

Abstract
The impact of stress on brain function is increasingly recognized. Various substances are released in response to stress and can influence distinct neuronal circuits, but the functional advantages of having such a diversity of stress mediators remain unclear. Individual neurotransmitter, neuropeptide and steroid stress mediators have specific spatial and temporal niches, but these niches also overlap. In addition, the effects of individual mediators on neuronal function and plasticity are integrated, and emerging evidence suggests that there is crosstalk between them. Together, this results in the stress instruments producing an orchestrated ‘symphony’ that enables fine-tuned responses to diverse challenges.
Any actual or potential disturbance of an individual's environment — a ‘stressor’ — is recognized or perceived by specific brain regions. The subjective state of sensing potentially adverse changes in the environment is called ‘stress’ and leads to the release of molecules that we here call ‘stress mediators’, which bind to receptor targets. Each of these mediators acts on specific neuronal populations, resulting in unique downstream effects. Together, these effects constitute the ‘stress response’, which enables the animal to adapt to the changing environment1-4.
A perplexing aspect of the stress response is its complexity. Various stress mediators have been described, including neurotransmitters (for example, noradrenaline and serotonin), peptides (for example, corticotropin-releasing hormone (CRH), other members of the CRH family and vasopressin) and steroid hormones (for example, cortisol in humans and corticosterone in rodents). What is the reason for the existence of these multiple instruments? Is there not a staggering degree of redundancy?
In this Perspective we initially discuss the characteristic spatial and temporal niches of key stress mediators and provide emerging evidence that these niches overlap, affording opportunities for interactions between mediators. We then highlight these interactions, showing that the stress mediators act in concert. We argue that the diversity of the instruments of stress, rather than being redundant, enables both optimal niches of action for each mediator and interactions between the multiple mediators that orchestrate our brain's remarkable ability to respond — and adapt — to a dynamic environment.
A need for multiple mediators
Stress signifies a potential or actual threat that requires immediate changes in behaviour as well as a modification of future behaviours. This is achieved through the modulation of neuronal functioning at several levels of the CNS — levels that govern decision making, learning and memory as well as hormonal, autonomic and emotional responses.
Different types of stressors require different responses (FIG. 1). The stressor type influences the repertoire of neuronal populations that perceive a potential threat, as well as the neurons and stress mediators that are engaged in the adaptive response. For example, physical stressors such as blood loss, trauma and cold rapidly recruit the brainstem and hypothalamic regions1,3, whereas psychological stressors such as social embarrassment, examinations or deadlines primarily engage stress mediators in brain regions that subserve emotion (the amygdala and the prefrontal cortex), learning and memory (the hippocampus) and decision making (the prefrontal cortex)2,4,5. These are not segregated systems, because physical stressors often have psychological aspects and vice versa.
Figure 1. Different stressors require different responses.

Many factors influence the pattern and magnitude of the response to stress, including the duration of stress exposure (acute versus chronic), the type of stress (physical versus psychological), the stress context (for example, time of day), the developmental stage of the animal (newborn, adult or aged) and the animal's sex and genetic background. The panoply of unique stress situations and neuronal populations that respond to them to affect neural and behavioural plasticity on a timescale from seconds to years is not well served by a single mediator — hence the need for multiple instruments, so that each combination of mediators addresses the specific aspects of a stressor. The molecules that convey the stress signal to the CNS and that contribute to the resulting functional changes in the CNS (stress mediators) include monoamines, neuropeptides and steroid hormones, examples of which are shown on the right. Each mediator has a preferred activity domain in space and time.
The duration of the stressor also greatly influences the nature of the neuronal responses (FIG. 1). Acute stressors — for example, a rapidly approaching car — cause a rapid surge of neurotransmission, neuronal activation and hormone release. This is followed by rapid return to baseline levels, although the temporary activation of, for example, hippocampal and hypothalamic neurons can ultimately lead to alterations in gene expression, which in turn change subsequent neuronal responses. By contrast, chronic stress (typically defined as stress that lasts a week or more) provokes sustained and/or progressive changes in the expression of particular genes, structural alterations in neurons and changes in neuronal firing patterns throughout the brain4,6. If such changes persist they can result in prolonged deviations from the original functioning of the network3.
In addition, the characteristics of the responding brain determine the pattern and magnitude of its response to a stressor. For example, the age of the animal influences the likelihood of a signal being perceived as a stressor. It also influences the mediators that are released by stressors, and their consequences3,7,8. For example, in rodents the molecular cascades that are activated by stress in the hypothalamus9,10 and the hippocampus9,11 during adulthood differ from those that are activated by stress in the infant and change again with aging7,12. In addition, sex and genetic factors interact with life events, such as environmental enrichment or isolation, to contribute to the diversity of the stressors and their impacts on an individual's brain.
Taken together, the large array of stress types and contexts, the genetic background, age and sex of the stressed animal, as well as additional factors such as the point in the circadian rhythm at which the stressful event takes place, create a rich and complex matrix of stressors. Each unique stress situation requires an efficient response from numerous neuronal ensembles throughout the CNS, a process that requires astute orchestration. This orchestration occurs through the deployment of a repertoire of signalling molecules that can bring about the temporal, spatial and context specificity of each individual stress response.
The diversity of stress mediators
Each stress mediator has its characteristic, ‘classic’ spatial and temporal domains. Below we elaborate on the preferred spatial and temporal domains of the three main classes of stress mediators and their individual contribution to the brain's stress response, as a preamble to discussing their coordinated and interactive effects.
Monoamines
Shortly after a stressful event, the release of monoamines, including noradrenaline, dopamine and serotonin, is increased in specific neuronal populations13-16. This release is triggered by brain circuits that are involved in the evaluation of the stressful event or is triggered indirectly, through activation of the sympathetic nervous system1,5. The extent to which monoaminergic systems are activated depends on many factors, including sex (for example, see REF. 17), the time of day18 and the controllability or recurrence of the stressor13. For instance, activation of raphe neurons (which produce serotonin) by shock exposure is only prominent when the situation is uncontrollable13,19.
An enhanced release of monoamines after stress has been demonstrated in the hippocampus, the amygdala, the prefrontal cortex and the nucleus accumbens, but it probably also takes place in many other brain regions. However, the spatial distribution of the consequences of this release depends also on the affinity and distribution of the monoamine receptor subtypes. Thus, the combination of release site and receptor distribution and affinity provides a single stress mediator with multiple spatial niches and different functions.
Stress-induced monoaminergic release and actions are fast. The release is generally induced within minutes after the onset of the stressor (although region-specific differences exist20) and seldom outlasts the duration of stressor exposure. Because monoamines generally act through G protein-coupled receptors, which rapidly transfer their activation to downstream effectors, the rapid rise in their level is quickly translated into altered functioning of neurons that express these receptors.
