

Abstract
We evaluated the anti-tumor activity and safety of erlotinib, a receptor tyrosine kinase inhibitor of the epidermal growth factor receptor, plus sirolimus, an inhibitor of the mammalian target of rapamycin, among patients with recurrent glioblastoma (GBM) in a phase 2, open-label, single-arm trial. Thirty-two patients received daily erlotinib and sirolimus. The doses of erlotinib and sirolimus were 150 mg and 5 mg for patients not on concurrent CYP3A-inducing anti-epileptics (EIAEDS), and 450 mg and 10 mg for patients on EIAEDS. Evaluations were performed every two months. The primary endpoint was 6-month progression-free survival and secondary endpoints included safety and overall survival. Archival tumor samples were assessed for EGFR, EGFRvIII, PTEN, pAKT and pS6. Enrolled patients were heavily pre-treated including 53% who had received three or more prior chemotherapy agents and 28% who had received prior bevacizumab therapy. The most common grade ≥2 adverse events were rash (59%), mucositis (34%) and diarrhea (31%). Grade 3 or higher events were rare. Best radiographic response included stable disease in 15 patients (47%); no patients achieved either a CR or PR. The estimated 6-month progression-free survival was 3.1% for all patients. Progression-free survival was better for patients not on EIAEDs (P = 0.03). Tumor markers failed to show an association with PFS except for increased pAKT expression which achieved borderline significance (P = 0.045). Although neither rash nor diarrhea had an association with outcome, hyperlipidemia was associated with longer PFS (P = 0.029). Erlotinib plus sirolimus was well tolerated but had negligible activity among unselected recurrent GBM patients.
Keywords: Malignant glioma, EGFR, mTOR, Erlotinib, Sirolimus
Introduction
The current standard of care for newly diagnosed glioblastoma (GBM) patients, maximal safe resection followed by radiation therapy and temozolomide, results in a median overall survival (OS) of only 14.6 months, and a median progression free survival (PFS) under 7 months [1]. Although bevacizumab with or without irinotecan chemotherapy has recently been associated with modest survival benefit for recurrent patients [2–4], death following progression is essentially universal as salvage therapies are ineffective [5–7]. Therefore, there remains a significant unmet need for innovative, more effective treatments to improve outcome for recurrent GBM patients.
Activation of the phosphatidylinositol 3′-kinase (PI3K) signal transduction pathway enhances tumor cell proliferation, migration, angiogenesis and survival. Dysregulated PI3K signaling occurs commonly in GBM [8, 9] due to activation of cell surface, growth factor receptors such as the epidermal growth factor receptor (EGFR), or functional loss of the phosphatase and tensin homolog (PTEN) tumor suppressor gene [10], and confers a poor prognosis following standard cytotoxic therapy [11]. The mammalian target of rapamycin (mTOR) is a key downstream mediator of the PI3K pathway, and provides critical regulation of several essential cellular processes in both normal and neoplastic cells including nutrient metabolism, cell-cycle progression and protein translation [12].
Erlotinib (Tarceva; Genentech, Inc., South San Francisco and OSI Pharmaceuticals, Melville, NY), an orally active, reversible EGFR tyrosine kinase inhibitor, is currently approved by the USA Food and Drug Administration for recurrent non-small cell lung cancer [13, 14] and in combination with gemcitabine for pancreatic cancer [15]. Sirolimus (Rapamycin; Rapamune®, Wyeth Pharmaceuticals, Ayerst, PA), first isolated from the soil bacteria Streptomyces hygoscopicus, was approved by the FDA in 1999 as an immunosuppressant to prevent rejection of solid organ transplants. Sirolimus binds to the FK binding protein complex (FKBP-12) thereby preventing mTOR activation. Sirolimus and its analogues can induce either G1 phase arrest or apoptosis in many human tumor cell lines including GBM [16–22] and have anti-tumor activity against malignant glioma and medulloblastoma xenografts [23, 24].
Although EGFR and mTOR are key PI3K signaling mediators, the anti-tumor benefit of single-agent antagonists against either EGFR or mTOR among unselected, recurrent GBM patients has been disappointing [25–32]. Rationally designed combinatorial regimens represent a potential strategy to improve efficacy of such compounds. Our group and others have demonstrated that combination of an EGFR antagonist with an mTOR inhibitor leads to synergistic anti-tumor activity in GBM xenografts [33, 34].
We conducted the current phase 2 study to determine the anti-tumor benefit of erlotinib combined with sirolimus as a strategy to simultaneously inhibit key upstream and downstream mediators of the PI3K signaling pathway among recurrent GBM patients.
Materials and methods
Protocol objectives
The primary objective was to define the anti-tumor activity as measured by progression-free survival at 6 months (PFS-6) of erlotinib plus sirolimus in adults with recurrent GBM. Secondary objectives included the assessment of additional efficacy endpoints including radiographic response rate, median PFS and median OS following treatment with this regimen, and to further define the toxicity of this combinatorial approach in recurrent GBM patients.
Patient eligibility criteria
Patients were required to have histologically confirmed GBM that was radiographically progressive following prior radiation or chemotherapy. Additional enrollment criteria included: age at least 18 years; KPS ≥ 70%; stable corticosteroid dose for at least 1 week prior to therapy initiation; hematocrit >29%; absolute neutrophil count >1,500 cells/μl; platelet count >100,000 cells/μl; and serum creatinine, aspartate aminotransferase and bilirubin <1.5 times the institutional upper limit of normal. Patients were also required to be at least 2 weeks from prior surgical resection, 12 weeks from prior radiotherapy (unless histopathologic confirmation of recurrent GBM was obtained or new enhancement outside the radiation field was demonstrated on MRI) and 4 weeks from prior chemotherapy (6 weeks for nitrosoureas). Patients treated with prior anti-angiogenic therapy were eligible, however a minimum of 6 weeks was required from prior bevacizumab dosing. All patients were also required to have recovered from toxicities of prior therapy and to provide informed consent.
Patients were excluded for any of the following: prior therapy with either an EGFR or mTOR antagonist; uncontrolled intercurrent illness including active infection requiring intravenous antibiotics, symptomatic congestive heart failure, unstable angina, grade 3 or greater hyperlipidemia or significant gastrointestinal, renal or liver disease; pregnancy or nursing; and refusal to use effective contraception if of reproductive potential.
Treatment design
Erlotinib and sirolimus were orally administered on a continuous daily dosing schedule of 28 day cycles. Erlotinib and sirolimus metabolism are enhanced by concurrent use of CYP3A-inducing anti-epileptic drugs (EIAEDs) including phenytoin, carbamazepine, phenobarbital, oxcarbazapine and primidone [26, 35–37]. Erlotinib was dosed at the MTD established previously for patients based on concurrent EIAED use [26] while the sirolimus dose was based on dosing that was safely administered with an EGFR inhibitor in prior studies [37] and that was associated with mTOR inhibition in patient tumor samples [28]. The daily erlotinib and sirolimus doses were 150 mg and 5 mg for patients not on EIAEDs, while those on EIAEDs took 450 mg and 10 mg/day, respectively. A study therapy continued for up to 12 cycles unless patients developed unacceptable toxicity, tumor progression, non-compliance with protocol guidelines or consent withdrawal.
Supportive care
Antiemetic therapy with ondansetron and dexamethasone was permitted. Loperamide was prescribed for diarrhea as previously described [38]. Hematopoietic growth factors and blood products were administered as indicated for hematologic toxicity. Lipid lowering agents were permitted for documented hyperlipidemia. Significant rash was treated with over the counter acne preparations, antihistamines as well as topical clindamycin and/or oral antibiotics (penicillins or cephalosporins) as needed.
Study assessments
Study investigators determined response by neurologic examination and contrast-enhanced MRI prior to the start of every other treatment cycle according to the Macdonald criteria [39]. A complete response (CR) was defined as disappearance of all enhancing tumor on consecutive MRIs at least 6 weeks apart, with corticosteroid discontinuation and neurologic stability or improvement. A partial response (PR) was defined as ≥50% reduction in size (product of largest perpendicular diameters) of enhancing tumor on consecutive MRIs at least 6 weeks apart, with stability or improvement of neurologic status and corticosteroid requirement. Progressive disease (PD) was defined as ≥25% increase of enhancing tumor, a new enhancing lesion or significant clinical decline. Stable disease (SD) was defined as any assessment not meeting CR, PR, or PD criteria.
