

Abstract
A central issue in cognitive neuroscience of aging research is pinpointing precise neural mechanisms that determine cognitive outcome in late adulthood as well as identifying early markers of less successful cognitive aging. One promising biomarker is beta amyloid (Aβ) deposition. Several new radiotracers have been developed that bind to fibrillar Aβ providing sensitive estimates of amyloid deposition in various brain regions. Aβ imaging has been primarily used to study patients with Alzheimer’s Disease (AD) and individuals with Mild Cognitive Impairment (MCI); however, there is now building data on Aβ deposition in healthy controls that suggest at least 20% and perhaps as much as a third of healthy older adults show significant deposition. Considerable evidence suggests amyloid deposition precedes declines in cognition and may be the initiator in a cascade of events that indirectly leads to age-related cognitive decline. We review studies of Aβ deposition imaging in AD, MCI, and normal adults, its cognitive consequences, and the role of genetic risk and cognitive reserve.
Keywords: Aging, Beta-amyloid, Brain, Cognitive reserve, fMRI, PET
Normal aging is associated with measurable declines in neural and cognitive systems. There is a wealth of literature documenting age-related declines in many aspects of cognitive behavior, including decreased speed of information processing, working memory capacity, and long-term memory function. At the same time, knowledge structures are relatively preserved with age (Ghisletta and Lindenberger 2003; Hedden and Gabrieli 2004; Park et al. 2002). Recently, significant advances have been made in our understanding of the structure and function of the aging brain utilizing new neuroimaging technologies (see Park and Reuter-Lorenz 2009; Park and Goh 2009; Raz and Kennedy 2009; Raz and Rodrigue 2006 for reviews). At present, a central issue in cognitive neuroscience of aging research is pinpointing the precise neural mechanisms that determine cognitive outcome in late adulthood as well as identifying markers of less successful cognitive aging as early in life as possible. Predicting successful vs. pathological aging trajectories requires early identification of sensitive behavioral or neural markers prior to actual neural pathology and cognitive decline. One such promising biomarker that has received recent attention is beta amyloid (Aβ), a protein that is deposited on the brain in some individuals as they age.
Significant amyloid deposition is a characteristic feature of all patients with Alzheimer’s disease (AD). However, it is also present in many normal adults; it is observed in individuals with Mild Cognitive Impairment (MCI) at a level higher than normal older adults; and it is a strong predictive factor in conversion to AD. At the same time that amyloid is associated with neural pathology, it is also surprisingly common to find that neuropsychologically normal, healthy older adults show significant neuropathology at autopsy in the form of amyloid deposition (Dickson et al. 1992). New imaging tracer ligands offer possibilities for measuring Aβ burden in the brain and for studying the time course of its progression in nondemented individuals as well as in AD. These radiotracer compounds bind to amyloid deposits in the living brain when injected into the bloodstream. When the injection is accompanied by Positron Emission Tomography (PET) scanning, a detailed assessment can be made of the amount of amyloid and its distribution throughout the brain. The purpose of the current review is to briefly explain the history of the study of beta-amyloid, review the amyloid imaging studies of normal aging and dementia, provide an integrative framework for how beta-amyloid may play a role in normal aging, and to offer some future directions for fruitful avenues for the study of healthy aging using this promising imaging technique.
Beta-Amyloid Measurement
The Beta-Amyloid Protein
One common, but not universally accepted view of the pathogenesis of Alzheimer’s Disease is the beta-amyloid hypothesis. Beta-amyloid is a protein fragment that is deposited in the brain in the form of sticky, starch-like plaques, in an increased manner in individuals with AD. While the exact pathogenesis of AD remains unknown and the role of Aβ in the brain is not entirely clear, one viewpoint is that the soluble form may cause synaptic dysfunction (Selkoe 2002; Nordberg 2008) as the amount of extracellular soluble Aβ in the brain is a better predictor of cognitive impairment in AD than the amount of plaques themselves (Nordberg 2008). The beta-amyloid protein fragments are snipped from amyloid precursor proteins (APP). One common view is that in an optimally-functioning brain, these protein fragments are broken down and eliminated, but in AD the problem is that the fragments are not broken down and accumulate in the brain to form plaques. Another possibility is that the plaques result from an overproduction of Aβ or APP, rather than a failure to clear these products, and this initiates the cytotoxic effects. Figure 1 illustrates varying levels of beta-amyloid plaques in postmortem samples ranging from sparse to moderate to frequent amyloid deposits (from Josephs et al. 2008).
Fig. 1.

Illustration of beta-amyloid plaques in fixed tissue at autopsy (from Josephs et al. 2008). a shows an example of sparse Aβ burden; b illustrates a brain with moderate Aβ; and c is an example of frequent Aβ deposition in the brain
Until the development of the amyloid-sensitive ligands, there have been a variety of other techniques used to measure amyloid plaque accumulation, including methods that indirectly estimated levels of brain amyloid plaques from Aβ levels in plasma or cerebral spinal fluid (CSF). The significance of amyloid deposits for disease specificity is uncertain, as deposits are often found in the cortex of non-demented older adults at autopsy, although these estimates are influenced by the age of the cohort sampled and the method of defining disease pathology (Bennett et al. 2006; Braak and Braak 1996; Thal et al. 2006). Postmortem studies, previously the only method of examining Aβ, have found that 25–30% of individuals with no clinical symptoms of dementia have levels of Aβ equal to the diagnostic level for AD (Katzman et al. 1988).
Beta-amyloid levels can also be measured in CSF and interestingly, higher levels of beta-amyloid deposits in the brain are correlated with lower levels of beta-amyloid in the CSF (Grimmer et al. 2009; Strozyk et al. 2003). This finding supports the notion that as Aβ plaques become less soluble and clearance of beta-amyloid declines, an aggregation of plaque is formed in the brain and less is broken down and observed in the circulating CSF. Further support for this process of Aβ deposition and clearance comes from studies that indicate CSF amyloid beta levels correlate positively with cognitive performance (Nordlund et al. 2008) and negatively with AD symptom severity (Samuels et al. 1999), predict conversion from MCI to AD (Andreasen et al. 1999; Diniz et al. 2008) and correlate positively with brain volume and negatively with ventricular size in persons with AD, suggesting that CSF Aβ levels drop as AD progresses (Tapiola et al. 2000; Wahlund and Blennow 2003).
PET Imaging of Beta Amyloid
In addition to measuring Aβ levels in the CSF, more recently beta amyloid deposits have become measureable using PET and radiotracer ligands that bind to the aggregated fibrillar form of Aβ. Radiotracers with a high affinity for amyloid (in extracellular Aβ plaques) have been developed recently for use in humans (Ichise et al. 2008; Klunk et al. 2004; Shoghi-Jadid et al. 2002). For example, Fig. 2 illustrates the binding pattern of a radiotracer in a patient with Alzheimer’s Disease and in a healthy control subject. Higher uptake rates, as evidenced by the warmer red and yellow color scale, can be seen in the patient compared to the normal older adult. These tracers are labeled with either carbon-11 or fluorine-18. The most important difference between these labeling isotopes is their half-lives. The half-life of an isotope refers to the amount of time that it takes for half of the atoms in the radioactive substance to decay. The half-life then depends on the rate of decay, which is exponential (see Fig. 3). Carbon-11 has a half-life of approximately 20 min, therefore every 20 min there is 50% less tracer available. Fluorine-18 has a half-life of approximately 110 min, allowing a longer time window before radioactive decay of the tracer.
Fig. 2.
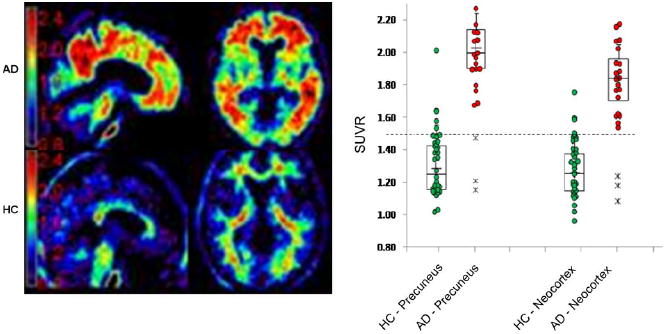
18F AV-45 uptake in an AD subject (left panel top) and a Healthy Control (left panel bottom). The right panel displays normalized signal uptake value (SUVr) in patients with AD and healthy controls in the precuneus and neocortex
Fig. 3.
