

Abstract
Interaction of the estrogen receptor/ligand complex with a DNA estrogen response element is known to regulate gene transcription. In turn, specific conformations of the receptor-ligand complex have been postulated to influence unique subsets of estrogen-responsive genes resulting in differential modulation and, ultimately, tissue-selective outcomes. The estrogen receptor ligands raloxifene and tamoxifen have demonstrated such tissue-specific estrogen agonist/antagonist effects. Both agents antagonize the effects of estrogen on mammary tissue while mimicking the actions of estrogen on bone. However, tamoxifen induces significant stimulation of uterine tissue whereas raloxifene does not. We postulate that structural differences between raloxifene and tamoxifen may influence the conformations of their respective receptor/ligand complexes, thereby affecting which estrogen-responsive genes are modulated in various tissues. These structural differences are 4-fold: (A) the presence of phenolic hydroxyls, (B) different substituents on the basic amine, (C) incorporation of the stilbene moiety into a cyclic benzothiophene framework, and (D) the imposition of a carbonyl “hinge” between the basic amine-containing side chain and the olefin. A series of raloxifene analogs that separately exemplify each of these differences have been prepared and evaluated in a series of in vitro and in vivo assays. This strategy has resulted in the development of a pharmacophore model that attributes the differences in effects on the uterus between raloxifene and tamoxifen to a low-energy conformational preference imparting an orthogonal orientation of the basic side chain with respect to the stilbene plane. This three-dimensional array is dictated by a single carbon atom in the hinge region of raloxifene. These data indicate that differences in tissue selective actions among benzothiophene and triarylethylene estrogen receptor modulators can be ascribed to discrete ligand conformations.
Recent advances in understanding the mechanisms by which nuclear hormone receptors exert their effects on gene transcription have greatly enhanced the prospects for tissue-selective regulation of these processes (1–4). The recognition that the estrogen receptor (ER) may interact with more than one DNA response element, that these interactions may be mediated by various coactivators and corepressors, and that the nature of these interactions may be dependent on the specific ligand that is bound to the receptor allows for the possibility of ligand-based transcriptional control (5–8). Indeed, it has been hypothesized that a series of ligand-dependent conformations exist and that each of these conformations may influence a unique subset of estrogen-responsive genes (9, 10). A logical extension of this hypothesis is the dependence of ER conformation on the structural features and molecular conformation of individual ligands (11–14). The recent characterization of a new ER isoform, ERβ, adds a further layer of complexity, because ligands with different structural characteristics may exhibit different binding profiles depending on which ER isoform is being evaluated (15, 16).
To date, ER ligands have been grouped into four classes on the basis of in vivo pharmacology, and representatives of these classes indeed have been shown to exhibit unique profiles with respect to transcriptional activation (9). Differential effects on mammary, uterine, cardiovascular, and skeletal tissues have been observed for each of the four classes (17). At opposing ends of the pharmacological spectrum are 17β-estradiol and ICI-164,384; the former is the natural ligand and a full agonist in target tissues whereas the latter appears to be essentially a pure antagonist in vivo (17, 18). In between these two extremes lie other ER ligands such as tamoxifen and raloxifene (Fig. 1), which exert tissue-selective estrogen-like effects. Both act as estrogen antagonists in mammary tissue, but mimic the agonist effects of estrogen on bone and in the cardiovascular system (19). In the uterus, however, tamoxifen induces significant stimulation and appears to function as a partial estrogen agonist, whereas raloxifene appears to function entirely as an antagonist. This uterine agonist activity has been associated with an increased risk of endometrial cancer for patients treated with tamoxifen (20). Because of its unique profile of tissue specificity, raloxifene has been characterized as a selective estrogen receptor modulator and is presently in clinical development for the prevention and treatment of postmenopausal osteoporosis (21, 22). Recently, a potential mechanism for this tissue specificity involving selective interaction of the ER-raloxifene complex with a response element distinct from the classical estrogen response element has been proposed (8). As part of our program to explore the pharmacology of raloxifene and related molecules, we have sought to uncover the molecular features that account for its unique profile of tissue specificity, particularly those features that differentiate it from tamoxifen. Four major structural differences are apparent on inspection (Fig. 1): (A) the presence of the phenolic hydroxyls, (B) the nature of the basic amine, (C) the incorporation of the stilbene into a cyclic benzothiophene framework, and (D) the imposition of a carbonyl “hinge” between the basic amine-containing side chain and the olefin. In this paper we directly compare the in vitro and in vivo pharmacology of a series of compounds that separately address each of these structural features. We demonstrate that individual elements of raloxifene’s molecular structure are responsible for its enhanced tissue selectivity relative to tamoxifen; that changes in these structural elements affect the profile of biological activities that previously have been described for raloxifene; and that one of these structural elements causes raloxifene to assume a distinct molecular conformation, which may, in turn, impact the conformation of the ER-raloxifene complex and in so doing be responsible for the unique tissue selectivity evidenced by this ER modulator.
Figure 1.