Toxicities were graded according to the National Cancer Institute's Common Toxicity Criteria Version 3.0 and classified as related to the study regimen unless attributable to either underlying tumor progression, concurrent medical condition or concomitant medication.
Dose modification and retreatment criteria
The daily erlotinib dose was reduced by 50 mg for patients not on EIAEDs, and 100 mg for those on EIAEDs who developed intolerable grade 3 rash, grade 3 diarrhea or other grade ≥3 attributable toxicity. Erlotinib was discontinued for grade 4 rash or diarrhea. The daily sirolimus dose was reduced by 50% for grade 4 neutropenia, grade 3 thrombocytopenia, grade 3 hyperlipidemia or other grade ≥3 attributable toxicity. If the event recurred, the sirolimus dose was further reduced to 3 times per week (Monday–Wednesday–Friday).
Initiation of each cycle required an ANC ≥1,000/mm3; a platelet count ≥ 1,000/mm3; AST, bilirubin and creatinine <twice the institutional upper limit of normal; resolution of any related grade ≥3 event to grade ≤1 except for rash or diarrhea which were required to resolve to grade ≤2.
Archival tumor biomarker assessment
Archival tumor samples from either initial diagnosis or after prior therapy, were analyzed for phosphorylated mitogen-activated protein kinase (p-MAPK), phosphorylated-S6 ribosomal protein (p-S6), phosphorylated-AKT (p-AKT), PTEN, EGFR and the EGFRvIII mutant receptor using immunohistochemistry (IHC) reagents and methods as previously described [37]. Similarly archival tumor samples were analyzed by fluorescence in situ hybridization (FISH) for EGFR and PTEN DNA locus copy number using reagents and methods as previously described [37]. Immunohistochemistry assays were scored for intensity of cytoplasmic/membranous staining detected by IHC was scored on a scale of 0–4+ and the distribution was defined as the percentage of cells with any level of expression. IHC staining was defined as “high” for tumors expressing 2–4+ intensity in 25% or more of tumor cells and as “low” for tumors expressing either 0–1+ staining in any percentage of tumor cells or 2–4+ intensity in less than 25% of tumor cells [8].
For FISH studies, the cutoff value for chromosomal gain was set at 20%, meaning that greater than 20% of the enumerated nuclei must show greater than two copies of the respective probe. For chromosomal loss, the cutoff value was set at 30% for definitive loss and 20–30% for indeterminate loss. EGFR gene amplification was defined as an EGFR/chromosome 7 centromere ratio of greater than 4.0. Definitive PTEN loss was defined as tumors in which ≥30% of nuclei exhibited <2 copies of PTEN locus and 2 copies of CEP 2 control. Indeterminate PTEN loss refers to tumors in which 20-30% of enumerated nuclei had <2 copies of PTEN locus and 2 copies of CEP 2 control.
Statistical considerations
The primary goal of this study was to evaluate the 6-month PFS rate of erlotinib plus sirolimus in the treatment of patients with recurrent or progressive GBM. Yung reported a median 6-month PFS of 21% (95% CI: 13.29) among recurrent GBM patients undergoing treatment with temozolomide at first recurrence [36]. The sample size goal of 32 patients for the current study provided 90% power with a test conducted at the 0.10 level of significance to differentiate between a 6-month PFS of 5 and 20%.
“Stopping rules” for unexpectedly poor efficacy and unacceptable toxicity were incorporated for each stratum. Specifically, if 10 or more of the first 16 patients per stratum experienced progression or death within 2 months of study initiation further accrual would be suspended. In addition, if six or more of the first 16 patients per stratum experienced unacceptable toxicity, defined as grade ≥4 non-hematologic events, further accrual would be suspended.
Progression-free survival (FFP) and overall survival (OS), measured from the date of cycle 1 treatment administration, were summarized using the Kaplan–Meier estimator including 95% CIs. A log-rank test was used to assess the relationship of adverse events (grade ≥ 2 rash, diarrhea, or hyperlipidemia) and tumor markers (EGFR, PTEN, pAKT, pS6 and pMAPK) on PFS.
Results
Patient characteristics
Thirty-two patients with recurrent GBM were enrolled at Duke University Medical Center between May 2007 and March 2008 (Table 1). Twenty-four patients (75%) were not on EIAEDs (stratum A) and 8 (25%) were on EIAEDs (stratum B). Patient characteristics did not differ substantially based on EIAED status. The median age was 54 years (range, 40–71 years). All patients had a KPS of at least 70%.
Table 1.
Patient demographics
| Characteristic | Not on EIAEDs (n = 24) |
On EIAEDs (n = 8) |
All (n = 32) |
|---|---|---|---|
| Age (years) | |||
| Median | 53 | 59 | 54 |
| Range | 40–71 | 45–66 | 40–71 |
| Gender | |||
| Male | 18 (75) | 7 (88) | 25 (78) |
| Female | 6 (25) | 1 (12) | 7 (22) |
| KPS | |||
| 90–100 | 12 (50) | 6 (75) | 18 (50) |
| 80 | 8 (33) | 1 (12) | 7 (22) |
| 70 | 4 (17) | 1 (12) | 5 (16) |
| Surgery prior to enrollment | |||
| GTR | 0 | 1 (13) | 1 (3) |
| STR | 0 | 0 | 0 |
| Biopsy | 4 (13) | 2 (25) | 6 (19) |
| None | 20 (63) | 5 (63) | 25 (78) |
| Prior treatment | |||
| XRT | 24 (100) | 8 (100) | 32 (100) |
| No. prior chemotherapy | |||
| Rx agents | |||
| 1 | 6 (25) | 2 (25) | 8 (25) |
| 2 | 6 (25) | 1 (12) | 7 (22) |
| 3 | 7 (29) | 1 (12) | 8 (25) |
| ≥4 | 5 (21) | 4 (50) | 9 (28) |
| Time from original diagnosis (weeks) | |||
| Median | 49.7 | 58 | 54.4 |
| Range | 19.6–158 | 31.9–153.9 | 19.6–158 |
| Prior Bevacizumab | |||
| Yes | 6 (25) | 3 (38) | 9 (28) |
| No | 18 (75) | 5 (62) | 23 (72) |
| Number Prior PD | |||
| 1 | 10 (42) | 3 (38) | 13 (41) |
| 2 | 6 (25) | 5 (62) | 11 (34) |
| 3 | 7 (29) | 0 | 7 (22) |
| ≥4 | 1 (4) | 0 | 1 (3) |
EIAEDs Enzyme-inducing anti-epileptic drug; GTR Gross total resection; KPS Karnofsky performance status; PD Progressive disease; STR Subtotal resection; XRT External beam radiotherapy
Numbers in parentheses indicate percentage unless otherwise indicated
All patients had received prior XRT and chemotherapy. Nineteen patients (59%) had more than one prior episode of progressive disease and 17 patients (53%) had received three or more prior chemotherapy agents. Eleven patients received prior anti-angiogenic therapy including 9 (28%) patients treated with bevacizumab and two patients treated with sorafenib. The median time from original diagnosis to initiation of study treatment was 54.4 weeks (range, 19.6–158 weeks).
As of 4/1/09, all patients have discontinued study therapy. Thirty patients have died due to progressive tumor.
Toxicity
Sixty-four courses of erlotinib plus sirolimus were administered to study patients including 54 courses to patients on stratum A and 10 courses to patients on stratum B. The most frequent grade ≥2 toxicities were rash (59%), mucositis (34%), diarrhea (31%), fatigue (28%) and hyperlipidemia (25%) (Table 2). Most toxicities were grade 2. Only one grade 4 event occurred––reversible thrombocytopenia in a patient who had received seven prior chemotherapeutic agents. There were no grade 5 events. Hematologic and electrolyte abnormalities were occasionally noted. Four patients developed infections; three of these were grade 2, while the remainder was a grade 3 urinary tract infection.
Table 2.