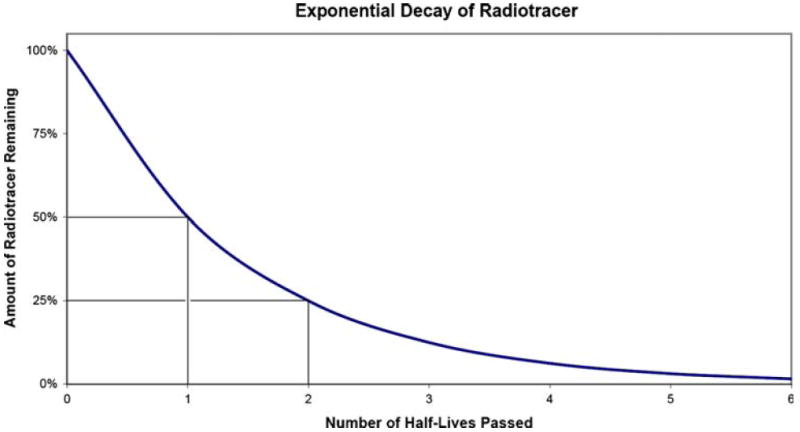
An illustration of the timecourse of the rate of the decay of a radiotracer. As each half-life is passed the radioactive material is reduced by 50% and follows an exponential function
The three most common ligands in use to image Aβ deposition with PET are the 11C-labeled PET tracer 6-OH-BTA also known as Pittsburgh compound B or PIB (Klunk et al. 2004), the 18F-labeled tracer FDDNP (Shoghi-Jadid et al. 2002), and 18Florpiramine, also known as 18F-AV-45 (Ichise et al. 2008; Zhang et al. 2007). While none of these compounds are FDA approved for clinical use, these amyloid imaging agents have been received with great interest in the research community and are in use in a number of clinical trials. The FDA has already approved the use of all three compounds as biomarkers to test the mechanisms of several putative amyloid-lowering drugs (Skovronsky 2008). All three ligands show higher amyloid uptake in the cortex of patients with AD compared to the cortex of healthy controls, reflecting the elevated accumulation of Aβ pathology and consequent binding of amyloid in the cortex of patients with AD (Lopresti et al. 2005; Skovronsky et al. 2008). Further, all three ligands demonstrate good test-retest reliability, especially when using the optimal method of correcting for cerebellar reference tissue uptake (Tolboom et al. 2009b).
The first ligand developed to image amyloid using carbon-11 was 11C-PIB (Klunk et al. 2004) and it has since been the most widely studied. Because this compound relies on the production of carbon-11 labeled tracers, which has only a 20-minute half life, the use of PIB requires an on-site cyclotron, available at less than 10% of PET scanner sites (Klunk and Mathis 2008). Therefore the availability of this compound for research use is limited to only those sites with a cyclotron. In contrast to PIB, fluorine-18 is used to label FDDNP and AV-45. Because 18F has a radioactive half-life of 110 min, it is quite feasible for regional preparation of the compound to occur with shipping of doses to research sites up to 4 h away (i.e., 22% of the radiotracer would remain and doses are adjusted accordingly). For this reason, more sites have begun using 18F labeled agents. Thus far, however, the vast majority of published studies have used PIB (Aizenstein et al. 2008; Buckner et al. 2005; Jack et al. 2008).
Studies conducted to date suggest that 18F-AV-45 labels amyloid plaques in a manner similar to PIB. Like PIB, AV-45 exhibits high affinity specific binding to amyloid plaques. In-vitro autoradiography studies further confirm that when applied at tracer concentrations AV-45 labels Aβ amyloid plaques in sections from patients with pathologically confirmed AD (Skovronsky et al. 2008; Wong et al. 2008; Ichise et al. 2008). The non-radioactive version of AV-45 can be prepared at high concentrations and shows very low to no affinity for all other central nervous system and cardiovascular receptors tested. Similarly, 18F-FDDNP also successfully labels amyloid plaques (Small et al. 2006), but may do so in a different manner than PIB (Tolboom et al. 2009a). For instance, in a study that imaged normal elderly, individuals with MCI, and AD patients, FDDNP had a higher binding level in MCI than did PIB, and thus, did not discriminate between MCI and AD groups as well as PIB. The regional uptake pattern differed between the two ligands as well, with almost no medial temporal lobe (MTL) binding with PIB, but a high affinity for MTL was found with FDDNP (Tolboom et al. 2009a), likely due to the binding of FDDNP to tau in the abundant neurofibrillary tangles found in the hippocampus in persons with AD (Schmidt et al. 1990). Although global brain uptake is moderately correlated (r=.45) between FDDNP and PIB, these two ligands likely capture related but different characteristics of AD. Some of this discrepancy may be explained by the fact that FDDNP binds to both Aβ and tau proteins as well as to prion proteins found in Aβ plaques in AD (Jagust 2009a).
Imaging Studies on Beta-Amyloid Deposition
Before reviewing the literature on studies of normal aging, it is helpful as a comparison to briefly summarize the findings of Aβ deposition in persons with Alzheimer’s Disease and Mild Cognitive Impairment.
Amyloid Deposition in AD and MCI
There is strong evidence that amyloid deposition (as measured by PIB, FDDNP, or AV-45) is found at a high level in essentially all patients with a clinical diagnosis of Alzheimer’s disease. Indeed, dementia in the absence of a high level of amyloid at autopsy would lead to exclusion of Alzheimer’s as a diagnosis and consideration of frontal-temporal dementia or some other cause for the observed cognitive symptoms. All studies using PIB, FDDNP, or AV-45 find evidence for greater amyloid deposition in AD patients (see Fig. 2 for an illustration; Buckner et al. 2005; Jack et al. 2008; Linazasoro 2008; Kemppainen et al. 2006; Rowe et al. 2008; Skovronsky et al. 2008; Small et al. 2006; Wong et al. 2008). Generally, in individuals with AD, Aβ binding is widely distributed across the cortex. Buckner et al. (2005) reported PIB binding prominently in the frontal regions along the midline and extending laterally, as well as medial and lateral posterior parietal cortex and precuneus extending into posterior cingulate and retrosplenial cortex. Braskie et al. (2008) reported FDDNP binding in frontal and parietal cortices, but also in lateral temporal regions. Similarly, Edison et al. (2007) found increased PIB uptake in AD relative to controls in frontal, cingulate, parietal, temporal and occipital regions, indicating a widespread distribution of Aβ in this population. The most consistent and uniform finding across AD studies is that early in the disease there is binding to the precuneus/posterior cingulate cortex (Jagust 2009b).
The other group that has been commonly imaged is individuals with mild cognitive impairment. MCI is a somewhat arbitrary category used to describe a population of individuals who do not display cognitive impairment severe enough for diagnosis of a neurodegenerative disorder, but who do display a mild level of cognitive impairment that normal older adults do not evidence (Petersen et al. 1999). It is typical to find a bivariate distribution for amyloid in the MCI population, with approximately half of the subjects evidencing a high level of amyloid that looks like the AD patients and half showing lower Aβ levels more similar to the healthy controls (Jack et al. 2008; Kemppainen et al. 2007; Li et al. 2008; Rowe et al. 2007).1 Level of Aβ binding in the MCI subjects with high levels is a strong predictor for later conversion to AD (Forsberg et al. 2008; Jack et al. 2008; Mormino et al. 2009; Pike et al. 2007). Small et al. (2009) recently reported that MCI subjects had greater FDDNP binding in the frontal, temporal, parietal, and posterior cingulate cortex than normal controls. Similarly, using principal component analysis, Fripp et al. (2008) found patterns of variation in Aβ deposition in MCI (low and high uptake) that distinguished between the normal controls and AD subjects. Forsberg et al. (2008) reported that the seven MCI converters evidenced greater PIB uptake than the six controls in the frontal, parietal, and temporal cortices, suggesting that a specific regional marker of normal-to-MCI has yet to be specified, although larger sample sizes may be more informative. Figure 4 (adapted from Pike et al. 2007) displays typical Aβ binding across different groups and stages of pathology. It can be seen in this figure that from normal aging to pathology there is an increase in Aβ deposition, where blue and purple regions represent little Aβ deposition and yellow and red areas evidence the highest levels of deposition.