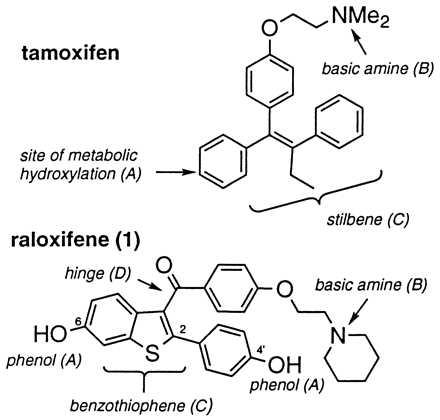
ER ligands tamoxifen and raloxifene. Differentiating structural features are highlighted.
MATERIALS AND METHODS
Chemistry.
The compounds shown in Table 1 were prepared as previously described (23). Analogs bearing side-chain variations, shown in Table 2, were prepared by Friedel–Crafts acylation of 2-[4-methoxyphenyl]-6-methoxybenzothiophene with anisoyl chloride followed by selective O-demethylation and alkylation of the resulting phenol with the appropriate electrophile under basic or Mitsunobu conditions (24, 25). Deprotection with ethanethiol/aluminum chloride provided the target compounds. Compound 2 (see Fig. 6) was prepared by a modification of the method of Crenshaw involving the acid-mediated cyclization of an appropriately substituted α-arylthiodeoxybenzoin (26). Compound 3 (see Fig. 6) was prepared from a suitably protected 11-thiacoumestrol by a method analogous to that described previously for coumestrol (27). Thus 3,9-dimethoxy-11-thiacoumestan (28) was demethylated and converted to the corresponding bis(tert-butyldimethylsilyl)ether. Low temperature reduction with diisobutylaluminum hydride followed by condensation of the resultant lactol with phenol provided the intermediate phenyl acetal. Displacement of the phenoxy moiety with 4-[2-(1-piperidinyl)ethoxy]phenylmagnesium bromide and removal of the silyl protecting groups with fluoride provided the desired compound 3 (see Fig. 6) in racemic form. All reported compounds were fully characterized by 1H-NMR, mass spectra, and/or elemental analysis. Tamoxifen, 17α-ethynyl estradiol, and 17β-estradiol were purchased from commercial sources.
Table 1.
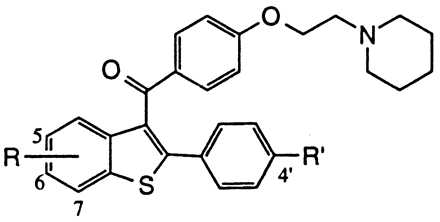
Effects on ER binding and inhibition of MCF-7 proliferation for a series of raloxifene analogs
| Compound | R | R′ | ER RBA | MCF-7 Inhib. IC50 (nM) |
|---|---|---|---|---|
| 17β-estradiol | 1.00 | NA | ||
| 4-OH Tam | 0.36 | 0.5 | ||
| Raloxifene (1) | 6-OH | OH | 0.34 | 0.2 |
| 1a | None | OH | .003 | 35 |
| 1b | 6-OMe | OH | .008 | 250 |
| 1c | 7-OH | OH | .020 | 300 |
| 1d | 5-OH | OH | 0.1 | 100 |
| 1e | 6-OH | H | .062 | 2.5 |
| 1f | None | H | .002 | >100 |
NA, not applicable.
Table 2.
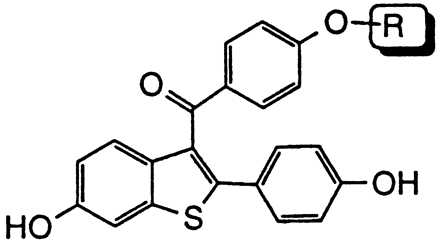
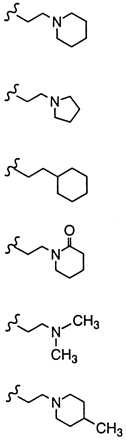
Comparative effects on in vivo estrogen antagonism for various basic side chain modifications in the raloxifene nucleus
| Compound | R | In Vivo estrogen antagonism, maximal percent inhibition (Uterine wt/body wt) |
|---|---|---|
| Tamoxifen | 48* | |
| 1 | 91* | |
| 1g | 81* | |
| 1h | 3.8 | |
| 1i | 10 | |
| 1j | 38* | |
| 1k | 36* |
Statistical significance relative to ethynyl estradiol-treated control (P ≤ 0.05).
Figure 6.
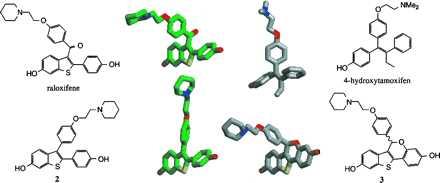
MOPAC/AM1 minimum energy conformations for raloxifene (green carbons), 4-hydroxytamoxifen (gray carbons), compound 2 (green carbons), and compound 3 (gray carbons). For the sake of clarity, a single rotomer of raloxifene and a single enantiomer of compound 3 are depicted. A preferred absolute orientation for the basic amine-containing side chain has not been determined.