Each monoamine contributes to specific behavioural aspects of the immediate response to the stressor. For instance, increased levels of noradrenaline supposedly cause a shift from focused processing of sensory information to a more general scanning of the environment, which may provide better solutions for challenging conditions21. Dopamine, which is released during moderate stress in the prefrontal cortex15, is thought to improve risk assessment and decision strategies, and serotonin is instrumental in reducing post-stress anxiety22. Thus, monoamines collectively promote important behavioural strategies that help the animal face and survive the initial phase of a stressful event.
Neuropeptides
A number of neuropeptides are released by stress in specific neuronal populations and contribute, often by activating multiple receptors, to the stress response11,23-26. Although several of these stress-induced neuropeptides also act on peripheral receptors, we focus here on their release and actions in the brain (TABLE 1). We confine the discussion to CRH and vasopressin, although other peptides also mediate the effects of stress (orexin, ghrelin and dynorphin) or counteract the stress response under specific contexts (oxytocin and neuropeptide Y); additional peptides may regulate stress-related anxiety and dysphoria (for example, galanin and substance P)26.
Table 1.
Examples of stress-activated neuropeptides in the brain
| Peptide | Release location | Receptor (and receptor location) | Functions related to stress |
|---|---|---|---|
| CRH | Central amygdala30,34,35 | CRHR1 (basolateral amygdala)30,34,38,78 CRHR2 (REF. 74) |
Stress/emotional memory, anxiety30,33,35,80 Termination of stress-related anxiety31-33 |
| Locus coeruleus28 | CRHR1 (locus coeruleus)60,78 | Interaction with noradrenaline systems28 | |
| Hippocampal interneurons27,56 | CRHR1 (pyramidal cells)11,39,48,56,78 | Stress-related learning and memory3,36,39 | |
| BnST26,81 | CRHR1 (REFS 48,81) (nucleus accumbens)81 and CRHR2 (REFS 74,78,81) |
Stress-related anxiety26,30 | |
| Urocortin | Non-ganglionic Edinger–Westphal nucleus41 |
CRHR1 and CRHR2 | Interaction with CRH systems during acute and chronic stress |
| Urocortin 2 and 3 | CRHR2 (REF. 41) | Role in stress not established | |
| Vasopressin | Hypothalamic dendrites BnST and amygdala25,26,43 |
V1A (septum, hippocampus, BnST and other areas) and V1B |
Stress-related and social memory, and perhaps emotionality25,26,43 |
| Orexin | Lateral hypothalamic area26 | OX1 and OX2 | Stress-related energy and sleep homeostasis |
| Dynorphin | Hippocampal dendrites82 | Several opioid receptors26 | Stress-related dysphoria26 |
BnST, bed nucleus of the stria terminalis; CRH, corticotropin-releasing hormone; CRHR, CRH receptor; OX, orexin receptor; V1, vasopressin receptor 1.
The canonical stress-activated neuropeptides include the CRH family and vasopressin, which were first discovered in the hypothalamus24,25. CRH is released in response to stress from axon terminals in the hypothalamic median eminence and acts on receptors in the pituitary24. However, it is now known to also be expressed in neuronal populations in the amygdala24,26, the hippocampus27 and the locus coeruleus28. Acting locally, the peptide exerts neuromodulatory effects on target neurons within seconds after its release9,29,30, through two G protein-coupled receptors, CRHR1 and CRHR2. CRH receptor occupancy influences neuronal firing patterns9,29,30, gene expression11,26 and behaviour11,26,31-33, in a CRH dose- and context-dependent manner. For example, release of CRH in the central nucleus of the amygdala during acute stress enhances memory consolidation34,35, and CRH released by modest stress from hippocampal interneurons11,56 primes long-term potentiation36 and improves memory37. By contrast, hippocampal release of large amounts of CRH in response to severe stress38,39 can lead to hyperexcitability and seizures9,40 and to rapid loss of dendritic spines in CA3 pyramidal cells39. CRH might also contribute to the structural changes in hippocampal pyramidal cells that are provoked by chronic stress in both adult and developing brains4,12,38. Other members of the CRH neuropeptide family, including the urocortins (UCN1, 2 and 3), can bind to CRH receptors and act in distinct spatial domains. Thus, UCN1-expressing neurons in the non-preganglionic Edinger–Westphal nucleus in the brainstem might contribute to stress adaptation41.
In the hypothalamus, vasopressin interacts with CRH, promoting adrenocortico-tropic hormone (ACTH) release from the pituitary in response to stress42. In the brain as a whole, vasopressin acts on a wide array of neurons25,43 (TABLE 1): in the amygdala, the excitatory actions of vasopressin, perhaps released from dendrites25, might contribute to the behavioural stress response. Vasopressin might also modulate emotional memory and anxiety26.
The ‘activating’ actions of stress-induced CRH release are mediated primarily through binding to CRHR1 (REFS 11,28,33,39), and the panoply of effects described above take place in a time frame of seconds to minutes. In addition, studies in knockout mice suggest that CRHR2 binding could exert effects at longer timescales and might function to shut down the stress response31,32. The yin–yang functions of the two CRH receptors described above are remarkably similar to the roles of the two corticosteroid receptors in mediating the early and late effects of stress-induced corticosteroid release on neurons, as described below.
Steroids
Corticosteroids are secreted in a pulsatile and circadian fashion; stress triggers the release of a large burst of corticosteroids that is superimposed on these rhythms44. Corticosteroids' access to the brain is regulated by p-glycoproteins45 (however, see REF. 46), but the circadian pattern and the stress-induced surge of corticosteroid levels are maintained in the brain (as was recently demonstrated for the hippocampus47), allowing peripheral and central aspects of the stress response to be integrated. All brain cells, including glia, are in principle exposed to corticosteroid hormones — this contrasts with the spatially restricted pathways through which monoamines and peptides exert their actions. Nevertheless, the sites in the brain where corticosteroids are effective are restricted by the distribution of corticosteroid receptors.
In the mammalian brain, corticosteroid hormones primarily act through mineralocorticoid and glucocorticoid receptors (MRs and GRs, respectively). MRs have a high affinity for corticosteroids, so they are mostly occupied even when circulating corticosteroid levels are low2. GRs have tenfold lower affinity; consequently, these receptors are only partially occupied under basal conditions and become more occupied as corticosteroid levels increase — for example, after stress. The two receptor types are differentially distributed in brain2. MRs are highly expressed in neurons of the hippocampal formation and the lateral septum and moderately expressed in subnuclei of the amygdala, the hypothalamic paraventricular nucleus (PVN) and the locus coeruleus (FIG. 2). These regions define a circuit that is involved in the cognitive, emotional and neuroendocrine processing of stressful events5. The distribution of MRs thus overlaps with the distribution of CRHR1 (REF. 48). GRs are ubiquitously expressed in the brain, but they are enriched in the hippocampus, the lateral septum and the PVN.