Fig. 4.
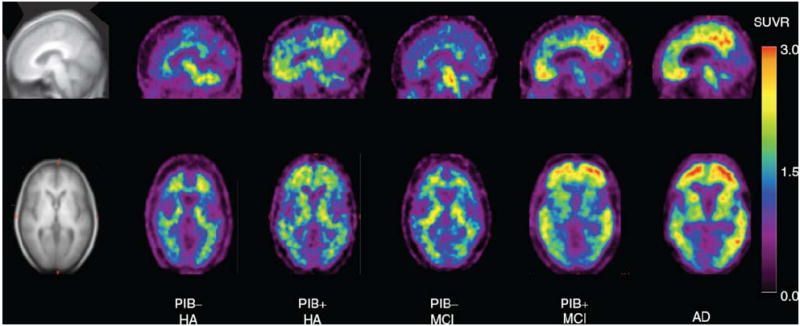
Illustration of typical Aβ deposition across varying groups and stages of pathology. Examples are from left to right, images of a PIB negative healthy older adult, a PIB positive healthy older adult, a PIB negative MCI subject, a PIB positive MCI subject, and an AD patient. Blues and purples represent little Aβ deposition and yellow and red the highest deposition (from Pike et al. 2007)
Genetics of Amyloid Deposition Risk
There is a large literature on the risk conveyed by carrying the APOE-ε4 allele for age-related decline in cognitive function in both normal adults and AD patients and this is the best characterized genetic polymorphism associated with AD (Corder et al. 1993; Farrer et al. 1997). Homozygosity for APOE-ε4 occurs in 1–2% of the population and confers a serious risk of AD, but even ε3/4 heterozygosity, which occurs in 25% of the population, results in an increased risk. In fact, inheritance of a single APOEε4 allele is associated with temporal and frontal brain atrophy in cognitively intact adults (Wishart et al. 2006). Further, APOE-ε4 homozygotes are at 10 to 30 times the risk of developing AD by the age of 75 compared to those with no ε4 alleles. Although the mechanism for this increased genetic risk is not clear, it is thought that it is due to an interaction with Aβ (Jiang et al. 2008). It has been reported that 80% of amyloid positive adults with MCI are APOE-ε4 carriers (Pike et al. 2007) and 40% of persons with MCI are homozygous for APOE-ε4 (Farlow et al. 2004). At liberal estimates, half of MCI individuals eventually convert to AD in 5 years (Grundman et al. 2002). Recent studies indicate that the APOE-ε4 genotype is a strong risk factor for amyloid deposition even in non-demented elderly individuals (Morris et al. 2009). Importantly, APOE-ε4 carriers have been found to have higher Aβ binding than the non-carriers (Rowe et al. 2008). Drzezga et al. (2009) found significantly increased amyloid deposition in temporoparietal and frontal cortex in AD patients who were APOE-ε4 carriers than AD patients who were non-carriers, suggesting that there is a genetic exacerbation even within Alzheimer’s disease. Small et al. (2009) recently compared MCI and normal adults’ FDDNP binding with respect to their APOE-ε4 genotype. Differential patterns of Aβ deposition were found among the groups, where MCI subjects who were ε4 carriers evidenced greater FDDNP binding in the medial temporal cortex, whereas normal elderly ε4 carriers displayed greater binding in the frontal cortex. Recently, Reiman et al. (2009) reported a gene dose-response effect for APOE-ε4 alleles, where higher Aβ burden was associated most highly with individuals possessing two copies of the ε4 allele, followed by those with one copy, compared to non-carriers in frontal, precuneus/posterior cingulate, temporal and parietal regions. However, there are older adults with high levels of Aβ who do not carry the APOE-ε4 genotype, indicating that this is a risk factor, but certainly not a determinant for Aβ deposition. Figure 5 illustrates the association between APOE genotype and PIB binding in the brain in cognitively normal older adults (from Reiman et al. 2009). There is only small to moderate PIB binding in the ε4 heterozygotes compared to non-carriers, whereas there is a greater increase in PIB binding in ε4 homozygotes compared to non-carriers, and a dose-related association of greater PIB binding with increasing ε4 alleles.
Fig. 5.
Illustration of association between APOE genotype and Aβ deposition in the brain in cognitively normal older adults (from Reiman et al. 2009). a displays a small to moderate increase in PIB binding in the individuals heterozygous for ε4 compared with non-carriers; b illustrates the greater increased PIB binding in individuals homozygous for ε4 compared with non-carriers; c depicts the association of greater PIB binding and APOE- ε4 dose for the whole sample
Beta-Amyloid and Normal Aging
Less is known about the role of Aβ in normal aging compared to the literature examining memory impairment or dementia. However, a few studies have measured amyloid deposition with respect to normal aging and the findings indicate the presence of deposits in frontal, cingulate, and parietal areas, with primary sensory/visual areas relatively protected from amyloid deposition. Studies with normal aging adult samples consistently report that approximately 20 to 30% of healthy older controls show significant amyloid deposition. This individual variability is illustrated in Fig. 6, which depicts a normal older adult with very high Aβ deposition and an adult with low deposition, both of whom are cognitively normal (Jack et al. 2008).
Fig. 6.

Illustration of normal variation in magnitude and extent of Aβ deposition in healthy controls (from Jack et al. 2008). a is the scan from the subject with the highest PIB retention in the study (despite being a normal control and cognitively normal) and b is the scan from a healthy control subject with low PIB retention, who was cognitively normal, but scored lower than the subject depicted in panel (a)
Of 20 healthy older controls (66–86 years old), four individuals (20%) evidenced PIB uptake equal to that of the AD subjects of similar age (Mintun et al. 2006). This increased uptake was primarily seen in the precuneus, a region that displays Aβ at an early stage in AD patients. Buckner et al. (2005) reported 2 of 8 healthy controls (25%) were PIB positive. Fotenos et al. (2008) reported that of 58 normal controls (aged 47–86), 9 individuals (16%) were classified as PIB positive, and these individuals evidenced smaller whole brain volumes than the PIB negative subjects. Nelissen et al (2007) found that 3 of 16 healthy controls (19%) had PIB uptake at the level of their AD patients. Of 34 elderly participants from the Melbourne Healthy Aging Study (Villemagne et al. 2008), 10 subjects were classified as “clinically declining” over time and these subjects showed a greater instance of Aβ binding than did the non-decliners (70% vs. 17%). In this study, Aβ burden was correlated with memory performance in the declining, but not the non-declining group.
The findings with respect to the relationship between amyloid deposition and measures of cognitive decline are somewhat mixed when amyloid is detected in a healthy older adult. For example, Rowe et al. (2007) reported that 6 (22%) of 27 older adult subjects were amyloid positive, but cognitive symptoms in this group were absent. Jack et al. (2008) tested 20 normal subjects and reported that 6 subjects (30%) were amyloid positive. No association between Aβ positivity and cognitive function was found, although some evidence for mediation of cognitive symptoms by hippocampal volume was reported (Jack et al. 2008). Thus, Jack et al. suggested that amyloid deposition is an early symptom of Alzheimer’s disease that is followed by shrinkage of the hippocampus.