Number of patients with grade ≥2 adverse events
| Toxicity grade | 2 | 3 | 4 | |||
|---|---|---|---|---|---|---|
| Stratum1 | A | B | A | B | A | B |
| Number of patients | 24 | 8 | 24 | 8 | 24 | 8 |
| Anemia | 1 (4) | 0 | 0 | 0 | 0 | 0 |
| Anorexia | 2 (8) | 1 (13) | 0 | 0 | 0 | 0 |
| Creatinine elevation | 0 | 0 | 0 | 1 (13) | 0 | 0 |
| Diarrhea | 5 (21) | 5 (63) | 0 | 0 | 0 | 0 |
| Fatigue | 3 (9) | 4 (50) | 2 (8) | 0 | 0 | 0 |
| Hypercholesterolemia | 4 (17) | 0 | 0 | 0 | 0 | 0 |
| Hypertriglyceridemia | 3 (13) | 0 | 1 (4) | 0 | 0 | 0 |
| Hypoalbuminemia | 3 (13) | 0 | 0 | 0 | 0 | 0 |
| Hypocalcemia | 1 (4) | 0 | 0 | 0 | 0 | 0 |
| Hypokalemia | 1 (4) | 0 | 1 (4) | 0 | 0 | 0 |
| Hypophosphatemia | 0 | 0 | 1 (4) | 0 | 0 | 0 |
| Infection | 2 (8) | 1 (13) | 1 (4) | 0 | 0 | 0 |
| Mucositis | 5 (21) | 4 (50) | 2 (8) | 0 | 0 | 0 |
| Nausea/emesis | 2 (8) | 1 (13) | 0 | 0 | 0 | 0 |
| Neutropenia | 1 (4) | 0 | 1 (4) | 0 | 0 | 0 |
| Rash | 7 (29) | 5 (63) | 6 (25) | 1 (13) | 0 | 0 |
| Thrombocytopenia | 0 | 1 (13) | 2 (8) | 1 (13) | 1 (4) | 0 |
| Transaminase elevation | 0 | 0 | 0 | 1 (13) | 0 | 0 |
| Weight loss | 1 (4) | 0 | 0 | 0 | 0 | 0 |
Numbers in parentheses refer to percentages
Stratum A: Patients not on CYP3A-inducing anti-epileptics (EIA-EDS) including phenytoin, carbamazepine, phenobarbitol, oxcarbazepine and primidone
Stratum B: Patients on EIAEDs
Archival tumor biomarker analysis
Archival tumor material was available for 22 patients (69%). Analysis by FISH revealed EGFR amplification in four of nine tumors (31%), whereas 11 of 22 tumors (50%) had evidence of PTEN loss. Analysis by IHC revealed that all evaluated tumors had “high” EGFR expression, including 7 of 15 tumors with 2–3+ staining in >90% of cells and 8 of 15 tumors with 2–3 staining in >25% of cells. Interpretation of IHC was consistent with PTEN loss in 12 of 20 (60%) evaluable tumors. Expression of EG-FRvIII, pAKT, pS6 and pMAPK was detected in 25%, 44%, 83% and 40% of assessed tumors, respectively (Fig. 1).
Fig. 1.

The immunohistochemical profile of patient A103 demonstrates a strong diffuse immunoreactivity for EGFR wild type, b 30% reactivity for PTEN that correlates with PTEN loss; c negative reactivity for EGFRvIII, d diffuse reactivity for pAKT, and e rare cells exhibiting pS6 immunoreactivity with a positive internal neuronal control (arrow). The internal bar represents 50 microns
Among tumors with PTEN loss by FISH that were also assessable for pAKT and pS6, five of eight (63%) had detectable pAKT and six of eight (75%) had detectable pS6. Among tumors with EGFRvIII expression that were also assessable for pAKT and pS6, only one of four (25%) had detectable pAKT while three of three had detectable pS6.
Outcome
Outcome is summarized in Table 3. All 32 patients were evaluable for response. No patients achieved a radiographic response however stable disease was achieved by 13 patients on stratum A (54%) and two patients on stratum B (25%).
Table 3.
Outcome
| Total | # Failed | Median (weeks) | 6 Months Survival (%) | 1 Year survival (%) | Logrank_p | |||
|---|---|---|---|---|---|---|---|---|
| Overall survival | ||||||||
| All Patients | 32 | 27 | 33.8 (21.9, 53.6) | 59.4 (40.5, 74) | 34.4 (18.8, 50.6) | |||
| Strata | ||||||||
| Non-EIAEDS | 24 | 19 | 32.5 (20, 57.6) | 58.3% (36.4, 75) | 33.3 (15.9, 51.9) | 0.43 | ||
| EIAEDS | 8 | 8 | 33.8 (15.4, 55.4) | 62.5% (22.9, 86.1) | 37.5 (8.7, 67.4) | |||
| Progression free survival | ||||||||
| All patients | 32 | 32 | 6.9 (3.9, 11) | 3.1% (0.2, 13.7) | ||||
| Strata | ||||||||
| Non-EIAEDS | 24 | 24 | 8.4 (3.9, 12.1) | 4.2% (0.3, 17.6) | 0.04 | |||
| EIAEDS | 8 | 8 | 4 (3.9, 7.4) | 0 | ||||
| Prognostic factors | ||||||||
| Adverse Event | ||||||||
| Grade 2 or higher rash | No | 15 | 15 | 6.7 (4, 11.1) | 0 | 0.64 | ||
| Yes | 17 | 17 | 7.3 (3.9, 11.9) | 5.9 (0.4, 23.5) | ||||
| Grade 3 rash | No | 25 | 25 | 6.7 (3.9, 11) | 4 (0.3, 17) | 0.59 | ||
| Yes | 7 | 7 | 7.3 (3.9, 15.9) | 0 | ||||
| Diarrhea | No | 23 | 23 | 6.7 (4, 11.9) | 4.3% (0.3, 18.2) | 0.59 | ||
| Yes | 9 | 9 | 7.3 (3.9, 9.4) | 0 | ||||
| Hyperlipidemia | No | 28 | 28 | 5.4 (3.9, 7.7) | 0 | 0.029 | ||
| Yes | 4 | 4 | 16.7 (3.6, 39.9) | 25 (0.9, 66.5) | ||||
| Tumor marker | ||||||||
| EGFR-FISH | Polysomy | 9 | 9 | 7.7 (3.9, 12.1) | 11.1% (0.6, 38.8) | 0.49 | ||
| Amplified | 4 | 4 | 5.7 (3.6, 12.7) | 0 | ||||
| EGFR-IHC | 30%–60% pos. | 4 | 4 | 3.9 (2.4, 7.7) | 0 | 0.35 | ||
| 80%–90% pos | 11 | 11 | 4 (3.9, 9.4) | 0 | ||||
| EGFR-IHC | <90% pos. | 8 | 8 | 4 (3.9, 7.7) | 0 | 0.64 | ||
| ≥90% pos. | 7 | 7 | 3.9 (3.6, 11.1) | 0 | ||||
| PTEN-FISH | Intact | 11 | 11 | 4 (3.9, 11.9) | 0 | 0.60 | ||
| Loss | 11 | 11 | 7.4 (3.9, 12.7) | 9.1 (0.5, 33.3) | ||||
| PTEN-IHC | Intact | 8 | 8 | 4 (3.6, 12.7) | 0 | 0.23 | ||
| Loss | 12 | 12 | 10.3 (7, 12.1) | 8.3 (0.5, 31.1) | ||||
| EGFRvIII-IHC | Negative | 16 | 16 | 8.6 (4, 12.1) | 6.3 (0.4, 24.7) | 0.36 | ||
| Positive | 4 | 4 | 5.5 (3.6, 12.7) | 0 | ||||
| pAKT-IHC | Negative | 9 | 9 | 4 (3.6, 7.4) | 0 | 0.04 | ||
| Positive | 7 | 7 | 9.4 (4, 12.7) | 14.3 (0.7, 46.5) | ||||
| pS6-IHC | Negative | 2 | 2 | 23.6 (7.4, 39.9) | 50 (0.6, 91) | 0.18 | ||
| Positive | 10 | 10 | 5.5 (3.9, 9.4) | 0 | ||||
| pMAPK-IHC | Negative | 3 | 3 | 4 (4, 7.7) | 0 | 0.49 | ||
| Positive | 2 | 2 | 5.5 (3.6, 7.4) | 0 |
Numbers in parentheses refer to 95% confidence intervals unless otherwise indicated
With a median follow-up of 69.3 weeks (95% CI, 62-87.4), the median PFS and 6-month PFS rate for all patients were 6.9 weeks (95% CI, 3.9-11.0 weeks) and 3.1% (95% CI, 0.2–13.7%), respectively (Fig. 2a). Median PFS and 6-month PFS for patients on stratum A were 8.4 weeks (95% CI, 3.9–12.1 weeks) and 4.2% (95% CI, 0.3–17.6%), respectively, and 4.0 weeks (95% CI, 3.9–7.4 weeks) and 0%, respectively for patients on stratum B. A comparison of stratum-specfic PFS was statistically significant (P = 0.03).
