

Abstract
Eosinophil migration in vivo is markedly attenuated in rats treated chronically with the NO synthase (NOS) inhibitor Nω-nitro-l-arginine methyl ester (l-NAME). In this study, we investigated the existence of a NOS system in eosinophils. Our results demonstrated that rat peritoneal eosinophils strongly express both type II (30.2 ± 11.6% of counted cells) and type III (24.7 ± 7.4% of counted cells) NOS, as detected by immunohistochemistry using affinity purified mouse mAbs. Eosinophil migration in vitro was evaluated by using 48-well microchemotaxis chambers and the chemotactic agents used were N-formyl-methionyl-leucyl-phenylalanine (fMLP, 5 × 10−8 M) and leukotriene B4 (LTB4, 10−8 M). l-NAME (but not d-NAME) significantly inhibited the eosinophil migration induced by both fMLP (54% reduction for 1.0 mM; P < 0.05) and LTB4 (61% reduction for 1.0 mM; P < 0.05). In addition, the type II NOS inhibitor 2-amino-5,6-dihydro-6-methyl-4H-1,3-thiazine and the type I/II NOS inhibitor 1-(2-trifluoromethylphenyl) imidazole also markedly (P < 0.05) attenuated fMLP- (52% and 38% reduction for 1.0 mM, respectively) and LTB4- (52% and 51% reduction for 1.0 mM, respectively) induced migration. The inhibition of eosinophil migration by l-NAME was mimicked by the soluble guanylate cyclase inhibitor 1H-[1,2,4] oxadiazolo [4,3,-a] quinoxalin-1-one (0.01 and 0.1 mM) and reversed by either sodium nitroprusside (0.1 mM) or dibutyryl cyclic GMP (1 mM). We conclude that eosinophils do express NO synthase(s) and that nitric oxide plays an essential role in eosinophil locomotion by acting through a cyclic GMP transduction mechanism.
Delayed eosinophil infiltration into sites of inflammation is usually observed by using a number of inflammatory agents and different animal species (1–6). Although the role of eosinophil accumulation in inflamed tissues is still unclear, these cells have been implicated in host defense mechanisms in parasitic infestation and in the pathogenesis of allergic, immunological, and malignant disorders (7). Eosinophils synthesize and release proinflammatory substances, including the preformed granular basic proteins (major basic protein, eosinophil cationic protein, eosinophil peroxidase, and protein X) and de novo synthesized products (eicosanoids, platelet-activating factor, oxygen metabolites, cytokines, and neuropeptides) (8–11).
NO release and synthesis by inflammatory cells such as macrophages (12), polymorphonuclear (13–16), and mononuclear (14, 16) cells is well documented, and the presence of inducible NO synthase has recently been shown in human eosinophils (17). In addition, the chronic treatment of rats with the NO synthase (NOS) inhibitor Nω-nitro-l-arginine methyl ester (l-NAME) inhibits eosinophil migration both in vivo and ex vivo in response to several stimuli, indicating that NO plays an essential role in eosinophil locomotion (18). Here we present immunohistochemical evidence for the existence of types II and III NOS in rat peritoneal eosinophils. In addition, by using both soluble guanylate cyclase modulators and a cyclic GMP analog, we demonstrate that this system is functionally coupled to the cyclic GMP transduction pathway, and plays a major role in eosinophil locomotion.
MATERIALS AND METHODS
Reagents.
l-NAME, Nω-nitro-d-arginine methyl ester (d-NAME), fMLP, LTB4, levamisole, dibutyryl cyclic GMP, chromotrope-2R, sodium nitroprusside, and metrizamide were obtained from Sigma. 2-Amino-5,6-dihydro-6-methyl-4H-1,3-thiazine (AMT), 1-(2-trifluoromethylphenyl) imidazole (TRIM), and 1H-[1,2,4] oxadiazolo [4,3,-a] quinoxalin-1-one were supplied by Tocris Cookson (St. Louis). NOS monoclonal and polyclonal antibodies were purchased from Transduction Laboratories (Lexington, KY). Biotinylated goat anti-mouse IgG and streptavidin-alkaline phosphatase were bought from Dako and GIBCO/BRL, respectively. Nitroblue tetrazolium, 5-bromo-4-chloro-3-indolyl-phosphate, and alkaline phosphatase-conjugated goat anti-rabbit IgG were provided by Promega. Eosin and Permount were supplied by Merck and Fisher, respectively.