In contrast to the aforementioned studies, Pike et al. (2007) reported in an early paper a significant link between amyloid deposition and cognitive function in apparently healthy elderly. Greater amyloid deposition was negatively related to episodic memory performance in 22% of 32 healthy controls who were amyloid positive. However, this group recently reported that in a larger sample (n=89), this association between PIB positivity and memory was nonsignificant in the normal control group (Bourgeat et al. 2009). Mormino et al. (2009) tested 37 normal controls and rather than simply characterizing subjects as amyloid positive or negative, they utilized amyloid deposition as a continuous variable. Increased amyloid deposition was related to both decreased hippocampal volume and poorer episodic memory. They suggested, in accord with Jack et al. (2008) that the critical event in determining cognitive health in late adulthood is amyloid deposition which results in a cascade of events—with amyloid initiating hippocampal shrinkage, followed by episodic memory decline and ultimately, over time, a diagnosis of Alzheimer’s Disease. Although, given the crucial role of the entorhinal cortex in the pathogenesis of AD, it is important for future studies to measure the volume of this structure. Interestingly, even though hippocampal volume measures from MRI are associated with AD and Aβ deposition, a recent study showed that overall brain atrophy measured at postmortem did not correlate with amount of Aβ plaques in the brain (Josephs et al. 2008), suggesting that rate of overall volume loss is not significantly influenced by amount of Aβ plaques. The effect may be specific to localized brain regions, such as the hippocampus or entorhinal cortex. For example, in a preliminary study Dickerson et al. (2009) compared cortical thickness of 9 PIB positive nondemented older adults to 35 PIB negative nondemented older adults and found thinner temporal pole cortex in the PIB positive adults and a nonsignificant trend for thinner superior frontal cortex. When pooling all regions to form an average thinning index, this did not differ significantly between the PIB positive and negative groups (p=.09). In a longitudinal study, Scheinin et al. (2009) report that PIB uptake at baseline predicted longitudinal change in hippocampus, precuneus, and temporal cortex volume in the group of normal control subjects (n=13) but not in the AD group (p=.07; n=14). Obviously, far larger samples are necessary to further test these relations.
There is some debate as to whether Aβ deposition proceeds in a linear or an accelerated fashion (Ingelsson et al. 2004; Jack et al. 2009). It seems clear that amyloid burden accumulates well before clinical symptoms. Then this deposition either plateaus with little to no further accumulation over time, or continues to slowly accumulate throughout the duration of the lifespan (Jack et al. 2009). Jack et al. (2009) found no difference in longitudinal change in Aβ progression among AD, MCI, and normal control groups, suggesting a linear decline, that when extrapolating back, would suggest that Aβ accumulation would have to begin in the 40s. Ingelsson et al. (2004) posits that Aβ deposits early and rapidly, and then slows with age. A theoretical model of Aβ deposition over the time course of normal or asymptomatic aging, MCI and AD is illustrated in Fig. 7 (from Jack et al. 2009).
Fig. 7.

Proposed model relating imaging, pathology and clinical presentation over an individual’s adult lifetime (from Jack et al. 2009). The lifetime course of progression from presymptomatic, prodromal (MCI), and dementia (AD) phases is plotted. Neurodegeneration, detected by MRI, is indicated by a dashed line. Cognitive function is indicated by a dot-dash line. Amyloid deposition is indicated by a solid line late in life (i.e. that portion of the disease for which data currently exist). The time course of amyloid deposition early in life is represented as two possible theoretical trajectories (dotted lines), reflecting uncertainty about the time course trajectory of early Aβ deposition
One of the earliest studies designed to investigate normal elderly was conducted by Aizenstein et al. (2008) on 43 normal elderly (aged 65–88). They reported that 21% of these subjects had significant amyloid and that amyloid positive subjects performed similarly on a cognitive battery to amyloid negative subjects. One theoretical explanation for these puzzling results is the notion of cognitive reserve (Stern 2002). The cognitive reserve hypothesis posits that some individuals are better equipped to cope with the physiological challenges to neural structure and function that occur with aging and/or disease processes, and is generally attributed to higher levels of education, socioeconomic status and intelligence in those individuals (Stern 2002). Thus, the negative cognitive consequences that are typically associated with declining structural integrity and reduced efficiency of neural function with aging are attenuated, delayed or masked in those with greater cognitive reserve capacity (Stern 2009). In accord with this hypothesis, the Aizenstein et al. (2008) study examined whether the amyloid positive subjects would have higher premorbid intelligence and greater levels of education that protected them from expressing negative cognitive symptoms. Contrary to predictions from the cognitive reserve hypothesis, the authors found the opposite result—that the amyloid negative individuals had higher levels of premorbid intelligence than the amyloid positive group, and the amyloid negative group (with higher premorbid intelligence and no amyloid) actually had lower episodic memory scores than the amyloid positive group. Other studies, however, are in agreement with the cognitive reserve hypothesis. For example, Kemppainen et al. (2008) compared AD patients with high (n=12) and low (n= 13) levels of education and found that the high education group had greater Aβ binding in the lateral frontal cortex than the lower education group, as well as lower glucose metabolism in the temporal-parietal cortex, demonstrating that the more highly educated group harbored greater levels of brain pathology at the same degree of cognitive decline as lower educated AD patients. For non-demented elderly, greater levels of education/socioeconomic status has been associated with smaller whole brain volumes and accelerated volume loss over time, suggesting that better educated, more privileged older adults may harbor premorbid dementia, or can hide the signs of brain changes longer than less privileged older adults (Fotenos et al. 2008). Other research has shown that in a group of MCI subjects, those who converted to AD had lower education. Moreover, the converters with the highest education had more severe amyloid pathology (as measured in CSF) but performed equally well on neuropsychological tests compared to MCI converters with lower education (Rolstad et al. 2009). This finding again suggests that greater brain pathology was associated with cognitively “privileged” older adults, in line with the cognitive reserve hypothesis.
To summarize this complex literature, early evidence tentatively suggests that greater reserve may stave off the cognitive expression of neural decline (i.e., amyloid burden), but the mechanisms though which this occurs are unknown. It is plausible that the apparent protection conferred by cognitive reserve is simply a masking effect where individuals with more developed cognitive abilities have a delayed behavioral expression of pathology. Additionally, a potential alternative explanation that has yet to be ruled out is that people with higher education perform better on neuropsychological tests at all stages (before, during and after they are diagnosed with dementia). Because dementia is diagnosed using neuropsychological tests, a diagnosis would come later to these higher performing, better educated individuals. Further, similar neuropsychological tests are used to both diagnosis dementia and to quantify cognitive reserve, possibly introducing a confound among diagnostic group, cognitive reserve score, and test performance. Finally, it is also possible that beta-amyloid plaques detected with PIB are not actually neurotoxic and are entirely independent (at a given timepoint) of cognitive preservation or decline, or are too early an event to be predictive of cognitive performance. Longitudinal studies will help disentangle these complicated relations. We suggest, in line with the STAC theory proposed by Park and Reuter-Lorenz (2009), that cognitive reserve may have an actual neural substrate in enhanced white matter connectivity and/or the propensity for more successful functional reorganization of the cortex which may compensate for neural insults such as accumulation of amyloid plaques.
Definitively evaluating cognitive performance in healthy adults in relation to Aβ deposition is difficult at this point, as the few extant studies have found inconsistent results. While finding no consistent differences in cognitive performance between normal elderly with high and low amyloid deposition, nor between high and low deposition in MCI subjects, Jack et al. (2008) did find moderate correlations (r=−.34 to −.36) with PIB binding and cognitive measures in the whole sample across three indices of memory performance. Pike et al. (2007) found stronger associations between episodic memory performance and Aβ binding in MCI (r=−.60) than in healthy older adults (r=−.38). Mormino et al. (2009) further found that the relation between memory performance and Aβ is mediated by hippocampal volume, suggesting an order for the cascade of events that build to affect memory decline. Aizenstein et al. (2008), however found that in general, most cognitive functions were unrelated to amyloid burden with the only significant finding being lower scores on a delayed recall task, but this was for the amyloid negative group. In a longitudinal study, in initially healthy elderly, those who declined clinically showed a significant association of memory performance and amyloid deposition (Fripp et al. 2008). Braskie et al. (2008) found in cognitively normal older adults higher FDDNP binding in the frontal and parietal cortex was associated with poorer cognitive performance, although in this study test performance from multiple domains was collapsed into one composite measure. Overall, it appears that at least memory performance may be negatively associated with Aβ deposition, even in relatively healthy older adults, although new, but growing evidence suggests that Aβ’s effects are carried out via structural and functional degradation, rather than a direct effect of amyloid deposition per se. Clearly, further research is needed to determine the specific cognitive effects of increased beta-amyloid deposition and by which markers of neural health these effects are mediated.