Fig. 2.
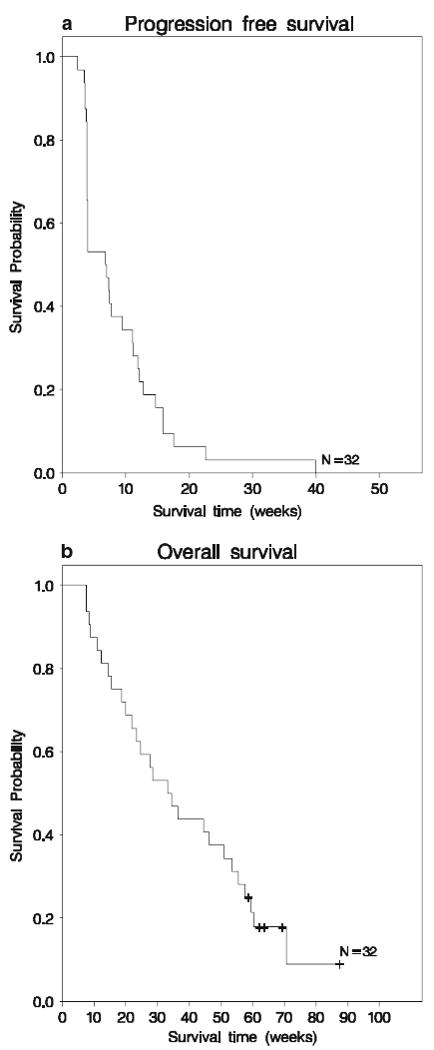
Kaplan–Meier plots of progression-free survival (Fig. 1a) and overall survival (Fig. 1b)
Median PFS for patients who received prior bevacizumab (n = 7) was 4.0 weeks (95% CI: 3.6, 7.0), while the median PFS for those who did not receive prior bevacizumab (n = 25) was 7.4 weeks (95% CI: 3.9, 11.9) (P = 0.18). None of the archival tumor sample markers showed an association with 6-month PFS except for expression of p-AKT, which achieved borderline significance (p = 0.045). However, tumor biomarker analysis was clearly limited by the small number of analyzed samples and the overall low activity of the study regimen. Among treatment-specific toxicities, PFS was associated with hyperlipidemia (P = 0.03) but not rash or diarrhea.
Median OS for all patients was 33.8 weeks (95% CI, 21.9-53.6 weeks) and did not differ significantly by EIAED status (Fig. 2b).
Discussion
Nearly all newly diagnosed GBM patients progress within months of diagnosis despite aggressive, multi-modality therapy. Historically, salvage therapies have been largely ineffective with most patients dying within months of recurrence. Many groups, including ours, have extensively evaluated salvage regimens incorporating recently developed therapeutics designed to inhibit key mediators of cell signaling pathways, including the PI3-Akt and ras-MAPK transduction pathways. These pathways are dysregulated in the majority of GBM tumor samples and are known to critically contribute to GBM pathophysiology including cell survival, proliferation, invasion, and angiogenesis. Furthermore, evidence of activation of these signaling pathways confers a poorer outcome [11, 40].
Unfortunately, clinical trials evaluating inhibitors of signal transduction pathways, administered as single-agents among unselected, recurrent GBM patients, have been largely disappointing [25–27, 29, 30, 41–43]. Several factors may underlie the poor outcome of these trials including heterogeneity of target expression within and across tumors, complexity of signaling cascades including redundancy and cross-talk, and resistance mediated by compensatory upregulation of alternative pathway mediators.
The current study was designed to evaluate erlotinib, an EGFR tyrosine kinase inhibitor, when combined with sirolimus, an mTOR inhibitor among recurrent GBM patients. The hypothesis underlying this combinatorial regimen is that simultaneously targeting upstream and downstream mediators is more likely to suppress PI3 K-AKT signaling and thereby induce greater tumor cell death than either agent alone. Furthermore, this combination may better overcome target heterogeneity, mitigate the impact of signaling pathway redundancy and cross-talk, and blunt resistance mediated by upregulation of alternative pathway mediators.
Results of preclinical studies demonstrate that combinatorial regimens can simultaneously inhibit dual targets within the PI3K-A pathway and provide enhanced anti-tumor efficacy. Combination of an EGFR inhibitor with inhibitors of either AKT or PI3K led to diminished cell proliferation [44, 45]. Similarly enhanced anti-glioma efficacy was observed with dual PI3 kinase/mTOR targeting [46]. We previously demonstrated that combination therapy with AEE788 (Novartis Pharma AG, Basel, Switzerland), an EGFR TKI, with the rapamycin-derivative, RAD001 (Novartis Pharma AG, Basel, Switzerland) increased cell cycle arrest and apoptosis with reduced proliferation in vitro compared to either agent against GBM cell lines. Furthermore, the combination improved tumor growth inhibition and overall survival in athymic mice with GBM xenografts [34]. Synergistic inhibition of glioma cell growth was also demonstrated following administration of sirolimus with the EGFR TKI EKI-785 (Wyeth Pharmaceuticals, Madision, New Jersey, USA) [33]. Finally, sirolimus and erlotinib achieved enhanced tumor cell death and growth inhibition, as well as blocking downstream PI3K pathway signaling in both PTEN-deficient and PTEN-intact GBM cells [47].
Clinical studies evaluating simultaneous administration of EGFR and mTOR inhibitors among recurrent GBM patients are limited. Doherty reported a retrospective review of 28 recurrent malignant glioma patients (GBM, n = 22; grade 3 anaplastic glioma, n = 6) treated with sirolimus plus either gefitinib or erlotinib. Of note, none of the patients were on EIAEDs. Partial responses were observed in five patients (19%) including four with GBM (18%), and stable disease was achieved in 14 patients (50%) including nine with GBM (41%). PFS-6 for the GBM patients was 25% [48]. Similarly, Kreisl noted partial radiographic response and stable disease in 3 (14%) and 8 (36%) of recurrent GBM patients treated with everolimus and gefitinib, however durability of disease control was poor with a median PFS of only 2.6 months and only one patient (4%) remaining progression-free at six months [49]. We recently demonstrated that the EGFR tyrosine kinase inhibitor gefitinib can be safely co-administered at standard single-agent dose levels in combination with sirolimus among recurrent malignant glioma patients. In that study, we also confirmed that gefitinib pharmacokinetics are not affected by sirolimus co-administration. Furthermore, despite the phase I design of this study, modest evidence of anti-tumor activity was observed including a PFS-6 of 23.5% [37]. Unfortunately, enthusiasm for further clinical development of gefitinib dampened upon revocation of accelerated approval status by the FDA following failure to demonstrate adequate anti-tumor activity in randomized phase 3 studies for recurrent non-small cell lung cancer [50, 51].