Eosinophil Isolation.
Eosinophils were obtained from the peritoneal cavity of 10–15 male Wistar rats (200–230 g) and purified on a discontinuous metrizamide gradient (19). Briefly, the animals were killed with an overdose of ether anesthesia and the peritoneal cavities were washed with 20 ml of Hanks’ balanced salt solution (pH 7.2) containing heparin (20 units/ml). The peritoneal washings obtained from the animals were pooled and centrifuged at 1,000 × g for 18 min at 20°C in a Hermle model Z 360 K centrifuge (Germany). Metrizamide was dissolved in MEM (pH 7.2) containing 0.1% gelatin. The discontinuous gradient was prepared by carefully layering 2.5 ml of decreasing concentrations of metrizamide solutions (23.5, 20, and 18%, wt/vol) into a conical propylene tube. The resulting leukocyte-rich pellet was gently resuspended in 18% metrizamide (2.5 ml) and layered over the top of the metrizamide gradient. The gradient tube was first centrifuged at 90 × g (11 min at 20°C) and then at 1,000 × g (14 min at 20°C). The gradient zone containing the eosinophils (between the 23.5% and 20% gradients) was removed and washed twice in MEM. The final cell suspension contained 80–90% eosinophils. Cell viability (>90%) was assessed by the trypan blue dye exclusion test. Before testing, the eosinophil suspension was resuspended in MEM to give a final concentration of 5 × 106 cells/ml.
Chemotaxis Assay.
The eosinophil migration assay was performed by using a 48-well microchemotaxis chamber (20). Briefly, 50 μl of the eosinophil suspension (5 × 106 cells/ml) were added to the upper compartment of the microchemotaxis chamber (Neuroprobe, Cabin John, MD) and separated from the chemotactic agents in the lower compartment by a polycarbonate filter with polyvinylpyrrolidone (3 μm pore size; Nucleopore, Pleasanton, CA). MEM was substituted for the chemotactic agent to measure random migration. The loaded chambers were incubated for 2 h at 37°C in a 5% CO2 atmosphere and the filters were removed, fixed in methanol for 1–2 min, stained with Diff-Quick (Baxter Health Care, Mundelein, IL), and mounted on a glass slide. Chemotaxis was quantified by counting the eosinophils that migrated completely through the filter in five random high-power fields (HPF; ×1,000) per well. Chemotactic activity was expressed as the mean number of migrated cells per HPF. The chemotactic agents used in this study were N-formyl-methionyl-leucyl-phenylalanine (fMLP; 5 × 10−8 M, dissolved in MEM) and leukotriene B4 (LTB4, 10−8 M, dissolved in MEM).
Eosinophil Fixation.
Eosinophils (2 × 105 cells per slide) were collected on gelatin-chromalum subbed slides by using a Cytospin (Revan Instrumentos Científicos, São Paulo, Brazil). The cells were fixed with freshly prepared 4% (wt/vol) paraformaldehyde in 0.1 M sodium phosphate buffer, pH 7.4, for 15 min. The cells were washed with 0.05 M Tris⋅HCl buffer, pH 7.4, containing 0.15 M NaCl, dipped in water thrice, and dried. All operations were carried out at room temperature.
Anti-NOS Antibodies.
