels. Appl. Phys. Lett. 82, 364–366. ( 10.1063/1.1537519) [DOI] [Google Scholar]
- Arkin A. 2008. Setting the standard in synthetic biology. Nat. Biotechnol. 26, 771–774. ( 10.1038/nbt0708-771) [DOI] [PubMed] [Google Scholar]
- Baker D., Church G., Collins J., Endy D., Jacobson J., Keasling J., Modrich P., Smolke C., Weiss R. 2006. Engineering life: building a fab for biology. Sci. Am. 294, 44–51. [DOI] [PubMed] [Google Scholar]
- Balagadde F. K., You L., Hansen C. L., Arnold F. H., Quake S. R. 2005. Long-term monitoring of bacteria undergoing programmed population control in a microchemostat. Science 309, 137–140. ( 10.1126/science.1109173) [DOI] [PubMed] [Google Scholar]
- Bange A., Halsall H. B., Heineman W. R. 2005. Microfluidic immunosensor systems. Biosens. Bioelectron. 20, 2488–2503. ( 10.1016/j.bios.2004.10.016) [DOI] [PubMed] [Google Scholar]
- Baroud C. N., Willaime H. 2004. Multiphase flows in microfluidics. Physique 5, 547–555. ( 10.1016/j.crhy.2004.04.006) [DOI] [Google Scholar]
- Baroud C. N., de Saint Vincent M. R., Deville J.-P. 2007. An optical toolbox for total control of droplet microfluidics. Lab Chip 7, 1029–1033. ( 10.1039/b702472j) [DOI] [PubMed] [Google Scholar]
- Beebe D. J., Mensing G. A., Walker G. M. 2002. Physics and applications of microfluidics in biology. Ann. Rev. Biomed. Eng. 4, 261–286. ( 10.1146/annurev.bioeng.4.112601.125916) [DOI] [PubMed] [Google Scholar]
- Bessoth F. G., deMello A. J., Manz A. 1999. Microstructure for efficient continuous flow mixing. Anal. Commun. 36, 213–215. [Google Scholar]
- Boyle P. M., Silver P. A. 2009. Harnessing nature's toolbox: regulatory elements for synthetic biology. J. R. Soc. Interface. 6, S535–S546( 10.1098/rsif.2008.0521.focus) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremond N., Thiam A. R., Bibette J. 2008. Decompressing emulsion droplets favor coalescence. Phys. Rev. Lett. 100, 024501 ( 10.1103/PhysRevLett.100.024501) [DOI] [PubMed] [Google Scholar]
- Bringer M. R., Gerdts C. J., Song H., Tice J. D., Ismagilov R. F. 2004. Microfluidic systems for chemical kinetics that rely on chaotic mixing in droplets. Phil. Trans. R. Soc. Lond. A 362, 1087–1104. ( 10.1098/rsta.2003.1364) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns M. A., et al. 1998. An integrated nanoliter DNA analysis device. Science 282, 484–487. ( 10.1126/science.282.5388.484) [DOI] [PubMed] [Google Scholar]
- Campbell C. J., Grzybowski B. A. 2004. Microfluidic mixers: from microfabricated to self-assembling devices. Phil. Trans.: Math. Phys. Eng. Sci. 362, 1069–1086. [DOI] [PubMed] [Google Scholar]
- Canton B., Labno A., Endy D. 2008. Refinement and standardization of synthetic biological parts and devices. Nat. Biotechnol. 26, 787–793. ( 10.1038/nbt1413) [DOI] [PubMed] [Google Scholar]
- Courtois F., Olguin L. F., Whyte G., Bratton D., Huck W. T. S., Abell C., Hollfelder F. 2008. An integrated device for monitoring time-dependent in vitro expression from single genes in picolitre droplets. ChemBioChem 9, 439–446. ( 10.1002/cbic.200700536) [DOI] [PubMed] [Google Scholar]