Despite the encouraging preclinical and clinical data supporting the rationale of combining erlotinib with sirolimus for recurrent GBM, results of the current study were disappointing. Several factors may have contributed. First, patients in the current study were heavily pretreated. Fifty-nine percent of patients enrolled after two or more prior episodes of progressive disease and 75% had received two or more prior chemotherapeutics, while 28% had failed prior bevacizumab therapy. Second, the dose of erlotinib used in this phase 2 study was based on the MTD of single-agent erlotinib [26], while we used a sirolimus dose that has been safely administered in prior studies [37] and associated with mTOR inhibition in patient tumor samples [28]. A phase I study of this regimen in this patient population has not been performed; thus the true MTD has not been defined. In addition, an optimal biologic dose has also not been determined. One strategy to determine an optimal biologic dose involves demonstration of target inhibition in GBM cells obtained from patients treated prior to planned debulking surgery. Of note, an analysis of tumor samples obtained from a sampling of recurrent GBM patients failed to identify an effect of pre-treatment with either gefitinib or erlotinib on EGFR activity in vivo [52]. These findings suggest that evaluation of alternative intra-tumoral pharmacodynamic molecular effects may be more informative for patients treated with a combination of EGFR and mTOR inhibitors. The critical importance of intratumoral pharmacodynamic assessment is underscored by the recent report of increased AKT activation observed among a subset of patients following mTOR inhibitor therapy [28]. Increased Akt activation and increased tumor cell survival pathway signaling is associated with mTOR inhibitor therapy [53, 54] and may contribute to failure of mTOR inhibitor therapy in some cancer patients.
Finally, pharmacokinetic analyses were not incorporated into this study. It is possible that since both erlotinib and sirolimus are CYP 3A substrates [35, 36], a detrimental pharmacokinetic interaction between these two agents may have occurred which led to limited anti-tumor efficacy. This interaction may have been further compounded by EIAEDs which were administered to 8 (25%) patients on this study. In fact, we noted that PFS was modestly improved among patients not on EIAEDs (P = 0.03). In addition, intra-tumoral pharmacokinetics are particularly relevant in malignant glioma patients because the blood brain barrier (BBB) may limit CNS penetration even when adequate systemic levels are achieved. In particular, it appears that several TKIs, including erlotinib, are substrates for BBB efflux proteins such as p-glycoprotein (P-gp), breast cancer resistance protein (BCRP, ABCG2) and multidrug resistance protein 2 (MRP2; ABCC2) [55, 56].
Although some studies have noted a correlation between tumor marker expression and outcome [32, 57–59], others studies have failed to detect such an association [25, 52, 60, 61]. In the current study, we did not observe a relationship between PI3/Akt pathway marker expression and outcome with the exception of increased pAKT, which achieved borderline significance. Paradoxically, others have noted an association between low pAKT and response to EGFR inhibitor therapy [58, 59]. An explanation for these discrepant findings is unclear, but may reflect differences in either immunohistochemistry assay methods, thresholds used to define expression levels or response criteria. Similarly, although increased sensitivity to mTOR inhibition has been reported among PTEN-deficient glioma lines compared to those with intact PTEN [62], others have reported that response to mTOR inhibition does not correlate with PTEN status [63], and a recently completed clinical trial of single-agent CCI-799 (temsirolimus) found no correlation between PTEN status and outcome [27]. An important factor limiting analyses that attempt to associate tumor biomarkers and outcome, including that performed in the current study, is the use of archival tumor material rather than tumor material obtained immediately prior to study treatment. While the latter accurately reflects the constellation of molecular genetic abnormalities that characterize a given tumor at the time of treatment, archival tumor material may do so less reliably.
Although some studies have noted an associaton between either rash [32] or diarrhea [25] and outcome following EGFR inhibitor therapy, our study was consistent with others that did not observe an association between outcome and either rash or diarrhea [61, 64]. However, we did observe a correlation between hyperlipidemia and improved outcome as was similarly noted among recurrent GBM patients treated with the mTOR inhibitor temsirolimus [27]. Although it is possible that this association occurred by chance due to multiple hypothesis testing, future studies should further evaluate the association between hyperlipidemia and response to mTOR inhibitor therapy.
Overall, the combination of erlotinib and sirolimus was well tolerated among patients treated on the current study. The range of toxicities was within that predicted to be observed with single-agent administration of each of these agents [26, 28], as well as within the spectrum of what we observed previously among recurrent GBM patients treated with gefitinib and sirolimus [37]. In contrast, significant toxicity was observed among patients treated with erlotinib and temsirolimus in a phase I dose escalation study, such that de-escalation of study agents was required. [65] An explanation for differences in tolerability between these regimens may be due to different pharmacokinetic interactions or undefined pharmacogenetic factors.
We report limited efficacy among heavily pre-treated, recurrent GBM patients treated with erlotinib and sirolimus. Due to limitations of the current study, it is not clear whether this regimen is truly inactive or if our dosing schedule was inadequate to achieve intra-tumoral concentrations sufficient to inhibit the intended PI3-AKT pathway targets. Although the rationale for combinatorial regimens that simultaneously target key upstream and downstream signal transduction pathway mediators is logical and supported preclinically, effective translation of this concept into the clinic will require further evaluation of clinical, tumor biomarker, pharmacokinetic and intra-tumoral pharmacodynamic measures.
Acknowledgments
This work was supported by NIH Grants NS20023 and CA11898; NIH Grant MO1 RR 30, GCRC Program, NCRR; and NCI SPORE 1 P20 CA096890
Abbreviations list
- ANC
Absolute neutrophil count
- AST
Aspartate aminotransferase
- BBB
Blood-brain barrier
- BUN
Blood urea nitrogen
- CBC
Complete blood count
- CI
Confidence intervals
- CNS
Central nervous system
- CR
Complete response
- CTC
Common Toxicity Criteria
- CYP
Cytochrome p450
- DLT
Dose-limiting toxicity
- EGFR
Epidermal growth factor receptor
- EIAEDs
Enzyme-inducing antieptileptic drugs
- EGFR
Epidermal growth factor receptor
- GBM
Glioblastoma multiforme
- GS
Gliosarcoma
- IRB
Institutional review board
- ITT
Intent-to treat
- KPS
Karnofsky performance status
- MG
Malignant glioma
- MTD
Maximum-tolerated dose
- mTOR
Mammalian target of rapamycin
- NCI
National Cancer Institute
- NE
Non-estimable
- pAKT
Phosphorylated akt murine thymomoa viral oncogene homologue 1
- PD
Progressive disease
- PFS
Progression-free survival
- P-gp
p-glycoprotein
- pMAPK
Phosphorylated mitogen activated protein kinase
- PR
Partial response
- pS-6
Phosphorylated S-6 ribosomal protein
- PTEN
Phosphatase and tensin homologue
- SD
Stable disease
- TKI
Tyrosine kinase inhibitor
- VEGF
Vascular endothelial growth factor
- XRT
External beam radiotherapy
Contributor Information
David A. Reardon, Department of Surgery, Duke University Medical Center, Durham, NC 27710, USA; Department of Pediatrics, Duke University Medical Center, Durham, NC 27710, USA
Annick Desjardins, Department of Medicine, Duke University Medical Center, Durham, NC 27710, USA.
James J. Vredenburgh, Department of Medicine, Duke University Medical Center, Durham, NC 27710, USA
Sridharan Gururangan, Department of Surgery, Duke University Medical Center, Durham, NC 27710, USA; Department of Pediatrics, Duke University Medical Center, Durham, NC 27710, USA.
Allan H. Friedman, Department of Surgery, Duke University Medical Center, Durham, NC 27710, USA
James E. Herndon, II, Cancer Center Biostatistics, Duke University Medical Center, Durham, NC 27710, USA.
Jennifer Marcello, Cancer Center Biostatistics, Duke University Medical Center, Durham, NC 27710, USA.