ER Binding.
Affinity to the ER was determined in MCF-7 cell lysates by standard methods (29). Relative binding affinities were calculated as IC50 17β-estradiol/IC50 compound.
MCF-7 Cell Proliferation.
Effects on cellular proliferation were determined by standard methods (29) in MCF-7 breast adenocarcinoma cells. Cell cultures were treated in triplicate with test compound and 10 pM 17β-estradiol, and incubated for 48 hr at 37°C. Proliferative effects were measured by 3H-thymidine incorporation.
Transforming Growth Factor (TGF)-β3 Induction.
Ligand effects on ERα-mediated TGF-β3 gene promoter activation were determined as previously described (8).
Ovariectomized Rat Assays.
Six-month-old Sprague–Dawley rats were ovariectomized and dosed orally with test compound, vehicle control, or 17α-ethynyl estradiol (0.1 mg/kg per day). After 4 days or 5 weeks of oral dosing animals were sacrificed, and blood and uteri were collected for analysis as previously described (23). Bone samples also were collected from the animals treated for 5 weeks, and bone mineral density was determined by the method of Sato (30).
Estrogen Antagonism Assay in Immature Rats.
Antagonist activity was determined in 21-day-old female Sprague–Dawley rats primed with 17α-ethynyl estradiol (0.1 mg/kg per day) as previously described (31). Test compounds were administered orally 15 min before the daily ethynyl estradiol challenge. After 3 days, animals were sacrificed, the uteri were collected and weighed, and uterine weight/body weight ratios were calculated for each animal.
Statistics.
All treatment groups for in vivo studies contained a minimum of five animals. Statistical evaluations were made by one-way ANOVA with posthoc Fisher’s probable least-squares difference analysis when indicated. Significance was ascribed at a P < 0.05.
Molecular Modeling.
MOPAC minimum energy conformations of raloxifene, 4-hydroxytamoxifen, and compounds 2 and 3 (see Fig. 6) were generated by using the AM1 parameter set with the mopac command line options of; AM1 GRADIENT POLAR GEO-OK HESS = 1 EF GNORM = 0.01 SCFCRT = 1D-12 MMOK PRECISE NOINTER and GRAPH (32). Least-squares superimpositions of the stilbene cores were performed by using the Insight-II version 95.0.4 (Molecular Simulations, San Diego, CA).
RESULTS AND DISCUSSION
To determine the structural components responsible for the tissue-specific actions of raloxifene and whether molecular conformation plays a role in these outcomes, a series of analogs in which these differences are directly comparable were systematically evaluated in vitro and in vivo. The assays ranged from sub-cellular (ER binding) to cell-based (MCF-7 proliferation, transforming growth factor-β3 induction) to whole animals (immature rat, ovariectomized rat). Biological activity across these assays then was assessed as a function of ligand structural characteristics.
ER Binding Region.
The affinity of selected raloxifene analogs for binding to the ER is shown in Table 1 along with the corresponding effects on MCF-7 human breast adenocarcinoma cell proliferation. Raloxifene (compound 1, see Fig. 1) binds the ER with a relative binding affinity of 0.34 (17β-estradiol = 1.0) and is a potent anti-proliferative agent (IC50 = 0.2 nM). The importance of the 6-OH group is illustrated by deletion of this group (compound 1a, Table 1) or blocking as a methyl ether, (compound 1b, Table 1) each of which results in ≥100-fold decreases in both receptor affinity (0.003 and 0.008, respectively) and MCF-7 growth inhibitory potency (35 and 250 nM, respectively). The 4′-hydroxy group plays a lesser role as shown by the approximately 10-fold decrease in binding and antagonist activity on elimination of this functional group (compound 1e, Table 1). Transposition of 6-OH group to the adjacent 7-position of the benzothiophene (compound 1c, Table 1) also significantly decreases receptor affinity and antiproliferative action. In the analogous 5-OH analog (compound 1d, Table 1), it is interesting to note that although this compound binds effectively to the ER (relative binding affinity = 0.1) this interaction does not translate to significant antagonistic effects on proliferation in MCF-7 cells (IC50 = 100 nM). Taken together, these data indicate that a key molecular component dictating ER binding is the 6-OH group, and that this interaction plays an important role in regulating the antiproliferative effects of these compounds (23). The significance of this group is supported further by studies of tamoxifen and 4-hydroxytamoxifen, which illustrate the importance of a hydroxy functionality, located at the corresponding position of the stilbene core, in dictating ER binding affinity and functional activity in MCF-7 cells (33).
Basic Side Chain.