Amyloid Burden and Functional Brain Imaging
The data relating amyloid deposition to functional imaging are very limited but expanding. There is some evidence that patients with amyloid deposition compensate for the amyloid burden through functional reorganization of neural activity, directing neural activity away from sites of amyloid deposits into healthier areas of the brain. Specifically, Nelissen et al. (2007) demonstrated in a fMRI study that AD subjects showed lesser activation than controls in superior temporal cortex, and this activation was associated with greater Aβ load in the same region. For the contralateral hemisphere, however, the pattern was reversed with greater activation in the AD subjects vs. controls, and this contralateral recruitment was positively correlated with cognitive measures, demonstrating that at least in early stage AD, functional reorganization can help overcome negative effects of Aβ deposition.
An alternative view comes from studies of default network activity. The default network consists of regions of the brain that are more active than others when the brain is at “rest” (i.e., periods when task-related activity are not required) compared to when the brain is engaged in active tasks. A core set of regions comprise the default network (Buckner et al. 2008; Greicius et al. 2003) that include the ventral medial and dorsal medial prefrontal, posterior cingulate/retrosplenial, inferior parietal, and lateral temporal cortices. Young adults suppress default activity when faced with cognitive challenge, but older adults display difficulty shifting out of the default mode to task-relevant modes when confronted with a cognitive task. Buckner et al. (2005) hypothesized that sustained engagement across the lifespan in default network activity might actually lead to amyloid deposition. Aβ deposition correlated with regions of deactivation in the default network, suggesting that regions used more frequently are the same ones that show increased Aβ deposition implying that repetitive use of these regions might promote plaque deposition. Support for this wear-and-tear hypothesis also comes from evidence that both Aβ production and amyloid precursor protein regulation are activity-dependent (Kamenetz et al. 2003; Nitsch et al. 1993), and therefore would be produced and expressed in greater quantities in the most metabolically active brain regions (Buckner et al. 2005). In a later study out of this lab, Andrews-Hanna et al. (2007) isolated nine older adults who were amyloid free. These individuals also showed default network disruption as well as white matter connectivity reduction, suggesting that the disruption of the default network is not solely related to amyloid pathology.
More recently, Buckner et al. (2009) have identified associative cortex regions that served as “cortical hubs” involved in both active and resting states during functional scanning. These hubs showed higher levels of Aβ deposition in AD patients compared to controls, consistent with the notion that “overuse” of the same regions of the brain is metabolically expensive and the resulting wear-and-tear may lead to adverse consequences such as increased Aβ deposition and cognitive decline or even development of AD. In their newest paper, Sperling et al (2009) investigated PIB- (n=22) vs PIB+ (n=13) groups in their patterns of overlapping fMRI default activity and PIB uptake. They found increased PIB binding in the precuneus/posterior cingulate and medial prefrontal cortices (both default network areas) in 30% of their older adults and PIB uptake in the precuneus/posterior cingulate correlated with increased default activity in this region. This pattern is similar to that found in AD patients and suggests that even in cognitively normal older adults, there is a failure to deactivate the regions of the default network necessary for memory function, perhaps due to increased Aβ burden in this system. PIB uptake did not, however correlate with memory performance on the functional task. The absolute number of observations in these studies was small, however, and the relationship of amyloid to functional imaging is still in its early stages, especially with regard to patterns of task-activation and functional reorganization. Resolving the nature of these associations will be a major goal of future research in the areas of both normal aging and AD.
Integrating Amyloid Deposition with a Model of Neurocognitive Aging
Thus far, it is clear from the literature that amyloid deposition in cognitively healthy individuals from age 65–90 is roughly 20–30%. Second, some studies find evidence that amyloid deposition is related to decreased hippocampal volume or decreased episodic memory, but the relationship is tentative, as a number of studies have failed to confirm this. Third, the limited data suggest that high premorbid ability may buffer against the impact of amyloid deposition, but much more research is required in this area. Fourth, while there is a high likelihood of mildly impaired individuals converting to AD over time, it appears that significant deposition of amyloid by no means guarantees conversion to AD. Finally, numerous authors indicate the importance of understanding whether amyloid deposition is occurring earlier in patients and whether amyloid deposition at a relatively young age is a harbinger of Alzheimer’s Disease at a later age. The issue of when amyloid deposition appears in healthy aging, particularly the changes that occur during middle age, becomes critically important as interventions become available to stave off both normal and pathological age-related cognitive decline (Linazasoro 2008; Small et al. 2008). At this time, there are no published lifespan studies of amyloid deposition. Thus, knowledge of the timecourse, extent and distribution of amyloid in normal adults remains limited, making this a much needed active area of future study.
The Scaffolding Theory of Aging and Cognition (STAC)
Given the central role that amyloid deposition appears to play in brain health and cognition in normally aging adults, it is helpful to think about Aβ deposition within an integrative model of neurocognitive aging, STAC (Park and Reuter-Lorenz 2009). The STAC model (Park and Reuter-Lorenz 2009) is a broad integration of the behavioral and neural data presently available with respect to cognitive aging and can be conceptualized as a means to theoretically integrate the amyloid data with the existing neurocognitive literature. The model is presented in Fig. 8. STAC posits that cognitive function in older adults can be understood in terms of the magnitude of neural insults that the brain has sustained (both structural and functional) as well as the compensatory neural activities (“scaffolding”) that operate to maintain cognitive behavior. According to this model, scaffolding is conceptualized as the recruitment of additional circuitry to compensate for declining structures whose functioning has become noisy, inefficient, or both. The pervasive finding of increased prefrontal activation in older adults across many different cognitive tasks reflects the engagement of “compensatory scaffolding.” The scaffolding is a direct response to amyloid deposition, regional brain shrinkage, white matter changes, as well as functional dedifferentiation of ventral visual cortex and deficient default network activity. STAC also provides for mechanisms that can enhance the development of compensatory scaffolding. The STAC model views amyloid deposition as one type of neural insult that will result in compensatory functional reorganization and decreased cognition. The data reviewed here are consistent with the predictions of STAC, but are increasingly suggestive that amyloid deposition may be an initiating event that leads to the hippocampal shrinkage (Rodrigue and Raz 2004) and decline in fMRI subsequent memory performance previously documented in aging studies (e.g., Gutchess et al. 2005).
Fig. 8.
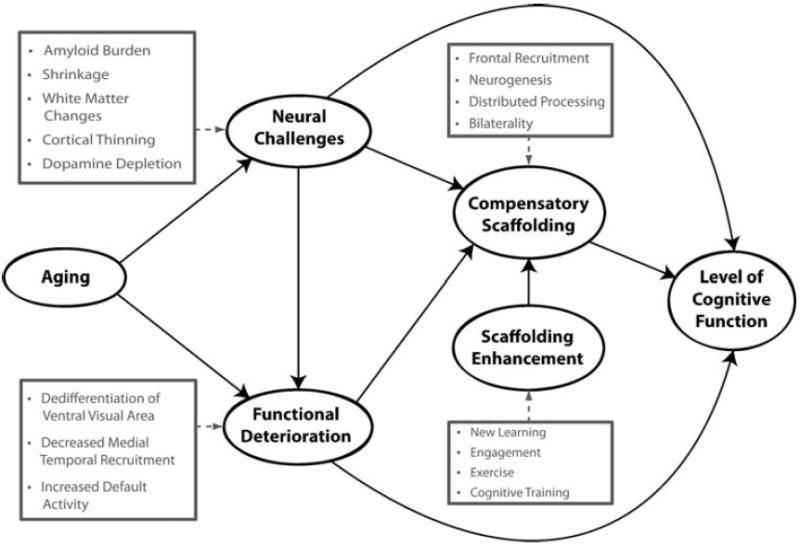
A conceptual model of the scaffolding theory of aging and cognition (STAC; adapted from Park and Reuter-Lorenz 2009)
Future Directions: What We Need to Know
To summarize, we believe that the existing evidence suggests that amyloid is a critical initiating event in a cascade of events that ultimately leads to cognitive decline. Because amyloid deposition is putatively the first event in this negative cascade, many normal old adults harbor amyloid burden but behave within normal cognitive limits. This may occur for two reasons. First, the pathology has yet to be expressed behaviorally because this is an early neuropathological event that has its detrimental effects via functional and structural degradation, and second, we hypothesize that the brain responds to the initial pathology by reorganizing and compensating functionally for the amyloid deposition to sustain normal cognitive performance. Given the likely importance of amyloid in determining an individual’s course of aging, filling in the knowledge gap about the presence and consequences of amyloid in normal populations of adults (particularly middle-aged participants) will advance our understanding of healthy aging and of disease progression. As larger samples of healthy adults are being amassed for the study of amyloid deposition, the middle age range needs to also be included to chart the timecourse of deposition. So far, there are only a handful of studies that integrate amyloid imaging with functional imaging and most of these focus on the default network system. Reasons for the sparcity of data are primarily the recency of the technology to conduct this type of research, the high cost of amyloid imaging, and the short half-life of the ligands, which makes it essential that a cyclotron be on site (for the PIB ligand) or within a few hours drive (for the AV-45 or FDDNP ligand). We believe that increasing amounts of data on this topic will facilitate the field’s ability to identify individuals early who are in need of interventions, as finding amyloid-reducing mechanisms is a joint goal of academic and pharmaceutical researchers. It appears increasingly likely that effective interventions for Alzheimer’s Disease (when they are developed) may need to occur in seemingly healthy middle-aged and elderly adults who harbor latent pathology.