Julie A. Norfleet, Department of Surgery, Duke University Medical Center, Durham, NC 27710, USA
Roger E. McLendon, Department of Pathology, Duke University Medical Center, Durham, NC 27710, USA
John H. Sampson, Department of Surgery, Duke University Medical Center, Durham, NC 27710, USA
Henry S. Friedman, Department of Surgery, Duke University Medical Center, Durham, NC 27710, USA; Department of Pediatrics, Duke University Medical Center, Durham, NC 27710, USA
References
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Vredenburgh JJ, Desjardins A, Herndon JE, II, Marcello J, Reardon DA, Quinn JA, Rich JN, Sathornsumetee S, Gururangan S, Sampson J, Wagner M, Bailey L, Bigner DD, Friedman AH, Friedman HS. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol. 2007;25:4722–4729. doi: 10.1200/JCO.2007.12.2440. [DOI] [PubMed] [Google Scholar]
- 3.Cloughesy TF, Prados MD, Mikkelsen T, Abrey LE, Schiff D, Yung WK, Maoxia Z, Dimery I, Friedman HS. A phase II, randomized, non-comparative clinical trial of the effect of bevacizumab (BV) alone or in combination with irinotecan (CPT) on 6-month progression free survival (PFS6) in recurrent, treatment-refractory glioblastoma (GBM) In: Grunberg SM, editor. Proceedings of the American Society for Clinical Oncology. Chicago, IL: 2008. p. 91. [Google Scholar]
- 4.Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I, Garren N, Mackey M, Butman JA, Camphausen K, Park J, Albert PS, Fine HA. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27:740–745. doi: 10.1200/JCO.2008.16.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wong ET, Hess KR, Gleason MJ, Jaeckle KA, Kyritsis AP, Prados MD, Levin VA, Yung WK. Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol. 1999;17:2572–2578. doi: 10.1200/JCO.1999.17.8.2572. [DOI] [PubMed] [Google Scholar]
- 6.Lamborn KR, Yung WK, Chang SM, Wen PY, Cloughesy TF, Deangelis LM, Robins HI, Lieberman FS, Fine HA, Fink KL, Junck L, Abrey L, Gilbert MR, Mehta M, Kuhn JG, Aldape KD, Hibberts J, Peterson PM, Prados MD. Progression-free survival: an important end point in evaluating therapy for recurrent high-grade gliomas. Neuro-oncology. 2008;10:162–170. doi: 10.1215/15228517-2007-062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ballman KV, Buckner JC, Brown PD, Giannini C, Flynn PJ, LaPlant BR, Jaeckle KA. The relationship between six-month progression-free survival and 12-month overall survival end points for phase II trials in patients with glioblastoma multiforme. Neuro-oncology. 2007;9:29–38. doi: 10.1215/15228517-2006-025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choe G, Horvath S, Cloughesy TF, Crosby K, Seligson D, Palotie A, Inge L, Smith BL, Sawyers CL, Mischel PS. Analysis of the phosphatidylinositol 3′-kinase signaling pathway in glioblastoma patients in vivo. Cancer Res. 2003;63:2742–2746. [PubMed] [Google Scholar]
- 9.Ruano Y, Mollejo M, Camacho FI, Rodriguez de Lope A, Fiano C, Ribalta T, Martinez P, Hernandez-Moneo JL, Melendez B. Identification of survival-related genes of the phosphatidylinositol 3′-kinase signaling pathway in glioblastoma multiforme. Cancer. 2008;112:1575–1584. doi: 10.1002/cncr.23338. [DOI] [PubMed] [Google Scholar]
- 10.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 11.Chakravarti A, Zhai G, Suzuki Y, Sarkesh S, Black PM, Muzikansky A, Loeffler JS. The prognostic significance of phosphatidylinositol 3-kinase pathway activation in human gliomas. J Clin Oncol. 2004;22:1926–1933. doi: 10.1200/JCO.2004.07.193. [DOI] [PubMed] [Google Scholar]
- 12.Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253–262. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- 13.Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel-Reid S, Squire J, Lorimer I, Zhang T, Liu N, Daneshmand M, Marrano P, da Cunha Santos G, Lagarde A, Richardson F, Seymour L, Whitehead M, Ding K, Pater J, Shepherd FA. Erlotinib in lung cancer––molecular and clinical predictors of outcome. N Engl J Med. 2005;353:133–144. doi: 10.1056/NEJMoa050736. [DOI] [PubMed] [Google Scholar]
- 14.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, van Kooten M, Dediu M, Findlay B, Tu D, Johnston D, Bezjak A, Clark G, Santabarbara P, Seymour L. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–132. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 15.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M, Parulekar W. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 16.Seufferlein T, Rozengurt E. Rapamycin inhibits constitutive p70s6k phosphorylation, cell proliferation, and colony formation in small cell lung cancer cells. Cancer Res. 1996;56:3895–3897. [PubMed] [Google Scholar]
- 17.Hosoi H, Dilling MB, Shikata T, Liu LN, Shu L, Ashmun RA, Germain GS, Abraham RT, Houghton PJ. Rapamycin causes poorly reversible inhibition of mTOR and induces p53-independent apoptosis in human rhabdomyosarcoma cells. Cancer Res. 1999;59:886–894. [PubMed] [Google Scholar]
- 18.Hosoi H, Dilling MB, Liu LN, Danks MK, Shikata T, Sekulic A, Abraham RT, Lawrence JC, Jr, Houghton PJ. Studies on the mechanism of resistance to rapamycin in human cancer cells. Mol Pharmacol. 1998;54:815–824. doi: 10.1124/mol.54.5.815. [DOI] [PubMed] [Google Scholar]
- 19.Grewe M, Gansauge F, Schmid RM, Adler G, Seufferlein T. Regulation of cell growth and cyclin D1 expression by the constitutively active FRAP-p70s6K pathway in human pancreatic cancer cells. Cancer Res. 1999;59:3581–3587. [PubMed] [Google Scholar]
- 20.Majewski M, Korecka M, Kossev P, Li S, Goldman J, Moore J, Silberstein LE, Nowell PC, Schuler W, Shaw LM, Wasik MA. The immunosuppressive macrolide RAD inhibits growth of human Epstein-Barr virus-transformed B lymphocytes in vitro and in vivo: a potential approach to prevention and treatment of posttransplant lymphoproliferative disorders. Proc Natl Acad Sci U S A. 2000;97:4285–4290. doi: 10.1073/pnas.080068597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dudkin L, Dilling MB, Cheshire PJ, Harwood FC, Hollingshead M, Arbuck SG, Travis R, Sausville EA, Houghton PJ. Biochemical correlates of mTOR inhibition by the rapamycin ester CCI-779 and tumor growth inhibition. Clin Cancer Res. 2001;7:1758–1764. [PubMed] [Google Scholar]
- 22.Jiang W, Cazacu S, Xiang C, Zenklusen JC, Fine HA, Berens M, Armstrong B, Brodie C, Mikkelsen T. FK506 binding protein mediates glioma cell growth and sensitivity to rapamycin treatment by regulating NF-kappaB signaling pathway. Neoplasia. 2008;10:235–243. doi: 10.1593/neo.07929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geoerger B, Kerr K, Tang CB, Fung KM, Powell B, Sutton LN, Phillips PC, Janss AJ. Antitumor activity of the rapamycin analog CCI-779 in human primitive neuroectodermal tumor/medulloblastoma models as single agent and in combination chemotherapy. Cancer Res. 2001;61:1527–1532. [PubMed] [Google Scholar]
- 24.Eshleman JS, Carlson BL, Mladek AC, Kastner BD, Shide KL, Sarkaria JN. Inhibition of the mammalian target of rapamycin sensitizes U87 xenografts to fractionated radiation therapy. Cancer Res. 2002;62:7291–7297. [PubMed] [Google Scholar]