Pioneering work on triphenylethylene anti-estrogens such as tamoxifen has demonstrated the importance of both the position and nature of the N,N-dimethylamine side chain with regard to blocking the uterine hypertrophic effects of coadministered estrogen in immature female rats (34). Likewise, studies with raloxifene indicate that a dialkylamine moiety is critical for antagonizing the in vivo effects of estrogen. As shown in Table 2, replacement of the nitrogen with a carbon (compound 1h) or a nonbasic nitrogen atom (compound 1i) results in complete loss of antagonist effects. Specific bases such as piperidine (compound 1) or pyrrolidine (compound 1f) are optimal and impart the highest degree of antagonist character. It is intriguing to note that although a basic nitrogen is required for optimal in vivo estrogen antagonism, the nature of the substituents on this heteroatom determine the relative level of antagonism. For example, N,N-dimethylamino analog 1j and 4-methylpiperidine analog 1k behave as partial agonists with ED50’s > 10 mg/kg and maximal inhibitory responses of only 38% and 36%, respectively. Indeed, these raloxifene analogs more closely resemble tamoxifen with respect to their effects on the uterus. Given these data, we initially postulated that the differences in estrogen antagonist activity between raloxifene and tamoxifen resulted primarily from the presence of different amine bases, i.e. piperidine vs. N,N-dimethylamine. However, in direct contrast to the benzothiophene series replacement of the N,N-dimethylamino base of tamoxifen with piperidine does not improve its anti-uterotrophic character (34). Thus, whereas the basic side-chain moiety of raloxifene is critical for its estrogen antagonistic properties, it is not the primary structural component differentiating the uterine pharmacology of raloxifene and tamoxifen.
Benzothiophene and “Hinge” Regions.
To establish whether the benzothiophene ring system and/or the carbonyl “hinge” of raloxifene is responsible for its enhanced tissue selectivity relative to tamoxifen, we have prepared compounds 2 and 3 (see Fig. 6). Both compounds maintain the benzothiophene core structure, hydroxylation pattern, and piperidine-containing basic side chain of raloxifene. In compound 2, however, the carbonyl “hinge” has been excised, whereas in compound 3 the orientation of the basic side chain has been rigidified by incorporation of the carbonyl moiety into a benzopyran ring system.
In standard in vitro models of estrogen action compounds 2 and 3 exhibited similar profiles. As expected, both compounds 2 and 3 bound ER (relative binding affinities 0.29 and 0.23, respectively) and were able to inhibit estrogen-stimulated MCF-7 cell proliferation (IC50 0.5 and 0.2 nM, respectively) at concentrations comparable to raloxifene and 4-hydroxytamoxifen (4-hydroxytamoxifen was used as the appropriate comparitor in in vitro assays, whereas tamoxifen was used in in vivo assays). Furthermore, all four compounds were able to stimulate transforming growth factor-β3 gene expression in cell culture 3–5 fold, at concentrations of 10−6–10−8 M, as previously has been described for raloxifene (8).
Differential tissue selectivities of raloxifene, tamoxifen, and compounds 2 and 3 have been demonstrated in ovariectomized rats treated for 4 days with endpoints of serum cholesterol lowering and uterine stimulation. Whereas all four compounds significantly lowered serum cholesterol at doses as low as 0.1 mg/kg (ED50s: tamoxifen, 0.21 mg/kg; raloxifene, 0.18 mg/kg; compound 2, 0.15 mg/kg; and compound 3, 0.29 mg/kg), tamoxifen and compound 2 produced significant increases in uterine eosinophilia (as measured by uterine eosinophil peroxidase activity, Fig. 2a) whereas raloxifene and compound 3 did not. Furthermore, tamoxifen and compound 2 also markedly increased uterine weight (Fig. 2b). Although raloxifene induced a modest increase in uterine wet weight, which previously has been ascribed to effects on water imbibition into the stromal compartment of the uterus (21) compound 3 produced no such increase.
Figure 2.
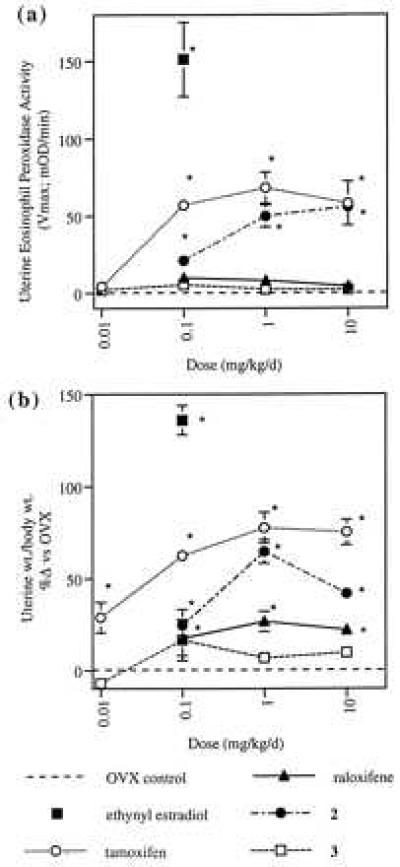
Effects of tamoxifen, raloxifene, compound 2, and compound 3 on uterine eosinophil peroxidase activity (a) and uterine weight (b) in ovariectomized rats. Eosinophil peroxidase activity is reported as the mean Vmax (± SEM) (23). Uterine weight/body weight values are reported as the mean percent increase relative to ovariectomized, vehicle-treated controls (± SEM). Statistical significance relative to ovariectomized controls is denoted by ∗ (P ≤ 0.05).