Figure 2. ‘Hot spots’ of receptors for key stress mediators.

The β1-adrenoceptors for noradrenaline (B1Rs), CRH receptor 1 (CRHR1), CRHR2 and the mineralocorticoid and glucocorticoid receptors (MRs and GRs, respectively) cluster in ‘hot spots’ in the brain. These hot spots include the prefrontal cortex, specific amygdala nuclei, the hippocampus (CA1, CA3 and the dentate gyrus (DG)), the paraventricular nucleus of the hypothalamus (PVN), the dorsal raphe nuclei (DR) and the locus coeruleus (LC). In these areas, receptors for at least two classes of mediators are highly expressed. The hot spots are strategic hubs that connect networks involved in diverse aspects of the brain's stress response, including learning and memory, decision making and hormonal, autonomic and emotional responses. BLA, basolateral amygdala; BnST, bed nucleus of the stria terminalis; CeA, central amygdala; CRH, corticotropin-releasing hormone; lat. septum, lateral septum; MA, medial amygdala; NTS, nucleus tractus solitarii. Data from REFS 48,74–78.
On binding of the hormone, corticosteroid receptors translocate to the nucleus, where they act as regulators of gene transcription49. Therefore, steroid effects on neuronal function usually require at least an hour to develop and last hours to days. In the hippocampus, MR activation is a prerequisite for maintaining the ongoing information flow6, whereas activation of GRs — for example, after stress — causes a delayed suppression of neuronal excitability and synaptic plasticity6,50, thus providing ‘negative-feedback regulation’ of behavioural aspects of the stress response. Moreover, the suppression of synaptic plasticity by GRs — preventing new input (not related to the earlier stressful situation) being encoded — could serve to protect the stress-related information that is being consolidated after stress exposure. The slow, gene-mediated consequences of GR activation depend on the life history; for example, adverse early-life experiences or chronic stress in adulthood influence the number and the functioning of GRs in the hippocampus6,51. Preliminary evidence suggests that in the basolateral amygdala — unlike in the hippocampus — GR activation has lasting excitatory rather than suppressive and normalizing effects on cell firing52. This illustrates the fact that one mediator acting through one receptor type can have multiple effects, depending on the life history and cellular context.
Acting in concert
As described above, each of the stress mediators has its own preferred spatial and temporal domains of release and action. In principle, this allows the brain to deal with the entire range of stress-provoked challenges — from immediate attention and strategy decisions that are important for survival in the short term, to storage of information about the stressful situation in case similar events are encountered in the future, which is advantageous in the long term. However, the actions of the different stress mediators need to be orchestrated into a coordinated stress response. This is enabled by overlap in the spatial and temporal niches of action of the different mediators, which allows their actions to converge. In addition, there is increasing evidence of direct interactions between the individual stress mediators.
Spatial niches
The fact that stress mediators are released only in certain areas and act only on those neurons that express specific receptor subtypes was discussed above. Interestingly, different stress mediator receptors are often expressed in ‘hot spots’ of the brain, such as the prefrontal cortex, the amygdala, the hippocampus and the neurotransmitter synthesis areas (for example, the dorsal raphe nucleus and the locus coeruleus) (FIG. 2), consistent with the concept of spatial convergence of their actions53. The hot spots are strategic hubs that connect networks involved in different aspects of the stress response. For example, neurons in the basolateral amygdala, which are affected by noradrenaline, dopamine, CRH and corticosteroids, are instrumental for sympathetic and neuroendocrine activation (through their output to the central amygdala), for fine-tuning of arousal (through reciprocal connections with the locus coeruleus) and for the processing of emotional and contextual aspects of stress (through the prefrontal cortex and hippocampus). A second hot spot is the hippocampus, where individual neurons can express receptors for corticosteroids, neuropeptides, monoamines (including serotonin and noradrenaline) and opioids (FIG. 3), enabling the fine-tuning of neuronal firing in response to a range of signals. Thus, although each stress mediator has a characteristic and unique spatial domain, their functional contribution to the stress response is subject to integration with that of other mediators, through convergence on interconnected networks (FIG. 2).
Figure 3. Subcellular localization of stress-mediator receptors.

The actions of different stress mediators on a single neuron are orchestrated through the distinct subcellular localization of the relevant receptors. This is illustrated in this cartoon of a hypothetical pyramidal cell (which includes features that exist on either or both CA3 and CA1 principal cells). For example, corticotropin-releasing hormone receptor 1 (CRHR1) is located in somata as well as on dendrites and dendritic spines3,39,48,56, whereas CRHR2 seems to reside on the axon initial segment (Y. Chen, personal communication). In addition, monoamine receptors that terminate on hippocampal pyramidal cells reside on specific dendritic domains57,58. Note that the subcellular distribution patterns of each receptor might differ between brain areas. For instance, glucocorticoid receptors (GRs) might be present on postsynaptic (dendritic) domains in certain amygdala neurons59. 5-HT, 5-hydroxytryptamine; MR, mineralocorticoid receptors; NA, noradrenaline.
Here we carry this concept further, by proposing that both the spatial specificity and the integration of the effects of distinct neuromodulators apply even at the level of a single neuron: receptors that bind a given mediator are not only localized to specific neuronal populations but also reside in selective subcellular neuronal domains, providing exquisite spatial resolution to a mediator's effects. In other words, the site of release coupled with receptor localization provides a fine-resolution spatial specificity regarding the actions of stress mediators. This is illustrated in the hypothetical hippocampal pyramidal neuron shown in FIG. 3. In this neuron, cytoplasmic receptors for corticosteroids — the MRs and GRs that, on binding, translocate to the nucleus — are localized in the soma54, whereas the so-called membrane MRs are found in both the pre- and the postsynaptic membrane55. Similarly, CRHR1 is located in somata, on dendrites38,39 and even in the postsynaptic density of dendritic spines56, whereas CRHR2 seems to reside on the axon initial segment (Y. Chen, personal communication). In addition, monoamine receptors in hippocampal pyramidal cells reside on specific dendritic domains57,58. It should be noted that the subcellular distribution of receptors is brain region-specific. For example, dendritically located GRs have been found in the lateral amygdala59, and presynaptically located CRHR1 has been found in locus coeruleus neurons28,60. This exquisite spatial control of receptor localization ensures that the net sum of the effects of stress on the activity of a single neuron will be a composite of the local effects of the different stress mediators.
Temporal niches
Stress responses take place at timescales that span milliseconds to days. As discussed, the different modulators have classically been considered to act at distinctive temporal profiles, allowing both activation and termination of the stress response. In general, stress is assumed to cause an initial, acute wave of effects that is carried by monoamines and peptides, starting within seconds after the onset of a stressor61.