- 25.Rich JN, Reardon DA, Peery T, Dowell JM, Quinn JA, Penne KL, Wikstrand CJ, Van Duyn LB, Dancey JE, McLendon RE, Kao JC, Stenzel TT, Ahmed Rasheed BK, Tourt-Uhlig SE, Herndon JE, 2nd, Vredenburgh JJ, Sampson JH, Friedman AH, Bigner DD, Friedman HS. Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol. 2004;22:133–142. doi: 10.1200/JCO.2004.08.110. [DOI] [PubMed] [Google Scholar]
- 26.Prados MD, Lamborn KR, Chang S, Burton E, Butowski N, Malec M, Kapadia A, Rabbitt J, Page MS, Fedoroff A, Xie D, Kelley SK. Phase 1 study of erlotinib HCl alone and combined with temozolomide in patients with stable or recurrent malignant glioma. Neuro-oncol. 2006;8:67–78. doi: 10.1215/S1522851705000451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galanis E, Buckner JC, Maurer MJ, Kreisberg JI, Ballman K, Boni J, Peralba JM, Jenkins RB, Dakhil SR, Morton RF, Jaeckle KA, Scheithauer BW, Dancey J, Hidalgo M, Walsh DJ. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group study. J Clin Oncol. 2005;23:5294–5304. doi: 10.1200/JCO.2005.23.622. [DOI] [PubMed] [Google Scholar]
- 28.Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, Hsueh T, Chen Y, Wang W, Youngkin D, Liau L, Martin N, Becker D, Bergsneider M, Lai A, Green R, Oglesby T, Koleto M, Trent J, Horvath S, Mischel PS, Mellinghoff IK, Sawyers CL. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS medicine. 2008;5:e8. doi: 10.1371/journal.pmed.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang SM, Wen P, Cloughesy T, Greenberg H, Schiff D, Conrad C, Fink K, Robins HI, De Angelis L, Raizer J, Hess K, Aldape K, Lamborn KR, Kuhn J, Dancey J, Prados MD. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Invest New Drugs. 2005;23:357–361. doi: 10.1007/s10637-005-1444-0. [DOI] [PubMed] [Google Scholar]
- 30.Chang SM, Kuhn J, Wen P, Greenberg H, Schiff D, Conrad C, Fink K, Robins HI, Cloughesy T, De Angelis L, Razier J, Hess K, Dancey J, Prados MD. Phase I/pharmacokinetic study of CCI-779 in patients with recurrent malignant glioma on enzyme-inducing antiepileptic drugs. Invest New Drugs. 2004;22:427–435. doi: 10.1023/B:DRUG.0000036685.72140.03. [DOI] [PubMed] [Google Scholar]
- 31.Brandes AA, Franceschi E, Tosoni A, Hegi ME, Stupp R. Epidermal growth factor receptor inhibitors in neuro-oncology: hopes and disappointments. Clin Cancer Res. 2008;14:957–960. doi: 10.1158/1078-0432.CCR-07-1810. [DOI] [PubMed] [Google Scholar]
- 32.van den Bent MJ, Brandes AA, Rampling R, Kouwenhoven MC, Kros JM, Carpentier AF, Clement PM, Frenay M, Campone M, Baurain JF, Armand JP, Taphoorn MJ, Tosoni A, Kletzl H, Klughammer B, Lacombe D, Gorlia T. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol. 2009;27:1268–1274. doi: 10.1200/JCO.2008.17.5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rao RD, Mladek AC, Lamont JD, Goble JM, Erlichman C, James CD, Sarkaria JN. Disruption of parallel and converging signaling pathways contributes to the synergistic antitumor effects of simultaneous mTOR and EGFR inhibition in GBM cells. Neoplasia. 2005;7:921–929. doi: 10.1593/neo.05361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goudar RK, Shi Q, Hjelmeland MD, Keir ST, McLendon RE, Wikstrand CJ, Reese ED, Conrad CA, Traxler P, Lane HA, Reardon DA, Cavenee WK, Wang XF, Bigner DD, Friedman HS, Rich JN. Combination therapy of inhibitors of epidermal growth factor receptor/vascular endothelial growth factor receptor 2 (AEE788) and the mammalian target of rapamycin (RAD001) offers improved glioblastoma tumor growth inhibition. Mol Cancer Ther. 2005;4:101–112. [PubMed] [Google Scholar]
- 35.Li J, Zhao M, He P, Hidalgo M, Baker SD. Differential metabolism of gefitinib and erlotinib by human cytochrome P450 enzymes. Clin Cancer Res. 2007;13:3731–3737. doi: 10.1158/1078-0432.CCR-07-0088. [DOI] [PubMed] [Google Scholar]
- 36.Kuhn JG, Chang SM, Wen PY, Cloughesy TF, Greenberg H, Schiff D, Conrad C, Fink KL, Robins HI, Mehta M, DeAngelis L, Raizer J, Hess K, Lamborn KR, Dancey J, Prados MD. Pharmacokinetic and tumor distribution characteristics of temsirolimus in patients with recurrent malignant glioma. Clin Cancer Res. 2007;13:7401–7406. doi: 10.1158/1078-0432.CCR-07-0781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reardon DA, Quinn JA, Vredenburgh JJ, Gururangan S, Friedman AH, Desjardins A, Sathornsumetee S, Herndon JE, 2nd, Dowell JM, McLendon RE, Provenzale JM, Sampson JH, Smith RP, Swaisland AJ, Ochs JS, Lyons P, Tourt-Uhlig S, Bigner DD, Friedman HS, Rich JN. Phase 1 trial of gefitinib plus sirolimus in adults with recurrent malignant glioma. Clin Cancer Res. 2006;12:860–868. doi: 10.1158/1078-0432.CCR-05-2215. [DOI] [PubMed] [Google Scholar]
- 38.Reardon DA, Quinn JA, Vredenburgh J, Rich JN, Gururangan S, Badruddoja M, Herndon JE, 2nd, Dowell JM, Friedman AH, Friedman HS. Phase II trial of irinotecan plus celecoxib in adults with recurrent malignant glioma. Cancer. 2005;103:329–338. doi: 10.1002/cncr.20776. [DOI] [PubMed] [Google Scholar]
- 39.Macdonald DR, Cascino TL, Schold SC, Jr, Cairncross JG. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990;8:1277–1280. doi: 10.1200/JCO.1990.8.7.1277. [DOI] [PubMed] [Google Scholar]
- 40.Pelloski CE, Lin E, Zhang L, Yung WK, Colman H, Liu JL, Woo SY, Heimberger AB, Suki D, Prados M, Chang S, Barker FG, 3rd, Fuller GN, Aldape KD. Prognostic associations of activated mitogen-activated protein kinase and Akt pathways in glioblastoma. Clin Cancer Res. 2006;12:3935–3941. doi: 10.1158/1078-0432.CCR-05-2202. [DOI] [PubMed] [Google Scholar]
- 41.Wen PY, Yung WK, Lamborn KR, Dahia PL, Wang Y, Peng B, Abrey LE, Raizer J, Cloughesy TF, Fink K, Gilbert M, Chang S, Junck L, Schiff D, Lieberman F, Fine HA, Mehta M, Robins HI, DeAngelis LM, Groves MD, Puduvalli VK, Levin V, Conrad C, Maher EA, Aldape K, Hayes M, Letvak L, Egorin MJ, Capdeville R, Kaplan R, Murgo AJ, Stiles C, Prados MD. Phase I/II study of imatinib mesylate for recurrent malignant gliomas: North American Brain Tumor Consortium study 99–08. Clin Cancer Res. 2006;12:4899–4907. doi: 10.1158/1078-0432.CCR-06-0773. [DOI] [PubMed] [Google Scholar]
- 42.Cloughesy TF, Wen PY, Robins HI, Chang SM, Groves MD, Fink KL, Junck L, Schiff D, Abrey L, Gilbert MR, Lieberman F, Kuhn J, DeAngelis LM, Mehta M, Raizer JJ, Yung WK, Aldape K, Wright J, Lamborn KR, Prados MD. Phase II trial of tipifarnib in patients with recurrent malignant glioma either receiving or not receiving enzyme-inducing antiepileptic drugs: a North American Brain Tumor Consortium study. J Clin Oncol. 2006;24:3651–3656. doi: 10.1200/JCO.2006.06.2323. [DOI] [PubMed] [Google Scholar]
- 43.Raymond E, Brandes AA, Dittrich C, Fumoleau P, Coudert B, Clement PM, Frenay M, Rampling R, Stupp R, Kros JM, Heinrich MC, Gorlia T, Lacombe D, van den Bent MJ. Phase II study of imatinib in patients with recurrent gliomas of various histologies: a European Organisation for Research and Treatment of Cancer Brain Tumor Group study. J Clin Oncol. 2008;26:4659–4665. doi: 10.1200/JCO.2008.16.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li B, Chang CM, Yuan M, McKenna WG, Shu HK. Resistance to small molecule inhibitors of epidermal growth factor receptor in malignant gliomas. Cancer Res. 2003;63:7443–7450. [PubMed] [Google Scholar]