Effects on serum cholesterol and uterine weight in ovariectomized rats treated with test compounds for 5 weeks were similar to those observed in the 4-day experiment (data not shown). Differential effects on uterine epithelial cell height also were observed, with tamoxifen and compound 2 inducing significant stimulation whereas raloxifene and compound 3 did not (Fig. 3). In this model, the abilities of raloxifene, tamoxifen, and compounds 2 and 3 to function as estrogen agonists by preventing ovariectomy-induced osteopenia also were demonstrated (Fig. 4).
Figure 3.
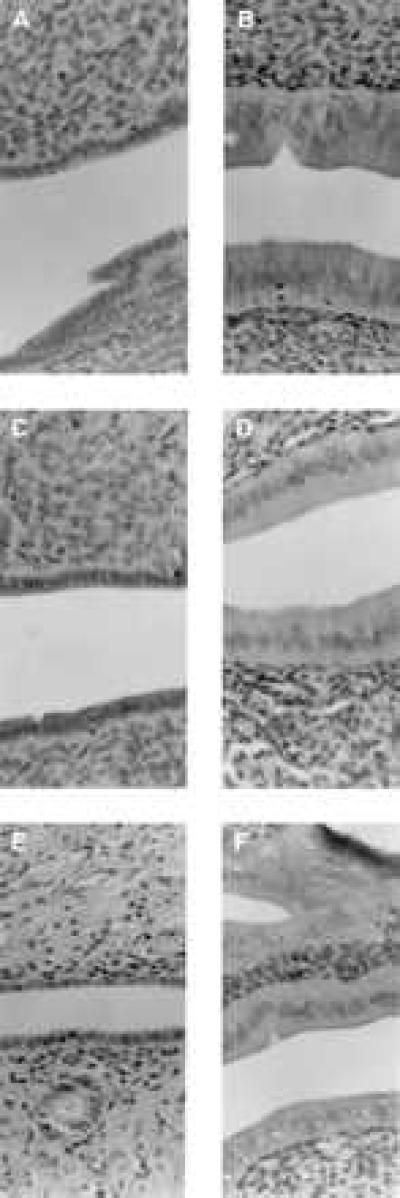
Hemotoxylin and eosin-stained sections of rat uteri, illustrating epthelial lining cells, obtained from ovariectomized rats treated for 35 days with vehicle (A), ethynyl estradiol (0.1 mg/kg per day; B), raloxifene (1.0 mg/kg per day; C), tamoxifen (1.0 mg/kg per day; D), compound 3 (1.0 mg/kg/per day; E), or compound 2 (1.0 mg/kg per day; F). Magnification is at 128×.
Figure 4.
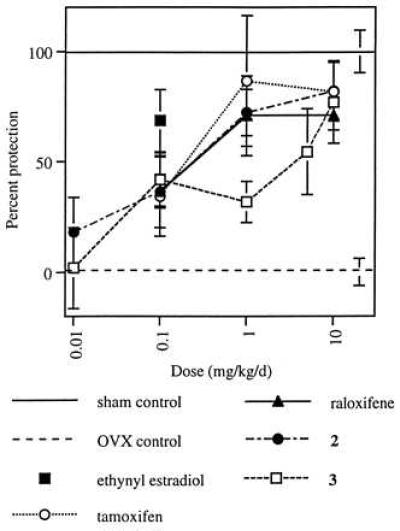
Effects of tamoxifen, raloxifene, compound 2, and compound 3 on bone mineral density in the ovariectomized rat. Bone mineral density values are reported as percent protection relative to ovariectomized, vehicle-treated controls (± SEM), with sham control values defined as 100% and ovariectomized controls defined as 0. All groups, except 0.01 mg/kg per day compounds 2 and 3 differ significantly from ovariectomized controls at P ≤ 0.05.
Finally, the ability of these compounds to function as estrogen antagonists in the uterus has been examined in an immature rat model (Fig. 5). In this model, raloxifene and compound 3, at a dose of 10 mg/kg per day, provided almost complete antagonism of the effects of ethynyl estradiol on uterine weight, whereas compound 2 and tamoxifen achieved only partial antagonism.
Figure 5.
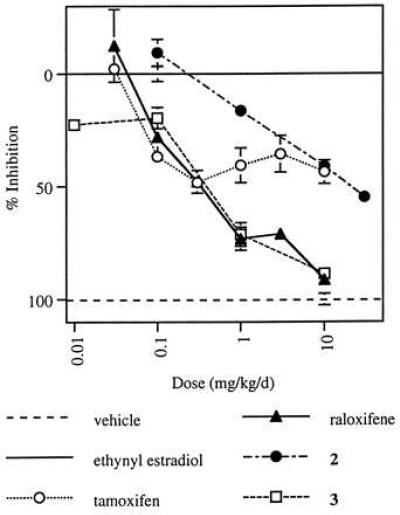
Effect of tamoxifen, raloxifene, compound 2, and compound 3 on ethynyl estradiol-induced uterine weight increase in immature rats. Data are reported as percent inhibition of uterine weight/body weight relative to ethynyl estradiol-treated controls (± SEM), with vehicle-treated control values defined as 100% and ethynyl estradiol-treated controls as 0.