A second wave of ‘molecular’ effects of stress is typically discernible starting one to two hours after stress exposure. These molecular effects are mediated primarily by GRs (acting as transcription factors) and CRHR2 signalling and inhibit the lingering effects of the first wave of stress-induced actions. This restores allostasis (that is, homeostatic control with a varying setpoint (governed by demand), which is thought to achieve stability through change4) for both cognitive and neuroendocrine functions.
Importantly, recent evidence concerning the full range of actions of the diverse stress mediators has demonstrated that the mediators exert more subtle and context-specific actions outside their classical temporal niches, and that these can be better considered within the framework of three temporal (and functional) domains (BOX 1). For example, monoamines can induce synaptic responses within seconds of stressor onset but also, through a different signalling cascade, activate immediate early genes such as ARC; this extends their actions (for example, on memory formation) to a more delayed time domain62. The consequences of such a delayed pathway for stress responsiveness are particularly significant when the downstream targets include genes that encode catecholamine-synthesizing enzymes63, as this would amplify the original response. In another example, activation of CRHR1 can within seconds alter neuronal afterhyper polarization29 and phosphorylate the transcription factor CRE-binding protein (CREB)11. The downstream effects of CREB phosphorylation — that is, transcriptional regulation of CRE (cAMP-responsive element)-containing genes, including c-FOS and CRH — take place over hours. Depending on which genes' transcription is affected by CREB, this contributes to the activation and/or resolution of the stress response.
Box 1. Temporal profiles of the orchestrated stress response.
The stress response has classically been characterized by two temporal ‘waves’ of stress mediator actions. The first includes rapid actions of noradrenaline, serotonin, dopamine and corticotropin-releasing hormone (CRH) (the latter being predominantly mediated by CRH receptor 1). These rapid actions of stress mediators promote vigilance, alertness, appraisal of the situation and the choice of an optimal strategy to face the challenge. Because local increases in stress mediator levels are short-lived, and because their actions typically quickly subside, this first wave of events is not optimal for provoking the sustained, adaptive components of a stress response, such as the consolidation of information associated with (that is, the memory of) the stressor. This is instead accomplished through alterations of gene expression and cell function, classically considered the second wave and attributed to corticosteroids acting through glucocorticoid receptors.
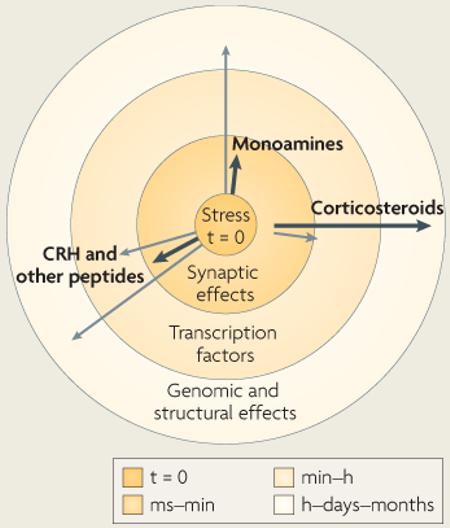
Recent work suggests that the temporal profile of the orchestrated stress response might fit better into a framework of three temporal domains that denote distinct mechanisms of operation of the stress mediators. For example, receptor activation by monoamines and peptides can, in addition to having rapid synaptic effects, regulate transcription factors (such as CRE-binding protein and AP1) within a seconds-to-minutes time frame3,11,63. This rapid activation (or deactivation) of transcription factors can lead to sustained genomic changes that help the organism to respond to stressors of different magnitudes and durations, as well as to recurrent stress. Conversely, corticosterone, through non-genomic pathways involving membrane-located mineralocorticoid receptors65, can act within minutes55. Taken together, these findings blur the classical distinction between rapid and delayed responses to stress.
In view of the above, the temporal action profiles of stress mediators might best be considered as a series of vectors commencing at the onset of stress, as shown in the figure. Each hormone has a preferred temporal vector (shown as black arrows) that is associated with a specific mode of cellular function (for example, synaptic or transcriptional). In addition, most modulators exert stress-related actions through alternative molecular and cellular pathways (shown as grey arrows), which take place in different temporal niches. The length of each vector in the figure corresponds to the temporal domain denoted by the concentric circles. Note that the non-genomic effects of corticosteroids develop slightly later than the rapid aminergic and peptidergic actions, because it takes time to transport the hormones from the adrenals (where they are secreted) to the brain. The combined actions of each stress mediator make up its compound temporal and functional profile.
Conversely, corticosteroids (which are classically thought to be slow-acting) also rapidly change brain functioning through non-genomic pathways — for example, in the hypothalamus64 and hippocampus65. In CA1 neurons, such rapid effects seem to be mediated by the binding of corticosterone to MRs in the pre- and postsynaptic plasma membrane55. The rapid MR activation facilitates increases in neuronal excitability by increasing the probability of glutamate release65, the trafficking of glutamate receptor subunits66 and the suppression of a K+ conductance55. Interestingly, the apparent affinity of this membrane MR for corticosterone is a tenth that of the nuclear MR65. This low-affinity MR supposedly enables the hippocampus to sense pulsatile corticosteroid fluctuations and, importantly, the surge in corticosterone levels that occurs during the initial response to stress67. The rapid actions of membrane MRs thus join those of other stress mediators like CRH and noradrenaline.
In summary, although each stress modulator contributes to the orchestrated stress response through a main role with a defined temporal niche, it is now clear that most modulators play additional, minor parts (BOX 1). These effects, which transcend the classical temporal niches, enable further gradation and integration of responses to stressors of diverse nature, magnitude and duration.
Direct interactions
Recently, several interactions between two or more stress mediators (and between stress mediators and other neurotransmitters) have emerged, showing that the total contribution of stress mediators to the stress response is more than just the sum of their individual effects (FIG. 4). For instance, the activity of noradrenergic neurons in the LC is modulated by interactions between CRH, a stress mediator, and opioids and glutamate so as to achieve optimal behavioural adaptation to a stressor28. Thus, as a result of stress exposure, LC noradrenergic cell firing shifts from a moderate, tonic activity that is permissive of a superimposed phasic activity to a high tonic firing that prevents phasic firing. A high ratio of phasic to tonic firing is optimal for responding to sensory stimuli and is associated with focused attention and maintenance of ongoing behaviour. By contrast, a low ratio, which results from stress exposure, is thought to promote arousal, scanning of the environment and decision making, thus increasing the chance of finding the optimal way to cope with the stressor21. Whereas glutamate is instrumental in enhancing the phasic firing, the stress-induced shift towards high tonic firing patterns is mediated by CRH projections from the central amygdala and by activation of CRHR1 in the rostrolateral dendritic zone of the LC28. In turn, opiates inhibit LC tonic firing by binding to κ- and μ-opioid receptors68. Thus, endogenous opioid release might be important for resetting the level and mode of LC noradrenergic neuron activity after stress, helping to terminate the central noradrenergic response to the stressor69.