NOS II and III expression was detected by using affinity purified mouse mAbs anti-types II (clone 3) and III (clone 6) NOS, diluted 1:10 (vol/vol) and 1:100 (vol/vol), respectively, in blocking buffer [0.02 M sodium phosphate buffer, pH 7.4, containing 0.45 M sodium chloride, 0.2% (wt/vol) Triton X-100 (New England Nuclear), 5% (wt/vol) defatted dry milk, and 15% (vol/vol) normal goat serum]. Type I NOS was detected by using an affinity purified rabbit polyclonal antibody diluted 1:10 (vol/vol) in blocking buffer.
Immunostaining.
Eosinophils were hydrated with 10 mM sodium citrate buffer, pH 6.0, for 30 min. NOS-like immunoreactivity was retrieved by boiling the hydrated cells in 10 mM sodium citrate buffer, pH 6.0, in a microwave oven (Sharp, model RB 4A33) for 15 min. Microwave settings were adjusted to obtain maximum power. The slides were cooled for 20 min at room temperature, and the cells then rinsed and incubated sequentially in 0.1 M Tris⋅glycine, pH 7.4, and in blocking buffer, for 30 min each. Subsequently, the cells were incubated with anti-NOS antibodies diluted as described above. In control sections, primary antibodies were substituted with blocking buffer. After incubation, the cells were washed with 0.02 M sodium phosphate buffer, pH 7.4, containing 0.45 M sodium chloride, 0.2% (wt/vol) Triton X-100 (buffer A). Detection of type I NOS antibody was carried out by using an alkaline phosphatase-conjugated goat anti-rabbit IgG, diluted 1:500 (vol/vol) in blocking buffer. Detection of types II and III NOS antibodies was carried out by using a biotinylated goat anti-mouse IgG, followed by an incubation with streptavidin-alkaline phosphatase. After each immunochemical incubation step, the cells were washed with buffer A. The alkaline phosphatase reaction was developed using nitroblue tetrazolium and 5-bromo-4-chloro-3-indolyl-phosphate in the presence of 2 mM levamisole, for 30 min (21). The reaction was stopped with water. The cells were then counterstained with eosin or chromotrope-2R (22), dehydrated, cleared with xylene, and coverslipped with Permount. All operations were carried out at room temperature.
Statistical Analysis.
Data are presented as the mean ± SEM and were analyzed by ANOVA for multiple comparisons followed by Duncan’s test. A P value of <0.05 was considered to indicate significance.
RESULTS
Expression of Types II and III NOS Immunoreactive-Like Isoforms in the Rat Eosinophil Preparations.
Types II and III NOS immunoreactivity were strongly expressed and localized in cytoplasmic granules of purified rat peritoneal eosinophils (Fig. 1). Types II and III NOS immunoreactivity were detected in 30.2 ± 11.6% and 24.7 ± 7.41% (mean ± SD) of at least 1,700 counted cells, respectively. In contrast, type I NOS immunoreactivity was detected in very few cells (<0.5%).
Figure 1.
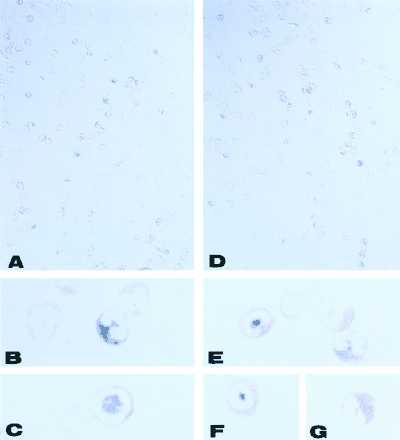
Immunolocalization of type II (A–C) and type III (D–G) NOS in cytoplasmic granules of purified rat peritoneal eosinophils. NOS was detected by using nitroblue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate (blue reaction product) and counterstained with eosin. Ring-like nuclear shape (A, C, D, and F) predominated over bilobular nuclei (A, B, D, E, and G) for both NOS isotypes. Nomarski micrographs. [Original magnifications: ×33 (A and D); ×333, (B, C, E–G).]
Involvement of NO in the Eosinophil Migration Induced by fMLP and LTB4 in vitro.