Molecular Determinants.
The results described above demonstrate how the structural differences between raloxifene and tamoxifen affect the biological activities of these compounds, both in vitro and in vivo. Clearly, the hydroxylation pattern is important for receptor binding and in vitro activity (35), and the presence and nature of the basic amine substituent is critical in determining estrogen antagonist activity. The shift from an acyclic olefin in tamoxifen to one contained within a benzothiophene ring system in raloxifene is one of the most striking structural differences between tamoxifen and raloxifene. This strategy of incorporating the olefin of a triphenylethylene into a fused ring system frequently has been used to confer configurational stability on the olefin, thus reducing metabolic conversion to potentially uterotrophic double bond isomers (36). Nevertheless, the well-established uterine stimulatory effects of nafoxidine, a nonisomerisable analog of tamoxifen, demonstrates that this strategy is an incomplete solution (19). That none of these characteristics uniquely provide satisfactory tissue selectivity is further suggested by results obtained with compound 2, which produces significant uterine stimulation in OVX rats (Figs. 2 and 3).
The final structural distinction between raloxifene and tamoxifen is the carbonyl hinge, which is imposed between the stilbene moiety and the basic side chain of raloxifene. Molecular modeling studies have indicated that this simple structural modification produces a drastic change in the orientation of the basic side chain, from a coplanar orientation in 4-hydroxytamoxifen to a nearly orthogonal orientation in raloxifene (Figs. 6 and 7) (14). (The two rotomers of raloxifene in which the side chain occupies this orientation are enantiomeric in nature and equivalent in energy.) Nevertheless, rotation about the carbonyl hinge can take place and higher energy conformations of raloxifene can be achieved in which the basic side chain and stilbene nucleus are coplanar. In compound 3, the basic side chain is locked in an orientation similar to that of raloxifene. (The individual enantiomers of compound 3 each overlay with an individual rotomer of raloxifene. It has not been determined whether a single absolute orientation of the side chain is preferred and therefore none is implied herein.) In compound 2, the side chain lies within the stilbene plane as is the case for 4-hydroxytamoxifen. Structural overlays of compound 3 with raloxifene and of compound 2 with 4-hydroxytamoxifen indicate that they are indeed distinguished by their side-chain orientations.
Figure 7.
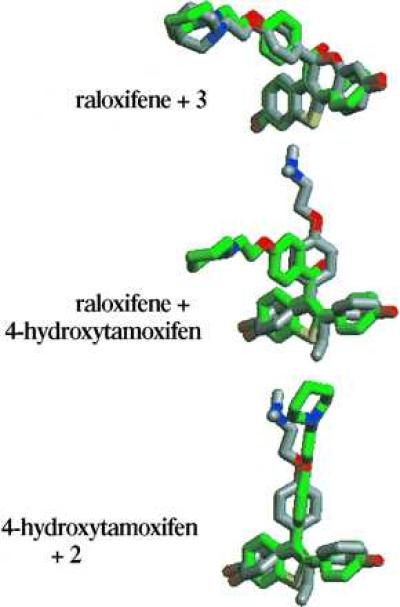
Overlay of minimized structures of raloxifene (green carbons) and compound 3 (gray carbons), raloxifene (green carbons) and 4-hydroxytamoxifen (gray carbons), and 4-hydroxytamoxifen (gray carbons) and compound 2 (green carbons). Note that overlay of the alternative rotomer of raloxifene with the alternative enantiomer of compound 3 produces a mirror image of that depicted here.
The biological effects described herein suggest that this side-chain orientation may play a role in the unique profile of tissue selectivity that is displayed by raloxifene and compound 3. Thus, the coplanar orientation of the side chain in tamoxifen and in compound 2 may contribute to the uterine stimulation that is observed with these compounds, whereas raloxifene and compound 3 produce little or no stimulation. Other compounds, in which the side chain is oriented orthogonally to a benzopyran ring system also have been reported to lack uterotrophic effects (14). The ability of raloxifene and compound 3 to function as tissue-selective estrogen agonists is evidenced by their effects on bone mineral density and serum cholesterol. That this tissue selectivity is not simply the result of selective tissue distribution is confirmed by the ability of raloxifene and compound 3 to completely antagonize the effects of estrogen on the immature rat uterus. In contrast, compound 2 and tamoxifen appear to be partial agonists in the uterus. Because the amine moieties of these compounds appear to be important for their ability to function as estrogen antagonists, it is reasonable to assume that they contribute an additional interaction with the ER, which is not present in estradiol. The topography of the amine functionality relative to the stilbene plane therefore may influence the manner in which a particular ligand interacts with the ER. The individual conformations of tamoxifen and raloxifene then may be reflected in different conformations of their respective ER-ligand complexes, with differential effects on the modulation of various estrogen-responsive genes, and different levels of tissue selectivity.