- Czar M. J., Anderson J. C., Bader J. S., Peccoud J. 2009. Gene synthesis demystified. Trends Biotechnol. 27, 63–72. ( 10.1016/j.tibtech.2008.10.007) [DOI] [PubMed] [Google Scholar]
- deMello A. J. 2003. Microfluidics: DNA amplification moves on. Nature 422, 28–29. ( 10.1038/422028a) [DOI] [PubMed] [Google Scholar]
- Dittrich P., Jahnz M., Schwille P. 2005. A new embedded process for compartmentalized cell-free protein expression and on-line detection in microfluidic devices. ChemBioChem 6, 811–814. ( 10.1002/cbic.200400321) [DOI] [PubMed] [Google Scholar]
- Dutta D., Ramachandran A., Leighton D. T. 2006. Effect of channel geometry on solute dispersion in pressure-driven, microfluidic systems. Microfluid. Nanofluid. 2, 275–290. ( 10.1007/s10404-005-0070-7) [DOI] [Google Scholar]
- Eid J., et al. 2009. Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138. [DOI] [PubMed] [Google Scholar]
- El-Ali J., Gaudet S., Gunther A., Sorger P. K., Jensen K. F. 2005. Cell stimulus and lysis in a microfluidic device with segmented gas–liquid flow. Anal. Chem. 77, 3629–3636. [DOI] [PubMed] [Google Scholar]
- Endy D. 2005. Foundations for engineering biology. Nature 438, 449–453. ( 10.1038/nature04342) [DOI] [PubMed] [Google Scholar]
- Garstecki P., Fuerstman M. J., Stone H. A., Whitesides G. M. 2006. Formation of droplets and bubbles in a microfluidic T-junction—scaling and mechanism of break-up. Lab Chip 6, 437–446. ( 10.1039/b510841a) [DOI] [PubMed] [Google Scholar]
- Gerdts C. J., Sharoyan D. E., Ismagilov R. F. 2004. A synthetic reaction network: chemical amplification using nonequilibrium autocatalytic reactions coupled in time. J. Am. Chem. Soc. 126, 6327–6331. ( 10.1021/ja031689l) [DOI] [PubMed] [Google Scholar]
- Goler J. A., Bramlett B. W., Peccoud J. 2008. Genetic design: rising above the sequence. Trends Biotechnol. 26, 538–544. ( 10.1016/j.tibtech.2008.06.003) [DOI] [PubMed] [Google Scholar]
- Griffiths A. D., Tawfik D. S. 2006. Miniaturising the laboratory in emulsion droplets. Trends Biotechnol. 24, 395–402. ( 10.1016/j.tibtech.2006.06.009) [DOI] [PubMed] [Google Scholar]
- Gunther A., Jensen K. F. 2006. Multiphase microfluidics: from flow characteristics to chemical and materials synthesis. Lab Chip 6, 1487–1503. ( 10.1039/b609851g) [DOI] [PubMed] [Google Scholar]
- Hasty J., McMillen D., Collins J. J. 2002. Engineered gene circuits. Nature 420, 224–230. ( 10.1038/nature01257) [DOI] [PubMed] [Google Scholar]
- He M., Edgar J. S., Jeffries G. D. M., Lorenz R. M., Shelby J. P., Chiu D. T. 2005. Selective encapsulation of single cells and subcellular organelles into picoliter- and femtoliter-volume droplets. Anal. Chem. 77, 1539–1544. [DOI] [PubMed] [Google Scholar]
- Heinemann M., Panke S. 2006. Synthetic biology—putting engineering into biology. Bioinformatics 22, 2790–2799. ( 10.1093/bioinformatics/btl469) [DOI] [PubMed] [Google Scholar]
- Huebner A., Srisa-Art M., Holt D., Abell C., Hollfelder F., deMello A. J., Edel J. B. 2007. Quantitative detection of protein expression in single cells using droplet microfluidics. Chem. Commun., 1218–1220. [DOI] [PubMed] [Google Scholar]
- Huebner A., Sharma S., Srisa-Art M., Hollfelder F., Edel J. B., deMello A. J. 2008. Microdroplets: a sea of application? Lab Chip 8, 1244–1254. ( 10.1039/b806405a) [DOI] [PubMed] [Google Scholar]