Figure 4. Direct interaction between different stress mediators.

Noradrenaline (NA), corticotropin-releasing hormone (CRH), opioids and corticosterone (CORT) interact in the locus coeruleus (LC) and its projection areas (including the hippocampus and the amygdala) to orchestrate exquisite tuning of neuronal firing patterns in response to stress. Exposure to stress shifts LC noradrenergic cell firing — which is normally kept at a moderate level by glutamatergic input, here represented by the top left arrow — from a moderate tonic activity to high tonic firing that prevents phasic firing. This shift is mediated by CRH projections from the central amygdala on to CRH 1 receptors in the LC28. In turn, LC noradrenergic cells project to the basolateral amygdala (BLA), hippocampal CA1 and the dentate gyrus (DG). Here noradrenaline, released shortly after stress exposure, enhances excitability, promoting the encoding of stress-related information. Glutamatergic output from the BLA to the DG is thought to provide a means to ‘emotionally tag’ information processed in the hippocampus, thus rendering it preference in storage71. The stress-induced enhancement in activity in the LC, the BLA, the DG and CA1 (which in the DG is aided by rapid corticosteroid actions; see main text) is gradually reversed, resulting in a return to the pre-stress activity level. In the LC, the level of tonic firing is reduced by opiates that bind to κ- and μ-opioid receptors68. In the BLA, the DG and CA1, these gradual normalizing effects are exerted by corticosterone, presumably through glucocorticoid receptor-mediated gene-dependent cascades6,70,79. The + signs indicate that the stress mediator enhances cell firing, whereas the–signs indicate decreased cell activity. CeA, central amygdala.
In the projection areas of the LC, direct interactions between stress mediators also fine-tune the activation and termination of noradrenergic signalling. Thus, in the hippocampal dentate gyrus noradrenaline enhances excitability and synaptic plasticity, and this effect can be augmented by rapidly acting corticosterone70. Behavioural observations in combination with local pharmacological interventions have led to the conclusion that there is also synergy between noradrenaline and glucocorticoids in the rodent basolateral amygdala71. This synergy is thought to promote consolidation of emotional information72. However, when corticosterone acts through the genomic pathway it can also suppress noradrenergic activity. In this slow temporal domain, the steroid effectively suppresses the facilitation of information transfer by noradrenaline6,70. This suppression of activity is supported by other slowly developing corticosteroid–monoamine interactions, including the steroid-induced enhancement of hyperpolarizing effects that are mediated by the serotonin 1A receptor6.
There are additional examples of interactions between monoamines and the neuropeptide CRH34,73. In the basolateral amygdala, CRH might interact with β-adrenergic activation accomplished by endogenous monoamines34. Such interactions also take place in amygdala projection areas. For instance, glutamate transmission in the pathway from the basolateral amygdala to the medial prefrontal cortex — which is important for emotional processing and decision making — is depressed by dopamine activation of D1 or D5 receptors, and CRH (which on its own does not affect glutamate transmission) augments this depression73. Importantly, this interaction is context dependent; for example, chronic cocaine administration switches the synergistic actions of dopamine and CRH from suppressing to facilitating glutamate transmission.
Note that interactions between stress mediators can take place in a single temporal domain (for example, the interaction between CRH and glutamate) or between the classic temporal niches: CRH ‘primes’ hippocampal neurons for subsequent excita-tory neurotransmission36, and fast-acting monoamines might prime neurons for subsequent effects of stress-induced, slower-acting mediators. The mechanisms by which these interactions take place require further study.
These examples illustrate that the spatial convergence and temporal overlap of the various stress mediators enable multiple, direct and rapid interactions, resulting in synergistic actions that orchestrate the acute stress response. Longer-lasting or delayed interactions between stress mediators might promote the termination of the stress response.
Conclusions and future directions
The ability to change neuronal activity (and hence behaviour and cognition) both rapidly and enduringly in response to threatening challenges is crucial for survival and has thus resulted in a highly coordinated, complex and evolutionarily conserved stress-response system. Because different challenges require distinct responses (for example, social decisions or flight) that involve different sets of neuronal populations acting in concert or sequentially, the brain has evolved a system that can produce such diverse alterations in neuronal activity. This system consists of the stress mediators described in this Perspective, which not only occupy characteristic niches of time, space and function, but also are exquisitely coordinated at multiple levels to create an orchestrated stress-response symphony. This ‘stress neuro-symphony’ provides the capacity to generate precise, focused alterations in neuronal activity in response to stress signals. These alterations can range spatially from individual synapses up to the whole animal and temporally from milliseconds to days.
Although our understanding of the individual instruments of this stress neuro-symphony has increased substantially, more studies are needed to examine the additional actions of and interactions between the stress mediators. New tools allow us to interfere with selected aspects of a stress media-tor's signalling pathway in a site-specific and inducible manner, and therefore let us examine the mediator's relevance to cellular activity in freely moving animals. Genome-wide screening tools will enable better study of the full spectrum of each individual mediator's actions, and of the coordinated effects of these mediators on stress-related programmes of gene expression, which in turn influence behaviour in the long term. Ultimately, the roles that interactions between stress mediators have in orchestrating human cognitive performance will need to be investigated.
Finally, in view of the complexity of this stress neuro-symphony, it is not surprising that many of its components can be deranged, leading to stress-related disorders including depression and anxiety. An improved understanding of how this system functions will enable better use of current therapeutic interventions and the development of new clinically useful drugs.
Acknowledgements
The authors thank E. R. de Kloet and A. Korosi for critical reading of the text and help with the figures. Supported by NWO grant #91204042 (M.J.) and grants NS29012 and MH 73136 from the National Institute of Health (T.Z.B.).
Footnotes
Competing financial interests
The authors declare no competing financial interests.
Contributor Information
Marian Joëls, SILS-CNS, University of Amsterdam, Kruislaan 320, 1098 SM Amsterdam, the Netherlands..
Tallie Z. Baram, University of California, Irvine, Departments of Pediatrics and Anatomy & Neurobiology, Medical Sciences I, ZOT 4475, Irvine, California 92697-4475, USA.