In conclusion, we propose that the ability of raloxifene to function as a selective estrogen receptor modulator is related to unique features of its molecular structure. In particular, the single molecular feature that distinguishes raloxifene from tamoxifen is the imposition of a carbonyl “hinge” between the basic amine-containing side chain and the olefin, inducing a molecular conformation that is significantly different from that of tamoxifen. We propose that this altered molecular conformation may, in turn, induce a unique conformation of the ER-ligand complex or it may selectively affect the interactions of raloxifene with the ERα or ERβ, and that these effects may be responsible for raloxifene’s unique profile of biological activities.
Acknowledgments
We wish to acknowledge the contributions of Dee Adrian, Michael Bell, Stephen Cho, Harlan Cole, Thomas Crowell, Kristen Marron, Kennan Fahey, Don Finley, Tina Fuson, Sushant Hardikar, Kenneth Hauser, Scott Jones, Chip Lugar, David Magee, Michael Martin, Brian Muehl, Lewis Pennington, Pamela Pennington, D. Lynn Phillips, Paul Pranc, Ellen Rowley, Pamela Shetler, Lorri Short, Mark Stocksdale, Kenneth Thrasher, Michelle Tokarz, Murali Venugopalan, and Kirk Wells to the work described herein. We also wish to thank Profs. Armen Tashjian (Harvard University), John Katzenellenbogen (University of Illinois), and Benita Katzenellenbogen (University of Illinois) for helpful discussions and suggestions.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviation: ER, estrogen receptor.
References
- 1.Katzenellenbogen J A, O’Malley B W, Katzenellenbogen B S. Mol Endocrinol. 1996;10:119–131. doi: 10.1210/mend.10.2.8825552. [DOI] [PubMed] [Google Scholar]
- 2.Katzenellenbogen J A, Katzenellenbogen B S. Chem Biol. 1996;3:529–536. doi: 10.1016/s1074-5521(96)90143-x. [DOI] [PubMed] [Google Scholar]
- 3.Rosen J, Day A, Jones T K, Turner Jones E T, Nadzan A M, Stein R B. J Med Chem. 1995;38:4855–4874. doi: 10.1021/jm00025a001. [DOI] [PubMed] [Google Scholar]
- 4.McDonnell D P, Clevenger B, Dana S, Santiso-Mere D, Tzukerman M T, Gleeson M A G. J Clin Pharmacol. 1993;33:1165–1172. doi: 10.1002/j.1552-4604.1993.tb03916.x. [DOI] [PubMed] [Google Scholar]
- 5.Berry M, Metzger D, Chambon P. EMBO J. 1990;9:2811–2818. doi: 10.1002/j.1460-2075.1990.tb07469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tzukerman M T, Esty A, Santiso-Mere D, Danielian P, Parker M G, Stein R B, Pike J W, McDonnell D P. Mol Endocrinol. 1994;8:21–30. doi: 10.1210/mend.8.1.8152428. [DOI] [PubMed] [Google Scholar]
- 7.Webb P, Lopez G N, Uht R M, Kushner P J. Mol Endocrinol. 1995;9:443–456. doi: 10.1210/mend.9.4.7659088. [DOI] [PubMed] [Google Scholar]
- 8.Yang N N, Venugopalan M, Hardikar S, Glasebrook A. Science. 1996;273:1222–1225. doi: 10.1126/science.273.5279.1222. [DOI] [PubMed] [Google Scholar]
- 9.McDonnell D P, Clemm D L, Hermann T, Goldman M E, Pike J W. Mol Endocrinol. 1995;9:659–669. doi: 10.1210/mend.9.6.8592512. [DOI] [PubMed] [Google Scholar]
- 10.Beekman J M, Allan G F, Tsai S Y, Tsai M-J, O’Malley B W. Mol Endocrinol. 1993;7:1266–1274. doi: 10.1210/mend.7.10.8264659. [DOI] [PubMed] [Google Scholar]
- 11.Klinge C M, Traish A M, Driscoll M D, Hilf R, Bambara R A. Steroids. 1996;61:278–289. doi: 10.1016/0039-128x(95)00219-g. [DOI] [PubMed] [Google Scholar]
- 12.McDonnell D P, Dana S L, Hoener P A, Lieberman B A, Imhof M O, Stein R B. Ann NY Acad Sci. 1995;761:121–137. doi: 10.1111/j.1749-6632.1995.tb31374.x. [DOI] [PubMed] [Google Scholar]
- 13.Christman J K, Nehls S, Polin L, Brooks S C. J Steroid Biochem Mol Biol. 1995;54:201–210. doi: 10.1016/0960-0760(95)00137-o. [DOI] [PubMed] [Google Scholar]