- 45.She QB, Solit D, Basso A, Moasser MM. Resistance to gefitinib in PTEN-null HER-overexpressing tumor cells can be overcome through restoration of PTEN function or pharmacologic modulation of constitutive phosphatidylinositol 3′-kinase/Akt pathway signaling. Clin Cancer Res. 2003;9:4340–4346. [PubMed] [Google Scholar]
- 46.Fan QW, Knight ZA, Goldenberg DD, Yu W, Mostov KE, Stokoe D, Shokat KM, Weiss WA. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell. 2006;9:341–349. doi: 10.1016/j.ccr.2006.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang MY, Lu KV, Zhu S, Dia EQ, Vivanco I, Shackleford GM, Cavenee WK, Mellinghoff IK, Cloughesy TF, Sawyers CL, Mischel PS. Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN-deficient and PTEN-intact glioblastoma cells. Cancer Res. 2006;66:7864–7869. doi: 10.1158/0008-5472.CAN-04-4392. [DOI] [PubMed] [Google Scholar]
- 48.Doherty L, Gigas DC, Kesari S, Drappatz J, Kim R, Zimmerman J, Ostrowsky L, Wen PY. Pilot study of the combination of EGFR and mTOR inhibitors in recurrent malignant gliomas. Neurology. 2006;67:156–158. doi: 10.1212/01.wnl.0000223844.77636.29. [DOI] [PubMed] [Google Scholar]
- 49.Kreisl TN, Lassman AB, Mischel PS, Rosen N, Scher HI, Teruya-Feldstein J, Shaffer D, Lis E, Abrey LE. A pilot study of everolimus and gefitinib in the treatment of recurrent glioblastoma (GBM) J Neurooncol. 2009;92:99–105. doi: 10.1007/s11060-008-9741-z. [DOI] [PubMed] [Google Scholar]
- 50.Herbst RS, Giaccone G, Schiller JH, Natale RB, Miller V, Manegold C, Scagliotti G, Rosell R, Oliff I, Reeves JA, Wolf MK, Krebs AD, Averbuch SD, Ochs JS, Grous J, Fandi A, Johnson DH. Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: a phase III trial–INTACT 2. J Clin Oncol. 2004;22:785–794. doi: 10.1200/JCO.2004.07.215. [DOI] [PubMed] [Google Scholar]
- 51.Giaccone G, Herbst RS, Manegold C, Scagliotti G, Rosell R, Miller V, Natale RB, Schiller JH, Von Pawel J, Pluzanska A, Gatzemeier U, Grous J, Ochs JS, Averbuch SD, Wolf MK, Rennie P, Fandi A, Johnson DH. Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer: a phase III trial––INTACT 1. J Clin Oncol. 2004;22:777–784. doi: 10.1200/JCO.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 52.Lassman AB, Rossi MR, Razier JR, Abrey LE, Lieberman FS, Grefe CN, Lamborn K, Pao W, Shih AH, Kuhn JG, Wilson R, Nowak NJ, Cowell JK, DeAngelis LM, Wen P, Gilbert MR, Chang S, Yung WA, Prados M, Holland EC. Molecular study of malignant gliomas treated with epidermal growth factor receptor inhibitors: tissue analysis from north american brain tumor consortium trials 01–03 and 00–01. Clin Cancer Res. 2005;11:7841–7850. doi: 10.1158/1078-0432.CCR-05-0421. [DOI] [PubMed] [Google Scholar]
- 53.Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, Khuri FR. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005;65:7052–7058. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- 54.O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ozvegy-Laczka C, Cserepes J, Elkind NB, Sarkadi B. Tyrosine kinase inhibitor resistance in cancer: role of ABC multidrug transporters. Drug Resist Updat. 2005;8:15–26. doi: 10.1016/j.drup.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 56.Marchetti S, de Vries NA, Buckle T, Bolijn MJ, van Eijndhoven MA, Beijnen JH, Mazzanti R, van Tellingen O, Schellens JH. Effect of the ATP-binding cassette drug transporters ABCB1, ABCG2, and ABCC2 on erlotinib hydrochloride (Tarceva) disposition in in vitro and in vivo pharmacokinetic studies employing Bcrp1-/-/Mdr1a/1b-/- (triple-knockout) and wild-type mice. Mol Cancer Ther. 2008;7:2280–2287. doi: 10.1158/1535-7163.MCT-07-2250. [DOI] [PubMed] [Google Scholar]
- 57.Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JH, Chute DJ, Riggs BL, Horvath S, Liau LM, Cavenee WK, Rao PN, Beroukhim R, Peck TC, Lee JC, Sellers WR, Stokoe D, Prados M, Cloughesy TF, Sawyers CL, Mischel PS. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012–2024. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 58.Haas-Kogan DA, Prados MD, Tihan T, Eberhard DA, Jelluma N, Arvold ND, Baumber R, Lamborn KR, Kapadia A, Malec M, Berger MS, Stokoe D. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst. 2005;97:880–887. doi: 10.1093/jnci/dji161. [DOI] [PubMed] [Google Scholar]
- 59.Prados MD, Chang SM, Butowski N, DeBoer R, Parvataneni R, Carliner H, Kabuubi P, Ayers-Ringler J, Rabbitt J, Page M, Fedoroff A, Sneed PK, Berger MS, McDermott MW, Parsa AT, Vandenberg S, James CD, Lamborn KR, Stokoe D, Haas-Kogan DA. Phase II study of erlotinib plus temozolomide during and after radiation therapy in patients with newly diagnosed glioblastoma multiforme or gliosarcoma. J Clin Oncol. 2009;27:579–584. doi: 10.1200/JCO.2008.18.9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Groot JF, Gilbert MR, Aldape K, Hess KR, Hanna TA, Ictech S, Groves MD, Conrad C, Colman H, Puduvalli VK, Levin V, Yung WK. Phase II study of carboplatin and erlotinib (Tarceva, OSI-774) in patients with recurrent glioblastoma. J Neurooncol. 2008;90:89–97. doi: 10.1007/s11060-008-9637-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brown PD, Krishnan S, Sarkaria JN, Wu W, Jaeckle KA, Uhm JH, Geoffroy FJ, Arusell R, Kitange G, Jenkins RB, Kugler JW, Morton RF, Rowland KM, Jr, Mischel P, Yong WH, Scheithauer BW, Schiff D, Giannini C, Buckner JC. Phase I/II trial of erlotinib and temozolomide with radiation therapy in the treatment of newly diagnosed glioblastoma multiforme: North Central Cancer Treatment Group study N0177. J Clin Oncol. 2008;26:5603–5609. doi: 10.1200/JCO.2008.18.0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Neshat MS, Mellinghoff IK, Tran C, Stiles B, Thomas G, Petersen R, Frost P, Gibbons JJ, Wu H, Sawyers CL. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci U S A. 2001;98:10314–10319. doi: 10.1073/pnas.171076798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang L, Clarke MJ, Carlson BL, Mladek AC, Schroeder MA, Decker P, Wu W, Kitange GJ, Grogan PT, Goble JM, Uhm J, Galanis E, Giannini C, Lane HA, James CD, Sarkaria JN. PTEN loss does not predict for response to RAD001 (Everolimus) in a glioblastoma orthotopic xenograft test panel. Clin Cancer Res. 2008;14:3993–4001. doi: 10.1158/1078-0432.CCR-07-4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krishnan S, Brown PD, Ballman KV, Fiveash JB, Uhm JH, Giannini C, Jaeckle KA, Geoffroy FJ, Nabors LB, Buckner JC. Phase I trial of erlotinib with radiation therapy in patients with glioblastoma multiforme: results of North Central Cancer Treatment Group protocol N0177. Int J Radiat Oncol Biol Phys. 2006;65:1192–1199. doi: 10.1016/j.ijrobp.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 65.Wen P, Kuhn J, Chang S, Lamborn K, Robbins H, Cloughesy T, Lieberman F, Mehta M, Gilbert M, Cooper J, Drappatz J, Kesari S, Norden AD, Groves M, Aldape K, Yung WKA, Dancey J, Prodos M. Phase I/II study of erlotinib and temsirolimus (CCI-779) for patients with recurrent malignant gliomas (NAB-TC 04-02). In: Yung WKA, editor. 13th Annual Meeting of the Society for Neuro-Oncology; Las Vegas, NV. 2008. p. 824. [Google Scholar]