References
- 1.Ulrich-Lai YM, Herman JP, et al. Neural regulation of endocrine and autonomic stress responses. Nature Rev. Neurosci. doi: 10.1038/nrn2647. in the press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Kloet ER, Joëls M, Holsboer F. Stress and the brain: from adaptation to disease. Nature Rev. Neurosci. 2005;6:463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- 3.Fenoglio KA, Brunson KL, Baram TZ. Hippocampal neuroplasticity induced by early-life stress: functional and molecular aspects. Front. Neuroendocrinol. 2006;27:180–192. doi: 10.1016/j.yfrne.2006.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol. Rev. 2007;87:873–904. doi: 10.1152/physrev.00041.2006. [DOI] [PubMed] [Google Scholar]
- 5.McGaugh JL. The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu. Rev. Neurosci. 2004;27:1–28. doi: 10.1146/annurev.neuro.27.070203.144157. [DOI] [PubMed] [Google Scholar]
- 6.Joëls M, Karst H, Krugers HJ, Lucassen PJ. Chronic stress: implications for neuronal morphology, function and neurogenesis. Front. Neuroendocrinol. 2007;28:72–96. doi: 10.1016/j.yfrne.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 7.Lupien SJ, et al. Stress hormones and human memory function across the lifespan. Psychoneuroendocrinology. 2005;30:225–242. doi: 10.1016/j.psyneuen.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 8.Shors TJ. Stressful experience and learning across the lifespan. Annu. Rev. Psychol. 2006;57:55–85. doi: 10.1146/annurev.psych.57.102904.190205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baram TZ, Hatalski CG. Neuropeptide-mediated excitability: a key triggering mechanism for seizure generation in the developing brain. Trends Neurosci. 1998;21:471–476. doi: 10.1016/s0166-2236(98)01275-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rice CJ, Sandman CA, Lenjavi MR, Baram TZ. A novel mouse model for acute and long-lasting consequences of early life stress. Endocrinology. 2008;149:4892–4900. doi: 10.1210/en.2008-0633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y, Fenoglio KA, Dube CM, Grigoriadis DE, Baram TZ. Cellular and molecular mechanisms of hippocampal activation by acute stress are age-dependent. Mol. Psychiatry. 2006;11:992–1002. doi: 10.1038/sj.mp.4001863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brunson KL, et al. Mechanisms of late-onset cognitive decline after early-life stress. J. Neurosci. 2005;25:9328–9338. doi: 10.1523/JNEUROSCI.2281-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maier SF, Watkins LR. Stressor controllability and learned helplessness: the roles of the dorsal raphe nucleus, serotonin, and corticotropin-releasing factor. Neurosci. Biobehav. Rev. 2005;29:829–841. doi: 10.1016/j.neubiorev.2005.03.021. [DOI] [PubMed] [Google Scholar]
- 14.Morilak DA, et al. Role of brain norepinephrine in the behavioral response to stress. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2005;29:1214–1224. doi: 10.1016/j.pnpbp.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 15.Goto Y, Otani S, Grace AA. The yin and yang of dopamine release: a new perspective. Neuropharmacology. 2007;53:583–587. doi: 10.1016/j.neuropharm.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Linthorst AC, Reul JM. Stress and the brain: solving the puzzle using microdialysis. Pharmacol. Biochem. Behav. 2008;90:163–173. doi: 10.1016/j.pbb.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 17.Mitsushima D, Yamada K, Takase K, Funabashi T, Kimura F. Sex differences in the basolateral amygdala: the extracellular levels of serotonin and dopamine, and their responses to restraint stress in rats. Eur. J. Neurosci. 2006;24:3245–3254. doi: 10.1111/j.1460-9568.2006.05214.x. [DOI] [PubMed] [Google Scholar]
- 18.Piazza PV, et al. Glucocorticoids have state-dependent stimulant effects on the mesencephalic dopaminergic transmission. Proc. Natl Acad. Sci. USA. 1996;93:8716–8720. doi: 10.1073/pnas.93.16.8716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amat J, et al. Medial prefrontal cortex determines how stressor controllability affects behavior and dorsal raphe nucleus. Nature Neurosci. 2005;8:365–371. doi: 10.1038/nn1399. [DOI] [PubMed] [Google Scholar]
- 20.Jackson ME, Moghaddam B. Stimulus-specific plasticity of prefrontal cortex dopamine neurotransmission. J. Neurochem. 2004;88:1327–1334. doi: 10.1046/j.1471-4159.2003.02205.x. [DOI] [PubMed] [Google Scholar]
- 21.Aston-Jones G, Cohen JD. An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu. Rev. Neurosci. 2005;28:403–450. doi: 10.1146/annurev.neuro.28.061604.135709. [DOI] [PubMed] [Google Scholar]
- 22.Adamec R, Holmes A, Blundell J. Vulnerability to lasting anxiogenic effects of brief exposure to predator stimuli: sex, serotonin and other factors-relevance to PTSD. Neurosci. Biobehav. Rev. 2008;32:1287–1292. doi: 10.1016/j.neubiorev.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathew SJ, Price RB, Charney DS. Recent advances in the neurobiology of anxiety disorders: implications for novel therapeutics. Am. J. Med. Genet. C Semin. Med. Genet. 2008;148:89–98. doi: 10.1002/ajmg.c.30172. [DOI] [PubMed] [Google Scholar]
- 24.Swanson LW, Sawchenko PE, Rivier J, Vale WW. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology. 1983;36:165–186. doi: 10.1159/000123454. [DOI] [PubMed] [Google Scholar]
- 25.Landgraf R, Neumann ID. Vasopressin and oxytocin release within the brain: a dynamic concept of multiple and variable modes of neuropeptide communication. Front. Neuroendocrinol. 2004;25:150–176. doi: 10.1016/j.yfrne.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 26.Koob GF. A role for brain stress systems in addiction. Neuron. 2008;59:11–34. doi: 10.1016/j.neuron.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y, Bender RA, Frotscher M, Baram TZ. Novel and transient populations of corticotropin-releasing hormone-expressing neurons in developing hippocampus suggest unique functional roles: a quantitative spatiotemporal analysis. J. Neurosci. 2001;21:7171–7181. doi: 10.1523/JNEUROSCI.21-18-07171.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valentino RJ, Van Bockstaele E. Convergent regulation of locus coeruleus activity as an adaptive response to stress. Eur. J. Pharmacol. 2008;583:194–203. doi: 10.1016/j.ejphar.2007.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aldenhoff JB, Gruol DL, Rivier J, Vale W, Siggins GR. Corticotropin releasing factor decreases postburst hyperpolarizations and excites hippocampal neurons. Science. 1983;221:875–877. doi: 10.1126/science.6603658. [DOI] [PubMed] [Google Scholar]