- 14.Grese T A, Sluka J P, Bryant H U, Cole H W, Kim J R, Magee D E, Rowley E R, Sato M. Bioorg Med Chem Lett. 1996;6:903–908. [Google Scholar]
- 15.Kuiper G G J M, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson J-A. Proc Natl Acad Sci USA. 1996;93:5925–5930. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mosselman S, Polman J, Dijkema R. FEBS Lett. 1996;392:49–53. doi: 10.1016/0014-5793(96)00782-x. [DOI] [PubMed] [Google Scholar]
- 17.Grese T A, Dodge J A. Annu Rep Med Chem. 1996;31:181–190. , and references therein. [Google Scholar]
- 18.Wakeling A E, Bowler J. J Steroid Biochem. 1988;31:645–653. doi: 10.1016/0022-4731(88)90014-3. [DOI] [PubMed] [Google Scholar]
- 19.Sato M, Rippy M K, Bryant H U. FASEB J. 1996;10:905–912. doi: 10.1096/fasebj.10.8.8666168. [DOI] [PubMed] [Google Scholar]
- 20.Kedar R P, Bourne T H, Powles T J, Collins W P, Ashley S E, Cosgrove D O, Campbell S. Lancet. 1994;343:1318–1321. doi: 10.1016/s0140-6736(94)92466-x. [DOI] [PubMed] [Google Scholar]
- 21.Black L J, Sato M, Rowley E R, Magee D E, Bekele A, Williams D C, Cullinan G J, Bendele R, Kauffman R F, Bensch W R, Frolik C A, Termine J D, Bryant H U. J Clin Invest. 1994;93:63–69. doi: 10.1172/JCI116985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Draper M W, Flowers D E, Huster W J, Neild J A, Harper K D, Arnaud C. J Bone Miner Res. 1996;11:835–842. doi: 10.1002/jbmr.5650110615. [DOI] [PubMed] [Google Scholar]
- 23.Grese T A, Cho S, Finley D R, Godfrey A G, Jones C D, Lugar C W, III, Martin M J, Matsumoto K, Pennington L D, Winter M A, Adrian M D, Cole H W, Magee D E, Phillips D L, Rowley E R, Short L L, Glasebrook A L, Bryant H U. J Med Chem. 1997;40:146–167. doi: 10.1021/jm9606352. [DOI] [PubMed] [Google Scholar]
- 24.Jones C D, Jevnikar M G, Pike A J, Peters M K, Black L J, Thompson A R, Falcone J F, Clemens J A. J Med Chem. 1984;27:1057–1066. doi: 10.1021/jm00374a021. [DOI] [PubMed] [Google Scholar]
- 25.Dodge J A, Stocksdale M G, Fahey K J, Jones C D. J Org Chem. 1995;60:739–741. [Google Scholar]
- 26.Crenshaw R R, Jeffries A T, Luke G M, Cheney L C, Biaiy G. J Med Chem. 1971;14:1185–1189. doi: 10.1021/jm00294a011. [DOI] [PubMed] [Google Scholar]
- 27.Grese T A, Cole H W, Magee D E, Phillips D L, Shetler P K, Short L L, Glasebrook A L, Bryant H U. Bioorg Med Chem Lett. 1996;6:2683–2686. [Google Scholar]
- 28.Conley R A, Heindel N D. J Org Chem. 1975;40:3169–3173. [Google Scholar]
- 29.Dodge J A, Glasebrook A L, Magee D E, Phillips D L, Sato M, Short L L, Bryant H U. J Steroid Biochem Mol Biol. 1996;59:155–161. doi: 10.1016/s0960-0760(96)00104-5. [DOI] [PubMed] [Google Scholar]
- 30.Sato M, Kim J, Short L L, Slemanda C W, Bryant H U. J Pharmacol Exp Ther. 1995;272:1252–1259. [PubMed] [Google Scholar]
- 31.Palkowitz A D, Glasebrook A L, Thrasher K J, Hauser K L, Short L L, Phillips D L, Muehl B S, Sato M, Shetler P K, Cullinan G J, Pell T R, Bryant H U. J Med Chem. 1997;40:1407–1416. doi: 10.1021/jm970167b. [DOI] [PubMed] [Google Scholar]
- 32.Stewart J P. Quantum Chemistry Program Exchange. Bloomington, IN: Indiana University; 1990. , Program No. 455, version 6.0 and references therein. [Google Scholar]
- 33.Jordan V C, Collins M M, Rowsby L, Prestwich G. J Endocrinol. 1977;75:305–316. doi: 10.1677/joe.0.0750305. [DOI] [PubMed] [Google Scholar]
- 34.Robertson D W, Katzenellenbogen J A, Hayes J R, Katzenellenbogen B S. J Med Chem. 1982;25:167–171. doi: 10.1021/jm00344a015. and references therein. [DOI] [PubMed] [Google Scholar]
- 35.Anstead G M, Carlson K E, Katzenellenbogen J A. Steroids. 1997;62:268–303. doi: 10.1016/s0039-128x(96)00242-5. [DOI] [PubMed] [Google Scholar]
- 36.McCague R, Jarman M, Leung O T, Foster A B, Leclercq G, Stoessel S. J Steroid Biochem. 1988;31:545–548. doi: 10.1016/0022-4731(88)90005-2. [DOI] [PubMed] [Google Scholar]