The chemotactic agent fMLP (5 × 10−8 M) caused significant eosinophil chemotaxis (7.6 ± 0.6 eosinophils/HPF) compared with random migration (3.1 ± 0.6 eosinophils/HPF; P < 0.05, n = 3). Fig. 2 shows that incubation of rat peritoneal eosinophils (30 min, 37°C) with the NO synthesis inhibitor l-NAME (0.1–1.0 mM; n = 3) dose-dependently inhibited the fMLP-induced chemotaxis whereas the inactive enantiomer d-NAME (0.1–1.0 mM; n = 3) had no significant effect on the chemotactic response.
Figure 2.
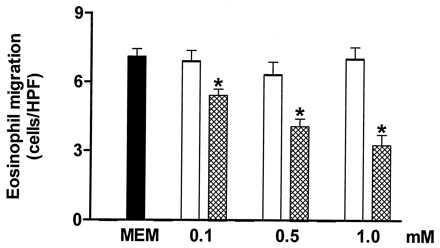
The inhibition by Nω-nitro-l-arginine methyl ester (l-NAME; cross-hatched columns) of in vitro rat eosinophil migration induced by fMLP (5 × 10−8 M) compared with either the control value (solid column) or the inactive enantiomer d-NAME (open columns). l-NAME (0.1–1.0 mM) or d-NAME (0.1–1.0 mM) was incubated with the rat eosinophil suspension for 30 min at 37°C. Each experiment was carried out in triplicate. Eosinophil migration is expressed as the mean number of migrated cells per HPF. The results are shown as the mean ± SEM (n = 3). ∗, P < 0.05 compared with either the control value or d-NAME.
The inhibition of fMLP-induced eosinophil chemotaxis by l-NAME (0.5 mM) was completely restored by coincubating the cells with either 0.1 mM sodium nitroprusside or 1 mM dibutyryl cyclic GMP (7.7 ± 0.4, 4.4 ± 0.3, 8.7 ± 0.9, and 8.2 ± 0.5 eosinophils/HPF for control, l-NAME, sodium nitroprusside + l-NAME, and dibutyryl cyclic GMP + l-NAME, respectively).
The selective soluble guanylate cyclase inhibitor 1H-[1,2,4]-oxidiazolo [4,3,-a] quinoxalin-1-one (0.01 and 0.1 mM; n = 3) significantly inhibited the fMLP-induced eosinophil chemotaxis (11.1 ± 0.7, 8.1 ± 0.5, and 2.8 ± 0.2 eosinophils/HPF for control, 1H-[1,2,4]-oxidiazolo [4,3,-a] quinoxalin-1-one 0.01 mM, and 0.1 mM, respectively; P < 0.05).
The compound AMT significantly reduced the fMLP-induced eosinophil chemotaxis in the concentration range of 0.1–1.0 mM (Fig. 3). At the same concentrations, TRIM also caused a significant inhibition of the fMLP-induced chemotaxis (Fig. 3).
Figure 3.
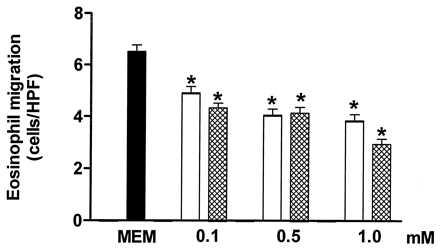
The inhibition by TRIM (open columns) and AMT (crosshatched columns) of in vitro rat eosinophil migration induced by fMLP (5 × 10−8 M). The control value (migration in absence of both TRIM and AMT) is shown by the solid bar. TRIM (0.1–1.0 mM) or AMT (0.1–1.0 mM) was incubated with the rat eosinophil suspension for 30 min at 37°C. Each experiment was carried out in triplicate. Eosinophil migration is expressed as the mean number of migrated cells per HPF. The results are shown as the mean ± SEM (n = 3). ∗, P < 0.05 compared with the control value.
l-NAME, AMT, and TRIM (0.1 and 1.0 mM each) also caused a significant inhibition of the LTB4 (10−8 M)-induced eosinophil chemotaxis (Fig. 4), whereas d-NAME (1.0 mM) had no significant effect (6.9 ± 0.3 and 6.0 ± 0.4 eosinophils/HPF in the absence and presence of d-NAME, respectively; n = 3).