- Joanicot M., Ajdari A. 2005. Droplet control for microfluidics. Science 309, 887–888. ( 10.1126/science.1112615) [DOI] [PubMed] [Google Scholar]
- Kaern M., Blake W. J., Collins J. J. 2003. The engineering of gene regulatory networks. Annu. Rev. Biomed. Eng. 5, 179–206. ( 10.1146/annurev.bioeng.5.040202.121553) [DOI] [PubMed] [Google Scholar]
- Kartalov E. P., Quake S. R. 2004. Microfluidic device reads up to four consecutive base pairs in DNA sequencing-by-synthesis. Nucleic Acids Res. 32, 2873–2879. ( 10.1093/nar/gkh613) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly B. T., Baret J.-C., Taly V., Griffiths A. D. 2007. Miniaturizing chemistry and biology in microdroplets. ChemComm 1773–1788. ( 10.1039/b616252e) [DOI] [PubMed] [Google Scholar]
- Khan S. A., Gunther A., Schmidt M. A., Jensen K. F. 2004. Microfluidic synthesis of colloidal silica. Langmuir 20, 8604–8611. ( 10.1021/la0499012) [DOI] [PubMed] [Google Scholar]
- Khandurina J., Guttman A. 2002. Bioanalysis in microfluidic devices. J. Chromatogr. A 943, 159–183. ( 10.1016/S0021-9673(01)01451-0) [DOI] [PubMed] [Google Scholar]
- Khnouf R., Beebe D. J., Fan Z. H. 2009. Cell-free protein expression in microchannel array with passive pumping. Lab Chip 9, 56–61. ( 10.1039/b808034h) [DOI] [PubMed] [Google Scholar]
- King K. R., Wang S., Irimia D., Jayaraman A., Toner M., Yarmush M. L. 2007. A high-throughput microfluidic real-time gene expression living cell array. Lab Chip 7, 77–85. ( 10.1039/b612516f) [DOI] [PMC free article] [PubMed] [Google Scholar]
- King K. R., Wang S., Jayaraman A., Yarmush M. L., Toner M. 2008. Microfluidic flow-encoded switching for parallel control of dynamic cellular microenvironments. Lab Chip 8, 107–116. ( 10.1039/b716962k) [DOI] [PubMed] [Google Scholar]
- Kiss M. M., Ortoleva-Donnelly L., Beer N. R., Warner J., Bailey C. G., Colston B. W., Rothberg J. M., Link D. R., Leamon J. H. 2008. High-throughput quantitative polymerase chain reaction in picoliter droplets. Anal. Chem. 80, 8975–8981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitney R. I., Freemont P. S., Rouilly V. 2007. Engineering a molecular predation oscillator. IET Synth. Biol. 1, 68–70. ( 10.1049/iet-stb:20070018) [DOI] [Google Scholar]
- Kong D. S., Carr P. A., Chen L., Zhang S., Jacobson J. M. 2007. Parallel gene synthesis in a microfluidic device. Nucleic Acids Res. 35, e61 ( 10.1093/nar/gkm121) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp M. U., deMello A. J., Manz A. 1998. Chemical amplification: continuous-flow PCR on a chip. Science 280, 1046–1048. ( 10.1126/science.280.5366.1046) [DOI] [PubMed] [Google Scholar]
- Krishnadasan S., Tovilla J., Vilar R., deMello A. J., deMello J. C. 2004. On-line analysis of CdSe nanoparticle formation in a continuous flow chip-based microreactor. J. Mater. Chem. 14, 2655–2660. ( 10.1039/b401559b) [DOI] [Google Scholar]
- Liau A., Karnik R., Majumdar A., Doudna J. H. 2005. Cate, mixing crowded biological solutions in milliseconds. Anal. Chem. 77, 7618–7625. ( 10.1021/ac050827h) [DOI] [PubMed] [Google Scholar]