- 30.Gallagher JP, Orozco-Cabal LF, Liu J, Shinnick-Gallagher P. Synaptic physiology of central CRH system. Eur. J. Pharmacol. 2008;583:215–225. doi: 10.1016/j.ejphar.2007.11.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coste SC, et al. Abnormal adaptations to stress and impaired cardiovascular function in mice lacking corticotropin-releasing hormone receptor-2. Nature Genet. 2000;24:403–409. doi: 10.1038/74255. [DOI] [PubMed] [Google Scholar]
- 32.Bale TL, et al. Mice deficient for corticotropin-releasing hormone receptor-2 display anxiety-like behaviour and are hypersensitive to stress. Nature Genet. 2000;24:410–414. doi: 10.1038/74263. [DOI] [PubMed] [Google Scholar]
- 33.Muller MB, et al. Limbic corticotropin-releasing hormone receptor 1 mediates anxiety-related behavior and hormonal adaptation to stress. Nature Neurosci. 2003;6:1100–1107. doi: 10.1038/nn1123. [DOI] [PubMed] [Google Scholar]
- 34.Roozendaal B, Brunson KL, Holloway BL, McGaugh JL, Baram TZ. Involvement of stress-released corticotropin-releasing hormone in the basolateral amygdala in regulating memory consolidation. Proc. Natl Acad. Sci. USA. 2002;99:13908–13913. doi: 10.1073/pnas.212504599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Merali Z, Khan S, Michaud DS, Shippy SA, Anisman H. Does amygdaloid corticotropin-releasing hormone (CRH) mediate anxiety-like behaviors? Dissociation of anxiogenic effects and CRH release. Eur. J. Neurosci. 2004;20:229–239. doi: 10.1111/j.1460-9568.2004.03468.x. [DOI] [PubMed] [Google Scholar]
- 36.Blank T, Nijholt I, Eckart K, Spiess J. Priming of long-term potentiation in mouse hippocampus by corticotropin-releasing factor and acute stress: implications for hippocampus-dependent learning. J. Neurosci. 2002;22:3788–3794. doi: 10.1523/JNEUROSCI.22-09-03788.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang HL, Wayner MJ, Chai CY, Lee EH. Corticotrophin-releasing factor produces a long-lasting enhancement of synaptic efficacy in the hippocampus. Eur. J. Neurosci. 1998;10:3428–3437. doi: 10.1046/j.1460-9568.1998.00352.x. [DOI] [PubMed] [Google Scholar]
- 38.Chen Y, et al. Modulation of dendritic differentiation by corticotropin-releasing factor in the developing hippocampus. Proc. Natl Acad. Sci. USA. 2004;101:15782–15787. doi: 10.1073/pnas.0403975101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Y, Dube CM, Rice CJ, Baram TZ. Rapid loss of dendritic spines after stress involves derangement of spine dynamics by corticotropin-releasing hormone. J. Neurosci. 2008;28:2903–2911. doi: 10.1523/JNEUROSCI.0225-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ehlers CL, et al. Corticotropin releasing factor produces increases in brain excitability and convulsive seizures in rats. Brain Res. 1983;278:332–336. doi: 10.1016/0006-8993(83)90266-4. [DOI] [PubMed] [Google Scholar]
- 41.Kozicz T. On the role of urocortin 1 in the non-preganglionic Edinger-Westphal nucleus in stress adaptation. Gen. Comp. Endocrinol. 2007;153:235–240. doi: 10.1016/j.ygcen.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 42.Harbuz MS, et al. Paradoxical responses of hypothalamic corticotropin-releasing factor (CRF) messenger ribonucleic acid (mRNA) and CRF-41 peptide and adenohypophysial proopiomelanocortin mRNA during chronic inflammatory stress. Endocrinology. 1992;130:1394–1400. doi: 10.1210/endo.130.3.1537299. [DOI] [PubMed] [Google Scholar]
- 43.Raggenbass M. Overview of cellular electrophysiological actions of vasopressin. Eur. J. Pharmacol. 2008;583:243–254. doi: 10.1016/j.ejphar.2007.11.074. [DOI] [PubMed] [Google Scholar]
- 44.Young EA, Abelson J, Lightman SL. Cortisol pulsatility and its role in stress regulation and health. Front. Neuroendocrinol. 2004;25:69–76. doi: 10.1016/j.yfrne.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 45.Karssen AM, et al. Multidrug resistance P-glycoprotein hampers the access of cortisol but not of corticosterone to mouse and human brain. Endocrinology. 2001;142:2686–2694. doi: 10.1210/endo.142.6.8213. [DOI] [PubMed] [Google Scholar]
- 46.Mason BL, Pariante CM, Thomas SA. A revised role for p-glycoprotein in the brain distribution of dexamethasone, cortisol, and corticosterone in wild type and ABCB1A/B-deficient mice. Endocrinology. 2008;149:5244–5253. doi: 10.1210/en.2008-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Droste SK, et al. Corticosterone levels in the brain show a distinct ultradian rhythm but a delayed response to forced swim stress. Endocrinology. 2008;149:3244–3253. doi: 10.1210/en.2008-0103. [DOI] [PubMed] [Google Scholar]
- 48.Chen Y, Brunson KL, Muller MB, Cariaga W, Baram TZ. Immunocytochemical distribution of corticotropin-releasing hormone receptor type-1 (CRF1)-like immunoreactivity in the mouse brain: light microscopy analysis using an antibody directed against the C-terminus. J. Comp. Neurol. 2000;420:305–323. doi: 10.1002/(sici)1096-9861(20000508)420:3<305::aid-cne3>3.0.co;2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu NZ, et al. International Union of Pharmacology. LXV. The pharmacology and classification of the nuclear receptor superfamily: glucocorticoid, mineralocorticoid, progesterone, and androgen receptors. Pharmacol. Rev. 2006;58:782–797. doi: 10.1124/pr.58.4.9. [DOI] [PubMed] [Google Scholar]
- 50.Kim JJ, Diamond DM. The stressed hippocampus, synaptic plasticity and lost memories. Nature Rev. Neurosci. 2002;3:453–462. doi: 10.1038/nrn849. [DOI] [PubMed] [Google Scholar]
- 51.Champagne DL, et al. Maternal care and hippocampal plasticity: evidence for experience-dependent structural plasticity, altered synaptic functioning, and differential responsiveness to glucocorticoids and stress. J. Neurosci. 2008;28:6037–6045. doi: 10.1523/JNEUROSCI.0526-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duvarci S, Paré D. Glucocorticoids enhance the excitability of principal basolateral amygdala neurons. J. Neurosci. 2007;27:4482–4491. doi: 10.1523/JNEUROSCI.0680-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bloom FE. The functional significance of neurotransmitter diversity. Am. J. Physiol. 1984;246:C184–C194. doi: 10.1152/ajpcell.1984.246.3.C184. [DOI] [PubMed] [Google Scholar]
- 54.Nishi M, Ogawa H, Ito T, Matsuda KI, Kawata M. Dynamic changes in subcellular localization of mineralocorticoid receptor in living cells: in comparison with glucocorticoid receptor using dual-color labeling with green fluorescent protein spectral variants. Mol. Endocrinol. 2001;15:1077–1092. doi: 10.1210/mend.15.7.0659. [DOI] [PubMed] [Google Scholar]
- 55.Olijslagers JE, et al. Rapid changes in hippocampal CA1 pyramidal cell function via pre- as well as postsynaptic membrane mineralocorticoid receptors. Eur. J. Neurosci. 2008;27:2542–2550. doi: 10.1111/j.1460-9568.2008.06220.x. [DOI] [PubMed] [Google Scholar]