Figure 4.
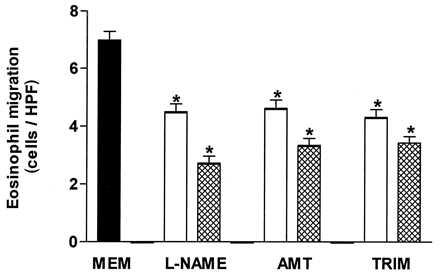
l-NAME, AMT, and TRIM significantly inhibit the in vitro rat eosinophil migration induced by LTB4 (10−8 M) compared with control values (MEM; solid bar). Each of these compounds was used at concentrations of 0.1 (open bars) and 1.0 (crosshatched bars) mM. Each experiment was carried out in triplicate. Eosinophil migration is expressed as the mean number of migrated cells per HPF. The results are shown as the mean ± SEM (n = 3). ∗, P < 0.05 compared with the control value.
DISCUSSION
Our results clearly demonstrate the existence of a functional NOS system in rat peritoneal eosinophils. This conclusion is based on the pharmacological results showing that NO synthesis blockade inhibited the in vitro fMLP- and LTB4-induced eosinophil chemotaxis and that the inhibition was reversed both by the NO-donor compound sodium nitroprusside and the cyclic GMP mimetic, dibutyryl cyclic GMP. The immunohistochemical results showing that eosinophils express immunoreactivity to NOS types II and III further reinforces the concept that eosinophils possess a functional NOS system.
The reported type I and type II NOS inhibitor TRIM (23) significantly decreased eosinophil migration and, in addition, type I NOS immunoreactivity was found in very few eosinophils. Although the latter observation would suggest the presence of type I NOS in some cells, it probably reflects some crossreactivity of the type I polyclonal antibody used with type II NOS.
Type II NOS expression is associated mainly with cell activation in pathological conditions (24), and it is possible that this expression may be because of the activation process that eosinophils undergo when migrating to the peritoneal cavity. The finding that the type II NOS inhibitor, AMT (25), significantly decreased eosinophil migration further supports the existence of the isoform in this cell type. Although the immunohistochemical data suggest the presence of two NOS isoforms in eosinophils, the possibility cannot be excluded that these cells express an alternative NOS isoform that reacts to both antibodies used here. Further purification of the eosinophil NOS(s) is desirable to fully characterize the isoform(s) involved: however, some difficulties are presented because we are dealing with a mixed cell population. This work is currently in progress.
Although NO has other actions independent of cGMP formation (26–28), the findings that the inhibition of eosinophil migration was mimicked by the soluble guanylate cyclase inhibitor 1H-[1,2,4]-oxidiazolo [4,3,-a] quinoxalin-1-one and that it was reversed by the cyclic GMP mimetic, dibutyryl cyclic GMP, indicate an essential role for this second messenger in eosinophil migration. This is further supported by evidence that unconventional NO donors such as azide and hydroxylamine inhibit apoptosis in isolated eosinophils, an effect mimicked by permeable cyclic GMP analogs (29).
Asthma is characterized by bronchial hyperreactivity and an increase in eosinophil migration (30). Use of inhaled corticosteroids has been associated with clinical improvement and a decrease in both of these parameters (31–32). Because glucocorticoids are known to inhibit type II NOS induction (33–34), our findings raise the exciting possibility that the clinical improvement experienced by patients with chronic treatment results from the inhibition of type II NOS expression in eosinophils. It will be important to evaluate whether type II NOS inhibitors are useful in the treatment of asthma.