- Link D. R., Anna S. L., Weitz D. A., Stone H. A. 2004. Geometrically mediated breakup of drops in microfluidic devices. Phys. Rev. Lett. 92, 054503 ( 10.1103/PhysRevLett.92.054503) [DOI] [PubMed] [Google Scholar]
- Lucks J., Qi L., Whitaker W. R., Arkin A. P. 2008. Toward scalable parts families for predictable design of biological circuits. Curr. Opin. Microbiol. 11, 567–573. ( 10.1016/j.mib.2008.10.002) [DOI] [PubMed] [Google Scholar]
- Margulies M., et al. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel R., Weiss R. 2005. Advances in synthetic biology: on the path from prototypes to applications. Curr. Opin. Biotechnol. 16, 476–483. ( 10.1016/j.copbio.2005.07.002) [DOI] [PubMed] [Google Scholar]
- Nakano M., Komatsu J., Matsuura S., Takashima K., Katsura S., Mizuno A. 2003. Single-molecule PCR using water-in-oil emulsion. J. Biotechnol. 102, 117–124. [DOI] [PubMed] [Google Scholar]
- Nguyen N.-T., Wu Z. 2005. Micromixers—a review. J. Micromech. Microeng. 15, 1–16. [Google Scholar]
- Nisisako T., Torii T., Higuchi T. 2002. Droplet formation in a microchannel network. Lab Chip 2, 24–26. ( 10.1039/b108740c) [DOI] [PubMed] [Google Scholar]
- Niu X., Gulati S., Edel J. B., deMello A. J. 2008. Pillar-induced droplet merging in microfluidic circuits. Lab Chip 8, 1837–1841. ( 10.1039/b813325e) [DOI] [PubMed] [Google Scholar]
- Priest C., Herminghaus S., Seemann R. 2006. Controlled electrocoalescence in microfluidics: targeting a single lamella. Appl. Phys. Lett. 89, 134101 ( 10.1063/1.2357039) [DOI] [Google Scholar]
- Quake S. R., Scherer A. 2000. From micro- to nanofabrication with soft materials. Science 290, 1536–1540. ( 10.1126/science.290.5496.1536) [DOI] [PubMed] [Google Scholar]
- Ronaghi M., Karamohamed S., Pettersson B., Uhlén M., Nyrén P. 1996. Real-time DNA sequencing using detection of pyrophosphate release. Anal. Biochem. 242, 84–89. [DOI] [PubMed] [Google Scholar]
- Schaerli Y., et al. 2009. Continuous-flow polymerase chain reaction of single-copy DNA in microfluidic microdroplets. Anal. Chem. 81, 302–306. ( 10.1021/ac802038c) [DOI] [PubMed] [Google Scholar]
- Schwarz M. A., Hauser P. C. 2001. Recent developments in detection methods for microfabricated analytical devices. Lab Chip 1, 1–6. ( 10.1039/b103795c) [DOI] [PubMed] [Google Scholar]
- Shendure J., et al. 2005. Accurate multiplex polony sequencing of an evolved bacterial genome. Science 309, 1728–1732. ( 10.1126/science.1117389) [DOI] [PubMed] [Google Scholar]
- Shestopalov I., Tice J. D., Ismagilov R. F. 2004. Multi-step synthesis of nanoparticles performed on millisecond time scale in a microfluidic droplet-based system. Lab Chip 4, 316–321. ( 10.1039/b403378g) [DOI] [PubMed] [Google Scholar]
- Shui L., Eijkel J. C. T., van der Berg A. 2007. Multiphase flow in microfluidic systems—control and applications of droplets and interfaces. Adv. Coll. Inter. Sci. 133, 35–49. ( 10.1016/j.cis.2007.03.001) [DOI] [PubMed] [Google Scholar]
- Sismour A. M., Benner S. A. 2005. Synthetic biology. Expert Opin. Biol. Ther. 5, 1409–1414. ( 10.1517/14712598.5.11.1409) [DOI] [PubMed] [Google Scholar]