- 56.Chen Y, et al. Hippocampal corticotropin releasing hormone: pre- and postsynaptic location and release by stress. Neuroscience. 2004;126:533–540. doi: 10.1016/j.neuroscience.2004.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Swanson LW, Kohler C, Björklund A. In: Handbook of Chemical Neuroanatomy. Björklund A, Hökfelt T, Swanson LW, editors. Vol. 5. Elsevier; Amsterdam; 1987. pp. 125–227. [Google Scholar]
- 58.Oleskevich S, Descarries L, Lacaille JC. Quantified distribution of the noradrenaline innervation in the hippocampus of adult rat. J. Neurosci. 1989;9:3803–3815. doi: 10.1523/JNEUROSCI.09-11-03803.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johnson LR, Farb C, Morrison JH, McEwen BS, LeDoux JE. Localization of glucocorticoid receptors at postsynaptic membranes in the lateral amygdala. Neuroscience. 2005;136:289–299. doi: 10.1016/j.neuroscience.2005.06.050. [DOI] [PubMed] [Google Scholar]
- 60.Reyes BA, Valentino RJ, Van Bockstaele EJ. Stress-induced intracellular trafficking of corticotropin-releasing factor receptors in rat locus coeruleus neurons. Endocrinology. 2008;149:122–130. doi: 10.1210/en.2007-0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Joëls M, Pu Z, Wiegert O, Krugers HJ. Learning under stress: how does it work? Trends Cogn. Sci. 2006;10:152–158. doi: 10.1016/j.tics.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 62.McIntyre CK, et al. Memory-influencing intrabasolateral amygdala drug infusions modulate expression of Arc protein in the hippocampus. Proc. Natl Acad. Sci. USA. 2005;102:10718–10723. doi: 10.1073/pnas.0504436102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sabban EL, Kvetnansky R. Stress-triggered activation of gene expression in catecholaminergic systems: dynamics of transcriptional events. Trends Neurosci. 2001;24:91–98. doi: 10.1016/s0166-2236(00)01687-8. [DOI] [PubMed] [Google Scholar]
- 64.Tasker JG, Di S, Malcher-Lopes R. Minireview: rapid glucocorticoid signaling via membrane-associated receptors. Endocrinology. 2006;147:5549–5556. doi: 10.1210/en.2006-0981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karst H, et al. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc. Natl Acad. Sci. USA. 2005;102:19204–19207. doi: 10.1073/pnas.0507572102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Groc L, Choquet D, Chaouloff F. The stress hormone corticosterone conditions AMPAR surface trafficking and synaptic potentiation. Nature Neurosci. 2008;11:868–870. doi: 10.1038/nn.2150. [DOI] [PubMed] [Google Scholar]
- 67.Joëls M, Karst H, DeRijk R, de Kloet ER. The coming out of the brain mineralocorticoid receptor. Trends Neurosci. 2008;31:1–7. doi: 10.1016/j.tins.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 68.Kreibich A, et al. Presynaptic inhibition of diverse afferents to the locus ceruleus by κ-opiate receptors: a novel mechanism for regulating the central norepinephrine system. J. Neurosci. 2008;28:6516–6525. doi: 10.1523/JNEUROSCI.0390-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Curtis AL, Bello NT, Valentino RJ. Evidence for functional release of endogenous opioids in the locus ceruleus during stress termination. J. Neurosci. 2001;21:RC152. doi: 10.1523/JNEUROSCI.21-13-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pu Z, Krugers HJ, Joëls M. Corticosterone time-dependently modulates beta-adrenergic effects on long-term potentiation in the hippocampal dentate gyrus. Learn. Mem. 2007;14:359–367. doi: 10.1101/lm.527207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Akirav I, Richter-Levin G. Mechanisms of amygdala modulation of hippocampal plasticity. J. Neurosci. 2002;22:9912–9921. doi: 10.1523/JNEUROSCI.22-22-09912.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roozendaal B, Okuda S, de Quervain DJ, McGaugh JL. Glucocorticoids interact with emotion-induced noradrenergic activation in influencing different memory functions. Neuroscience. 2006;138:901–910. doi: 10.1016/j.neuroscience.2005.07.049. [DOI] [PubMed] [Google Scholar]
- 73.Orozco-Cabal L, et al. Dopamine and corticotropin-releasing factor synergistically alter basolateral amygdala-to-medial prefrontal cortex synaptic transmission: functional switch after chronic cocaine administration. J. Neurosci. 2008;28:529–542. doi: 10.1523/JNEUROSCI.2666-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chalmers DT, Lovenberg TW, De Souza EB. Localization of novel corticotropin-releasing factor receptor (CRF2) mRNA expression to specific subcortical nuclei in rat brain: comparison with CRF1 receptor mRNA expression. J. Neurosci. 1995;15:6340–6350. doi: 10.1523/JNEUROSCI.15-10-06340.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rainbow TC, Parsons B, Wolfe BB. Quantitative autoradiography of β1- and β2-adrenergic receptors in rat brain. Proc. Natl Acad. Sci. USA. 1984;81:1585–1589. doi: 10.1073/pnas.81.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reul JMHM, de Kloet ER. Anatomical resolution of two types of corticosterone receptor sites in rat brain with in vitro autoradiography and computerized image analysis. J. Steroid Biochem. 1986;24:269–272. doi: 10.1016/0022-4731(86)90063-4. [DOI] [PubMed] [Google Scholar]
- 77.Härfstrand A, et al. Glucocorticoid receptor immunoreactivity in monoaminergic neurons of rat brain. Proc. Natl Acad. Sci. USA. 1986;83:9779–9783. doi: 10.1073/pnas.83.24.9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Van Pett K, et al. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J. Comp. Neurol. 2000;428:191–212. doi: 10.1002/1096-9861(20001211)428:2<191::aid-cne1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 79.Pu Z, Krugers HJ, Joëls M. Beta-adrenergic facilitation of synaptic plasticity in the rat basolateral amygdala in vitro is gradually reversed by corticosterone. Learn. Mem. 2009;16:155–160. doi: 10.1101/lm.1272409. [DOI] [PubMed] [Google Scholar]
- 80.Avishai-Eliner S, Brunson KL, Sandman CA, Baram TZ. Stressed-out, or in (utero)? Trends Neurosci. 2002;25:518–524. doi: 10.1016/s0166-2236(02)02241-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lim MM, et al. CRF receptors in the nucleus accumbens modulate partner preference in prairie voles. Horm. Behav. 2007;51:508–515. doi: 10.1016/j.yhbeh.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chavkin C. Dynorphins are endogenous opioid peptides released from granule cells to act neurohumorally and inhibit excitatory neurotransmission in the hippocampus. Prog. Brain Res. 2000;125:363–367. doi: 10.1016/S0079-6123(00)25025-5. [DOI] [PubMed] [Google Scholar]