Acknowledgments
We thank Dr. Lucia H. Faccioli for helpful discussions on eosinophil morphology and Mr. Hildeberto Caldo for skillful technical assistance, both from the Faculty of Medicine of Ribeirão Preto, University of São Paulo. This work was supported by grants from Fundaçao de Amparo à Pesquisa do Estado de São Paulo and Conselho Nacional de Pesquisas, Brazil.
ABBREVIATIONS
- l-NAME
Nω-nitro-l-arginine methyl ester
- d-NAME
Nω-nitro-d-arginine methyl ester
- HPF
high-power field
- AMT
2-amino-5,6-dihydro-6-methyl-4H-1,3-thiazine
- TRIM
1-(2-trifluoromethylphenyl) imidazole
- NOS
nitric oxide synthase
- fMLP
N-formyl-methionyl-leucyl-phenylalanine
- LTB4
leukotriene B4
References
- 1.Martins M A, Silva P M R, Faria-Neto H C, Bozza P, Dias P M F L, Lima M C R, Cordeiro R S B, Vargaftig B B. Br J Pharmacol. 1989;96:363–371. doi: 10.1111/j.1476-5381.1989.tb11826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanjar S, Aoki S, Kristersson A, Smith D, Morley J. Br J Pharmacol. 1990;99:679–686. doi: 10.1111/j.1476-5381.1990.tb12989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossi G A, Crimi E, Lantero S, Gianiorio P, Oddera S, Crimi P, Brusasco V. Am Rev Respir Dis. 1991;144:379–383. doi: 10.1164/ajrccm/144.2.379. [DOI] [PubMed] [Google Scholar]
- 4.Bozza P T, Castro-Faria-Neto H C, Martins M A, Larangeira A P, Perales J E, Silva P M R, Cordeiro R S B. Eur J Pharmacol. 1993;248:41–47. doi: 10.1016/0926-6917(93)90023-j. [DOI] [PubMed] [Google Scholar]
- 5.Teixeira M M, Williams T J, Hellewell P G. Br J Pharmacol. 1993;110:1515–1521. doi: 10.1111/j.1476-5381.1993.tb13994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fornhem C, Kumlin M, Lundberg J M, Alving K. Eur Respir J. 1995;8:1100–1109. doi: 10.1183/09031936.95.08071100. [DOI] [PubMed] [Google Scholar]
- 7.Kroegel C, Virchow J-C, Jr, Luttmann W, Walker C, Warner J A. Eur Respir J. 1994;7:519–543. doi: 10.1183/09031936.94.07030519. [DOI] [PubMed] [Google Scholar]
- 8.Gleich G J, Loegering D A, Bell M P, Checkel J L, Ackerman S J, McKean D J. Proc Natl Acad Sci USA. 1986;83:3146–3150. doi: 10.1073/pnas.83.10.3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wasmoen T L, Bell M P, Loegering D A, Gleich G J, Prendergast F G, McKean D J. J Biol Chem. 1988;263:12559–12563. [PubMed] [Google Scholar]
- 10.Weller P F, Ackerman S J, Smith J A. J Leukocyte Biol. 1988;43:1–4. doi: 10.1002/jlb.43.1.1. [DOI] [PubMed] [Google Scholar]
- 11.Weller P F. N Engl J Med. 1991;324:1110–1118. doi: 10.1056/NEJM199104183241607. [DOI] [PubMed] [Google Scholar]
- 12.Marletta M A, Yoon P S, Leaf C D, Wishnok J S. Biochemistry. 1988;27:8706–8711. doi: 10.1021/bi00424a003. [DOI] [PubMed] [Google Scholar]
- 13.Rimele T J, Sturm R J, Adams L M. J Pharmacol Exp Ther. 1988;245:102–111. [PubMed] [Google Scholar]
- 14.Salvemini D, de Nucci G, Gryglewski R, Vane J R. Proc Natl Acad Sci USA. 1989;86:6328–6332. doi: 10.1073/pnas.86.16.6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt H W, Seifert R, Bohme E. FEBS Lett. 1989;244:357–360. doi: 10.1016/0014-5793(89)80562-9. [DOI] [PubMed] [Google Scholar]