- Song H., Ismagilov R. F. 2003. Millisecond kinetics on a microfluidic chip using nanoliters of reagants. J. Am. Chem. Soc. 125, 14613–14619. ( 10.1021/ja0354566) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H., Tice J. D., Ismagilov R. F. 2003. A microfluidic system for controlling reaction networks in time. Angew. Chem. Int. Ed. 42, 767–772. ( 10.1002/anie.200390203) [DOI] [PubMed] [Google Scholar]
- Song H., Chen D. L., Ismagilov R. F. 2006. Reactions in droplets in microfluidic channels. Angew. Chem. Int. Ed. 45, 7336–7356. ( 10.1002/anie.200601554) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squires T. M., Quake S. R. 2005. Microfluidics: fluid physics at the nanoliter scale. Rev. Mod. Phys. 77, 977–1026. [Google Scholar]
- Stone H. A., Kim S. 2001. Microfluidics: basic issues, applications, and challenges. AIChE J. 47, 1250–1254. ( 10.1002/aic.690470602) [DOI] [Google Scholar]
- Tan Y.-C., Fisher J. S., Lee A. I., Cristini V., Lee A. P. 2004. Design of microfluidic channel geometries for the control of droplet volume, chemical concentration, and sorting. Lab Chip 4, 292–298. ( 10.1039/b403280m) [DOI] [PubMed] [Google Scholar]
- Tawfik D. S., Griffiths A. D. 1998. Man-made cell-like compartments for molecular evolution. Nat. Biotechnol. 16, 652–656. ( 10.1038/nbt0798-652) [DOI] [PubMed] [Google Scholar]
- Thompson D. M., King K. R., Wieder K. J., Toner M., Yarumush M. L., Jayaraman A. 2004. Dynamics gene expression profiling using a microfabricated living cell array. Anal. Chem. 76, 4098–4103. ( 10.1021/ac0354241) [DOI] [PubMed] [Google Scholar]
- Tice J. D., Song H., Lyon A. D., Ismagilov R. F. 2003. Formation of droplets and mixing in multiphase microfluidics at low values of the Reynolds and the capillary numbers. Langmuir 19, 9127–9133. ( 10.1021/la030090w) [DOI] [Google Scholar]
- Tice J. D., Lyon A. D., Ismagilov R. F. 2004. Effects of viscosity on droplet formation and mixing in microfluidic channels. Anal. Chim. Acta 507, 73–77. ( 10.1016/j.aca.2003.11.024) [DOI] [Google Scholar]
- Viskari P. J., Landers J. P. 2006. Unconventional detection methods for microfluidic devices. Electrophoresis 27, 1797–1810. ( 10.1002/elps.200500565) [DOI] [PubMed] [Google Scholar]
- Wang J. 2002. Electrochemical detection for microscale analytical systems: a review. Talanta 56, 223–231. [DOI] [PubMed] [Google Scholar]
- Xu S., et al. 2001. Generation of monodisperse particles by using microfluidics: control over size, shape, and composition. Angew. Chem. Int. Ed. 44, 724–728. ( 10.1002/anie.200462226) [DOI] [PubMed] [Google Scholar]
- Yehezkel T. B., Linshiz G., Buaron H., Kaplan S., Shabi U., Shapiro E. 2008. De novo DNA synthesis using single molecule PCR. Nucleic Acids Res. 36, e107 ( 10.1093/nar/gkn457) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Xu J., Ma W., Zheng W. 2006. PCR microfluidic devices for DNA amplification. Biotechnol. Adv. 24, 243–284. [DOI] [PubMed] [Google Scholar]
- Zheng B., Roach S., Ismagilov R. F. 2003. Screening of protein crystallization conditions on a microfluidic chip using nanoliter-size droplets. J. Am. Chem. Soc. 125, 11170–11171. ( 10.1021/ja037166v) [DOI] [PubMed] [Google Scholar]