Adding amyloid imaging to cognitive neuroscience of aging studies to predict structural, neural, and cognitive function will yield valuable information to the research community and will provide significant insight into the role of subtle neural pathology in predicting changes in cognitive function. What is missing from this early body of literature to date is a characterization of beta-amyloid deposition across the entire adult lifespan and large-scale longitudinal follow-up of its development and progression. These studies will allow us to assess across each decade how amyloid deposition, neural structure and neural function predict cognition, as well as to better understand the role of amyloid deposition in predicting patterns of neuronal function. This approach will open up new possibilities for understanding critical dissociations between normal and pathological aging.
Conclusion
Several new radiotracers have been developed that bind to fibrillar amyloid beta providing sensitive estimates of amyloid deposition in various brain regions. Aβ imaging has been primarily used to study patients with AD and MCI; however, there is now building, but limited data on Aβ deposition in healthy controls, and these Aβ imaging studies suggest that approximately 20–30% of healthy older adults show significant deposition. The most consistent and uniform finding across AD studies is that early in the disease there is binding to the precuneus/posterior cingulate cortex and early evidence suggests that this also seems to be a prevalent region for deposition in nondemented elderly in addition to medial prefrontal regions. There is considerable evidence suggesting that amyloid deposition precedes declines in cognition and may be the initiator in a cascade of other neural events that ultimately lead to age-related cognitive decline. What is less clear is why there is a relative lack of association between amyloid binding and cognitive performance in these studies. It seems likely that Aβ deposition is such an early event in the cascade that its ultimate detrimental effect on cognitive performance is carried out via mediating effects of structural and functional disruption. Adding amyloid imaging to cognitive neuroscience of aging studies to predict structural, neural, and cognitive function will enable us to disentangle these associations and understand the mediating neural effects and will provide significant insight into the role of subtle neural pathology in predicting distal changes in cognitive function. This approach will open up new possibilities for understanding critical dissociations between normal and pathological aging. Detailed characterization of the most salient predictors of successful versus unsuccessful aging will provide much needed information for the development of preventative approaches to optimize cognitive health in late adulthood.
Acknowledgments
Preparation of this paper was supported by National Institutes of Health grant AG-006265-23 to Denise Park.
Footnotes
Disclosures The authors have no financial disclosures to report.
This bimodal distribution may serve to highlight the arbitrary nature of a categorical MCI group and emphasizes the continuum of cognitive performance from normal to AD, with MCI being a midpoint of this continuum.
References
- Aizenstein HJ, Nebes RD, Saxton JA, Price JC, Mathis CA, Tsopelas ND, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Archives of Neurology. 2008;65:1509–1517. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen N, Minthon L, Vanmechelen E, Vanderstichele H, Davidsson P, Winblad B, et al. Cerebrospinal fluid tau and Abeta42 as predictors of development of Alzheimer’s disease in patients with mild cognitive impairment. Neuroscience Letters. 1999;273:5–8. doi: 10.1016/s0304-3940(99)00617-5. [DOI] [PubMed] [Google Scholar]
- Andrews-Hanna JR, Snyder AZ, Vincent JL, Lustig C, Head D, Raichle ME, et al. Disruption of large-scale brain systems in advanced aging. Neuron. 2007;56:924–935. doi: 10.1016/j.neuron.2007.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66:1837–1844. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- Bourgeat P, Villemagne VL, Fripp J, Pike KE, Raniga P, Acosta O, et al. Relation between amyloid burden, brain atrophy and memory in Alzheimer’s disease. Alzheimer’s Association 2009 International Conference on Alzheimer’s Disease (ICAD); 2009, July.2009. [Google Scholar]
- Braak H, Braak E. Evolution of the neuropathology of Alzheimer’s disease. Acta Neurologica Scandinavica. 1996;165:3–12. doi: 10.1111/j.1600-0404.1996.tb05866.x. [DOI] [PubMed] [Google Scholar]
- Braskie MN, Klunder AD, Hayashi KM, Protas H, Kepe V, Miller KJ, et al. Plaque and tangle imaging and cognition in normal aging and Alzheimer’s disease. Neurobiology of Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, et al. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. Journal of Neuroscience. 2005;25:7709–7717. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Andrews-Hanna JR, Schacter DL. The brain’s default network: anatomy, function, and relevance to disease. Annals of the New York Academy of Sciences. 2008;1124:1–38. doi: 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- Buckner RL, Sepulcre J, Talukdar T, Krienen FM, Liu H, Hedden T, et al. Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer’s disease. Journal of Neuroscience. 2009;29:1860–1873. doi: 10.1523/JNEUROSCI.5062-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Dickerson BC, Bakkour A, Salat DH, Feczko E, Pacheco J, Greve DN, et al. The cortical signature of Alzheimer’s disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cerebral Cortex. 2009;19:497–510. doi: 10.1093/cercor/bhn113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, et al. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiology of Aging. 1992;13:179–189. doi: 10.1016/0197-4580(92)90027-u. [DOI] [PubMed] [Google Scholar]
- Diniz BS, Pinto JA, Forlenza OV. Do CSF total tau, phosphorylated tau, and beta-amyloid 42 help to predict progression of mild cognitive impairment to Alzheimer’s disease? A systematic review and meta-analysis of the literature. World Journal of Biological Psychiatry. 2008;9:172–182. doi: 10.1080/15622970701535502. [DOI] [PubMed] [Google Scholar]
- Drzezga A, Grimmer T, Henriksen G, Mühlau M, Perneczky R, Miederer I, et al. Effect of APOE genotype on amyloid plaque load and gray matter volume in Alzheimer disease. Neurology. 2009;72:1487–1494. doi: 10.1212/WNL.0b013e3181a2e8d0. [DOI] [PubMed] [Google Scholar]
- Edison P, Archer HA, Hinz R, Hammers A, Pavese N, Tai YF, et al. Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology. 2007;68:501–508. doi: 10.1212/01.wnl.0000244749.20056.d4. [DOI] [PubMed] [Google Scholar]
- Farlow MR, He Y, Tekin S, Xu J, Lane R, Charles HC. Impact of APOE in mild cognitive impairment. Neurology. 2004;63:1898–1901. doi: 10.1212/01.wnl.0000144279.21502.b7. [DOI] [PubMed] [Google Scholar]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis APOE and Alzheimer Disease meta analysis consortium. Journal of the American Medical Association. 1997;278:1349–1356. [PubMed] [Google Scholar]
- Forsberg A, Engler H, Almkvist O, Blomquist G, Hagman G, Wall A, et al. PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiology of Aging. 2008;29:1456–1465. doi: 10.1016/j.neurobiolaging.2007.03.029. [DOI] [PubMed] [Google Scholar]
- Fotenos AF, Mintun MA, Snyder AZ, Morris JC, Buckner RL. Brain volume decline in aging: evidence for a relation between socioeconomic status, preclinical Alzheimer disease, and reserve. Archives of Neurology. 2008;65:113–120. doi: 10.1001/archneurol.2007.27. [DOI] [PubMed] [Google Scholar]