- 16.Condino-Neto A, Muscará M N, Grumach A, Carneiro-Sampaio M S, de Nucci G. Br J Clin Pharmacol. 1993;35:485–490. doi: 10.1111/j.1365-2125.1993.tb04174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Del Pozo V, Arruda-Chaves E, Andrés B, Cárdaba B, López-Farré A, Gallardo S, Cortegano I, Vidarte L, Jurado A, Sastre J, Palomino P, Lahoz C. J Immunol. 1997;158:859–864. [PubMed] [Google Scholar]
- 18.Ferreira H H A, Medeiros M V, Lima C S P, Flores C A, Sannomiya P, Antunes E, de Nucci G. Eur J Pharmacol. 1996;310:201–207. doi: 10.1016/0014-2999(96)00379-2. [DOI] [PubMed] [Google Scholar]
- 19.Vadas M A, David R J, Butterworth A, Pisani N T, Siongok T A. J Immunol. 1979;122:1228–1236. [PubMed] [Google Scholar]
- 20.Richards K L, McCullough J M. Immunol Commun. 1984;13:49–62. doi: 10.3109/08820138409025449. [DOI] [PubMed] [Google Scholar]
- 21.Harrow E, Lane D, editors. Antibodies: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. [Google Scholar]
- 22.Kay A B. Clin Exp Immunol. 1970;6:75–86. [PMC free article] [PubMed] [Google Scholar]
- 23.Handy R L C, Wallace P, Gaffen Z A, Whitehead K J, Moore P K. Br J Pharmacol. 1995;116:2349–2350. doi: 10.1111/j.1476-5381.1995.tb15078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Southan G J, Szabó C. Biochem Pharmacol. 1996;51:383–394. doi: 10.1016/0006-2952(95)02099-3. [DOI] [PubMed] [Google Scholar]
- 25.Nakane M, Klinghofer V, Kuk J E, Donnelly J L, Budzik G P, Pollock J S, Basha F, Carter G W. Mol Pharmacol. 1995;47:831–834. [PubMed] [Google Scholar]
- 26.Brune B, Lapetina E G. J Biol Chem. 1989;264:8455–8458. [PubMed] [Google Scholar]
- 27.Bolotina V M, Najibi S, Palacino J J, Pagano P J, Cohen R A. Nature (London) 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- 28.Clancy R M, Leszczynska-Piziak J, Amin A, Levertovsky D J, Abramson S B. J Leukocyte Biol. 1994;58:196–202. doi: 10.1002/jlb.58.2.196. [DOI] [PubMed] [Google Scholar]
- 29.Beauvais F, Michel L, Dubertret L. FEBS Lett. 1995;361:229–232. doi: 10.1016/0014-5793(95)00188-f. [DOI] [PubMed] [Google Scholar]
- 30.Bousquet J, Chanez P, Lacoste J Y, Barneon G, Chavanian N, Enander I, Venge P, Ahistedt S, Simony-Lafontaine J, Godard P, Michel F B. N Engl J Med. 1990;323:1033–1039. doi: 10.1056/NEJM199010113231505. [DOI] [PubMed] [Google Scholar]
- 31.Schleimer R P. Annu Rev Respir Dis. 1990;141:S59–S69. [PubMed] [Google Scholar]
- 32.Barnes P J. N Engl J Med. 1995;332:868–875. doi: 10.1056/NEJM199503303321307. [DOI] [PubMed] [Google Scholar]
- 33.Radomski M W, Palmer R M J, Moncada S. Proc Natl Acad Sci USA. 1990;87:10043–10047. doi: 10.1073/pnas.87.24.10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Knowles R G, Salter M, Brooks S L, Moncada S. Biochem Biophys Res Commun. 1990;172:1042–1048. doi: 10.1016/0006-291x(90)91551-3. [DOI] [PubMed] [Google Scholar]