- Fripp J, Bourgeat P, Acosta O, Raniga P, Modat M, Pike KE, et al. Appearance modeling of 11C PiB PET images: characterizing amyloid deposition in Alzheimer’s disease, mild cognitive impairment and healthy aging. Neuroimage. 2008;43:430–439. doi: 10.1016/j.neuroimage.2008.07.053. [DOI] [PubMed] [Google Scholar]
- Ghisletta P, Lindenberger U. Age-based structural dynamics between perceptual speed and knowledge in the Berlin aging study: direct evidence for ability dedifferentiation in old age. Psychology and Aging. 2003;18:696–713. doi: 10.1037/0882-7974.18.4.696. [DOI] [PubMed] [Google Scholar]
- Greicius MD, Krasnow B, Reiss AL, Menon V. Functional connectivity in the resting brain: a network analysis of the default mode hypothesis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:253–258. doi: 10.1073/pnas.0135058100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimmer T, Riemenschneider M, Förstl H, Henriksen G, Klunk WE, Mathis CA, et al. Beta amyloid in Alzheimer’s disease: increased deposition in brain is reflected in reduced concentration in cerebrospinal fluid. Biological Psychiatry. 2009;65:927–934. doi: 10.1016/j.biopsych.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundman M, Sencakova D, Jack CR, Jr, Petersen RC, Kim HT, Schultz A, et al. Brain MRI hippocampal volume and prediction of clinical status in a mild cognitive impairment trial. Journal of Molecular Neuroscience. 2002;19:23–27. doi: 10.1007/s12031-002-0006-6. [DOI] [PubMed] [Google Scholar]
- Gutchess AH, Welsh RC, Hedden T, Bangert A, Minear M, Liu LL, et al. Aging and the neural correlates of successful picture encoding: frontal activations compensate for decreased medial-temporal activity. Journal of Cognitive Neuroscience. 2005;17:84–96. doi: 10.1162/0898929052880048. [DOI] [PubMed] [Google Scholar]
- Hedden T, Gabrieli JDE. Insights into the ageing mind: a view from cognitive neuroscience. Nature Reviews Neuroscience. 2004;5:87–97. doi: 10.1038/nrn1323. [DOI] [PubMed] [Google Scholar]
- Ichise M, Plett S, Joshi A, Stern Y, van Heertum R, Lowe V, et al. Quantitative comparison of three novel 18F-labeled ligands for PET imaging of brain amyloid-β plaques in Alzheimer’s disease. Journal of Nuclear Medicine. 2008;49:214. [Google Scholar]
- Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, et al. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- Jack CR, Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, Shiung MM, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer’s disease and amnestic mild cognitive impairment. Brain. 2008;131:665–680. doi: 10.1093/brain/awm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: implications for sequence of pathological events in Alzheimer’s disease. Brain. 2009;132:1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust W. Mapping brain beta-amyloid. Current Opinion in Neurology. 2009a;22:356–361. doi: 10.1097/WCO.0b013e32832d93c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust W. Amyloid + activation = Alzheimer’s? Neuron. 2009b;63:141–143. doi: 10.1016/j.neuron.2009.07.008. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, et al. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Whitwell JL, Ahmed Z, Shiung MM, Weigand SD, Knopman DS, et al. Beta-amyloid burden is not associated with rates of brain atrophy. Annals of Neurology. 2008;63:204–212. doi: 10.1002/ana.21223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, et al. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Katzman R, Terry R, DeTeresa R, Brown T, Davies P, Fuld P, et al. Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Annals of Neurology. 1988;23:138–144. doi: 10.1002/ana.410230206. [DOI] [PubMed] [Google Scholar]
- Kemppainen NM, Aalto S, Wilson IA, Någren K, Helin S, Brück A, et al. Voxel-based analysis of PET amyloid ligand [11C]PIB uptake in Alzheimer disease. Neurology. 2006;67:1575–1580. doi: 10.1212/01.wnl.0000240117.55680.0a. [DOI] [PubMed] [Google Scholar]
- Kemppainen NM, Aalto S, Wilson IA, Någren K, Helin S, Brück A, et al. PET amyloid ligand [11C]PIB uptake is increased in mild cognitive impairment. Neurology. 2007;68:1603–1606. doi: 10.1212/01.wnl.0000260969.94695.56. [DOI] [PubMed] [Google Scholar]
- Kemppainen NM, Aalto S, Karrasch M, Någren K, Savisto N, Oikonen V, et al. Cognitive reserve hypothesis: Pittsburgh Compound B and fluorodeoxyglucose positron emission tomography in relation to education in mild Alzheimer’s disease. Annals of Neurology. 2008;63:112–118. doi: 10.1002/ana.21212. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Mathis CA. The future of amyloid-beta imaging: a tale of radionuclides and tracer proliferation. Current Opinion in Neurology. 2008;21:683–687. doi: 10.1097/WCO.0b013e3283168e1a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Annals of Neurology. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Li Y, Rinne JO, Mosconi L, Pirraglia E, Rusinek H, DeSanti S, et al. Regional analysis of FDG and PIB-PET images in normal aging, mild cognitive impairment, and Alzheimer’s disease. European Journal of Nuclear Medicine and Molecular Imaging. 2008;35:2169–2181. doi: 10.1007/s00259-008-0833-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linazasoro G. Imaging beta-amyloid burden in aging and dementia. Neurology. 2008;70:1649–1650. doi: 10.1212/01.wnl.0000318046.06992.24. [DOI] [PubMed] [Google Scholar]
- Lopresti BJ, Klunk WE, Mathis CA, Hoge JA, Ziolko SK, Lu X, et al. Simplified quantification of Pittsburgh Compound B amyloid imaging PET studies: a comparative analysis. Journal of Nuclear Medicine. 2005;46:1959–1972. [PubMed] [Google Scholar]
- Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446–452. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- Mormino EC, Kluth JT, Madison CM, Rabinovici GD, Baker SL, Miller BL, et al. Episodic memory loss is related to hippocampal-mediated {beta}-amyloid deposition in elderly subjects. Brain. 2009;132:1310–1323. doi: 10.1093/brain/awn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC, Roe CM, Mintun MA. APOE status predicts PiB-positivity in nondemented aging: evidence for Preclinical Alzheimer’s disease. Neurology. 2009;72:A92. [Google Scholar]
- Nelissen N, Vandenbulcke M, Fannes K, Verbruggen A, Peeters R, Dupont P, et al. Abeta amyloid deposition in the language system and how the brain responds. Brain. 2007;130:2055–2069. doi: 10.1093/brain/awm133. [DOI] [PubMed] [Google Scholar]
- Nitsch RM, Farber SA, Growdon JH, Wurtman RJ. Release of amyloid beta-protein precursor derivatives by electrical depolarization of rat hippocampal slices. Proceedings of the National Academy of Sciences. 1993;90:5191–5193. doi: 10.1073/pnas.90.11.5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordberg A. Amyloid imaging in Alzheimer’s disease. Neuropsychologia. 2008;46:1636–1641. doi: 10.1016/j.neuropsychologia.2008.03.020. [DOI] [PubMed] [Google Scholar]
- Nordlund A, Rolstad S, Klang O, Lind K, Pedersen M, Blennow K, et al. Episodic memory and speed/attention deficits are associated with Alzheimer-typical CSF abnormalities in MCI. Journal of the International Neuropsychological Society. 2008;14:582–590. doi: 10.1017/S135561770808079X. [DOI] [PubMed] [Google Scholar]
- Park DC, Goh JO. Successful aging. In: Cacioppo J, Berntson G, editors. Handbook of Cognitive Neuroscience for the Behavioral Sciences. Ch. 61. Hoboken: Wiley; 2009. pp. 1203–1219. [Google Scholar]
- Park DC, Reuter-Lorenz PA. The adaptive brain: aging and neurocognitive scaffolding. Annual Review of Psychology. 2009;60:173–196. doi: 10.1146/annurev.psych.59.103006.093656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DC, Lautenschlager G, Hedden T, Davidson NS, Smith AD, Smith PK. Models of visuospatial and verbal memory across the adult life span. Psychology and Aging. 2002;17:299–320. [PubMed] [Google Scholar]
- Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Archives of Neurology. 1999;56:303–308. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- Pike KE, Savage G, Villemagne VL, Ng S, Moss SA, Maruff P, et al. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer’s disease. Brain. 2007;130:2837–2844. doi: 10.1093/brain/awm238. [DOI] [PubMed] [Google Scholar]
- Raz N, Rodrigue KM. Differential aging of the brain: patterns, cognitive correlates and modifiers. Neuroscience & Biobehavioral Reviews. 2006;30:730–748. doi: 10.1016/j.neubiorev.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz N, Kennedy KM. A systems approach to age-related change: Neuroanatomical changes, their modifiers, and cognitive correlates. In: Jagust W, D’Esposito M, editors. Imaging the aging brain. Ch 4. Oxford UP; NYC: 2009. pp. 43–70. [Google Scholar]
- Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proceedings of the National Academy of Sciences. 2009;106:6820–6825. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigue KM, Raz N. Shrinkage of the entorhinal cortex over five years predicts memory performance in healthy adults. Journal of Neuroscience. 2004;24:956–963. doi: 10.1523/JNEUROSCI.4166-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolstad S, Nordlund A, Eckerström C, Gustavsson MH, Zetterberg H, Wallin A. Biomarkers in relation to cognitive reserve in patients with mild cognitive impairment—proof of concept. Dementia and Geriatric Cognitive Disorders. 2009;27:194–200. doi: 10.1159/000203130. [DOI] [PubMed] [Google Scholar]
- Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, et al. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68:1718–1725. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- Rowe CC, Ackerman U, Browne W, Mulligan R, Pike KL, O’Keefe G, et al. Imaging of amyloid beta in Alzheimer’s disease with 18F-BAY94-9172, a novel PET tracer: proof of mechanism. Lancet Neurology. 2008;7:129–135. doi: 10.1016/S1474-4422(08)70001-2. [DOI] [PubMed] [Google Scholar]
- Samuels SC, Silverman JM, Marin DB, Peskind ER, Younki SG, Greenberg DA, et al. CSF beta-amyloid, cognition, and APOE genotype in Alzheimer’s disease. Neurology. 1999;52:547–551. doi: 10.1212/wnl.52.3.547. [DOI] [PubMed] [Google Scholar]
- Scheinin NM, Aalto S, Koikkalainen J, Lötjönen J, Karrasch M, Kemppainen N, et al. Follow-up of [11C]PIB uptake and brain volume in patients with Alzheimer disease and controls. Neurology. 2009;73:1186–1192. doi: 10.1212/WNL.0b013e3181bacf1b. [DOI] [PubMed] [Google Scholar]
- Schmidt ML, Lee VM, Trojanowski JQ. Relative abundance of tau and neurofilament epitopes in hippocampal neurofibrillary tangles. American Journal of Pathology. 1990;136:1069–1075. [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shoghi-Jadid K, Small GW, Agdeppa ED, Kepe V, Ercoli LM, Siddarth P, et al. Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. American Journal of Geriatric Psychiatry. 2002;10:24–35. [PubMed] [Google Scholar]
- Skovronsky D. Use of eINDs for evaluation of multiple related PET amyloid plaque imaging agents. Journal of Nuclear Medicine. 2008;49:47N–48N. [PubMed] [Google Scholar]
- Skovronsky D, Coleman RE, Frey K, Garg P, Ichise M, Lowe V, et al. Results of multi-center clinical trials comparing four 18F PET amyloid-imaging agents: preclinical to clinical correlations. Journal of Nuclear Medicine Meeting Abstracts. 2008;49(1):34P. [Google Scholar]
- Small GW, Kepe V, Ercoli LM, Siddarth P, Bookheimer SY, Miller KJ, et al. PET of brain amyloid and tau in mild cognitive impairment. New England Journal of Medicine. 2006;355:2652–2663. doi: 10.1056/NEJMoa054625. [DOI] [PubMed] [Google Scholar]
- Small GW, Bookheimer SY, Thompson PM, Cole GM, Huang S, Kepe V, et al. Current and future uses of neuroimaging for cognitively impaired patients. Lancet Neurology. 2008;7:161–172. doi: 10.1016/S1474-4422(08)70019-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small GW, Siddarth P, Burggren AC, Kepe V, Ercoli LM, Miller KJ, et al. Influence of cognitive status, age, and APOE-4 genetic risk on brain FDDNP positron-emission tomography imaging in persons without dementia. Archives of General Psychiatry. 2009;66:81–87. doi: 10.1001/archgenpsychiatry.2008.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Laviolette PS, O’Keefe K, O’Brien J, Rentz DM, Pihlajamaki M, et al. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63:178–188. doi: 10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern Y. What is cognitive reserve? Theory and research application of the reserve concept. Journal of the International Neuropsychological Society. 2002;8:448–460. [PubMed] [Google Scholar]
- Stern Y. Cognitive reserve. Neuropsychologia. 2009;47:2015–2028. doi: 10.1016/j.neuropsychologia.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strozyk D, Blennow K, White LR, Launer LJ. CSF Abeta 42 levels correlate with amyloid-neuropathology in a population-based autopsy study. Neurology. 2003;60:652–656. doi: 10.1212/01.wnl.0000046581.81650.d0. [DOI] [PubMed] [Google Scholar]
- Tapiola T, Pirttilä T, Mikkonen M, Mehta PD, Alafuzoff I, Koivisto K, et al. Three-year follow-up of cerebrospinal fluid tau, beta-amyloid 42 and 40 concentrations in Alzheimer’s disease. Neuroscience Letters. 2000;280:119–122. doi: 10.1016/s0304-3940(00)00767-9. [DOI] [PubMed] [Google Scholar]
- Thal DR, Capetillo-Zarate E, Del Tredici K, Braak H. The development of amyloid beta protein deposits in the aged brain. Science of Aging Knowledge Environment: SAGE KE. 2006;2006(6):re1. doi: 10.1126/sageke.2006.6.re1. [DOI] [PubMed] [Google Scholar]
- Tolboom N, Yaqub M, van der Flier WM, Boellaard R, Luurtsema G, Windhorst AD, et al. Detection of Alzheimer pathology in vivo using both 11C-PIB and 18F-FDDNP PET. Journal of Nuclear Medicine. 2009a;50:191–197. doi: 10.2967/jnumed.108.056499. [DOI] [PubMed] [Google Scholar]
- Tolboom N, Yaqub M, Boellaard R, Luurtsema G, Windhorst AD, Scheltens P, et al. Test-retest variability of quantitative [(11) C]PIB studies in Alzheimer’s disease. European Journal of Nuclear Medicine and Molecular Imaging. 2009b;36:1629–1638. doi: 10.1007/s00259-009-1129-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemagne VL, Pike KE, Darby D, Maruff P, Savage G, Ng S, et al. Abeta deposits in older non-demented individuals with cognitive decline are indicative of preclinical Alzheimer’s disease. Neuropsychologia. 2008;46:1688–1697. doi: 10.1016/j.neuropsychologia.2008.02.008. [DOI] [PubMed] [Google Scholar]
- Wahlund LO, Blennow K. Cerebrospinal fluid biomarkers for disease stage and intensity in cognitively impaired patients. Neuroscience Letters. 2003;339:99–102. doi: 10.1016/s0304-3940(02)01483-0. [DOI] [PubMed] [Google Scholar]
- Wishart HA, Saykin AJ, McAllister TW, Rabin LA, McDonald BC, Flashman LA, et al. Regional brain atrophy in cognitively intact adults with a single APOE epsilon4 allele. Neurology. 2006;67:1221–1224. doi: 10.1212/01.wnl.0000238079.00472.3a. [DOI] [PubMed] [Google Scholar]
- Wong D, Rosenberg P, Zhou Y, Kumar A, Ravert H, Brasic J, et al. In vivo imaging of amyloid deposition in Alzheimer’s disease using the novel radioligand [18F] AV-45. Journal of Nuclear Medicine Meeting Abstracts. 2008;49(1):214P. doi: 10.2967/jnumed.109.069088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Kung M, Oya S, Hou C, Kung HF. 18F-labeled styrylpyridines as PET agents for amyloid plaque imaging. Nuclear Medicine and Biology. 2007;34:89–97. doi: 10.1016/j.nucmedbio.2006.10.003. [DOI] [PubMed] [Google Scholar]