

Abstract
Nitric-oxide synthases (NOS) are highly regulated heme-thiolate enzymes that catalyze two oxidation reactions that sequentially convert the substrate l-Arg first to Nω-hydroxyl-l-arginine and then to l-citrulline and nitric oxide. Despite numerous investigations, the detailed molecular mechanism of NOS remains elusive and debatable. Much of the dispute in the various proposed mechanisms resides in the uncertainty concerning the number and sources of proton transfers. Although specific protonation events are key features in determining the specificity and efficiency of the two catalytic steps, little is known about the role and properties of protons from the substrate, cofactors, and H-bond network in the vicinity of the heme active site. In this study, we have investigated the role of the acidic proton from the l-Arg guanidinium moiety on the stability and reactivity of the ferrous heme-oxy complex intermediate by exploiting a series of l-Arg analogues exhibiting a wide range of guanidinium pKa values. Using electrochemical and vibrational spectroscopic techniques, we have analyzed the effects of the analogues on the heme, including characteristics of its proximal ligand, heme conformation, redox potential, and electrostatic properties of its distal environment. Our results indicate that the substrate guanidinium pKa value significantly affects the H-bond network near the heme distal pocket. Our results lead us to propose a new structural model where the properties of the guanidinium moiety finely control the proton transfer events in NOS and tune its oxidative chemistry. This model may account for the discrepancies found in previously proposed mechanisms of NOS oxidation processes.
Keywords: Enzymes/Catalysis, Enzymes/Heme, Enzymes/Mechanisms, Enzymes/Nitric-oxide Synthase, Methods/FTIR, Methods/Raman Spectroscopy
Introduction
Nitric oxide (NO) 2 is a ubiquitous physiological mediator involved in a large number of signaling processes ranging from neural communication to vascular tone regulation (1–3). NO is synthesized in mammals by a family of highly regulated enzymes called NO synthase (NOS) that exist in three known isoforms (neuronal, endothelial, and inducible). In the last decade, however, NOS have been increasingly linked to oxidative stress phenomena and to the development of several pathological conditions, such as cardiovascular and neurodegenerative diseases (4–7). For this reason, the elucidation of the intricate NOS mechanism has urgently become a major challenge for the biomedical research community. Since their discovery in the early 1990s (8–12), NOS have been the focus of numerous structure-function investigations that quickly yielded valuable information concerning NOS catalytic activity (13–15) as follows: NOS consist of homodimeric hemoproteins, whose monomers contain an NH2-terminal oxygenase domain and a COOH-terminal reductase domain (16). The oxygenase domain harbors the catalytic active site, which includes a proximal cysteine-bound protoporphyrin IX heme prosthetic group, the l-Arg substrate, and the crucial redox-active cofactor (6R)-5,6,7,8-tetrahydro-l-biopterin (H4B) cofactor (17). The reductase domain provides electrons from NADPH to the heme group via two intermediate flavins, FAD and FMN (18–20). The oxygenase and reductase domains are linked together by a calmodulin-binding subunit that triggers the above electron transfer events based on local increases of Ca2+ concentrations (21, 22). NOS catalyze the activation of dioxygen (O2) to ultimately convert the substrate l-Arg to l-citrulline and NO via two oxidation steps with the formation of Nω-hydroxyl-l-arginine (NOHA) as an intermediate (Scheme 1) (23). Despite numerous structure-function investigations (reviewed in Ref. 24), the detailed molecular mechanism of NOS chemistry remains a subject of controversy. By analogy with the accepted cytochrome P450 mechanism, it has been proposed that the first step of the NOS mechanism (i.e. the hydroxylation of l-Arg to NOHA) would involve the following steps (see Scheme 2) (25). In the NOS resting state, the ferric (FeIII) heme is initially reduced to the ferrous (FeII) state by one electron provided by the reductase domain. Dioxygen binding to the ferrous heme then leads to the formation of a ferrous-dioxygen (FeII-O2) complex that is isoelectronic with a ferric-superoxo FeIII-OO˙̄ species (Scheme 2). To avoid the autoxidation of the heme FeII-O2 species (i.e. formation of heme FeIII and the release of free superoxide O2˙̄), and thus the uncoupling of electron transfer from the reductase domain, the H4B cofactor should rapidly provide an electron to the ferrous FeII-O2 species to promote the formation of a heme ferric-peroxo FeIII-OO− species (24, 26). The subsequent double protonation of this latter peroxo species would trigger heterolytic cleavage of the O–O bond resulting in an oxo-ferryl species (Por+-FeIV=O) (25) believed to be responsible for the hydroxylation of the guanidine moiety of l-Arg to NOHA (24–26). The second catalytic step (oxidation of NOHA) is believed to also involve the formation of the ferric-peroxo FeIII-OO− species (27, 28), as described above, but at this point there ensues a nucleophilic attack of the peroxo group upon the NOHA hydroxyguanidinium carbon atom followed by a rearrangement of the resulting tetrahedral complex, ultimately leading to the release of NO (24, 29). Although this analogous P450 model has been the working paradigm for the NOS mechanism, alternative models have been proposed (30–34) to address serious deficiencies. The main discrepancy between all the putative models proposed so far resides in the nature of the oxidative species, which directly results from differences in the proposed sequences of electron and proton transfer (30, 32–35). However, on top of controlling the specificity of NOS oxidative chemistry, the nature of electron and proton transfer events determines NOS catalytic efficiency, leading either to the specific formation of NO or to the release of other reactive oxygen and/or nitrogen species (ROS/RNS). NOS isoforms indeed have the capacity to generate ROS such as superoxide anion (O2˙̄) and hydrogen peroxide (H2O2) when electron and proton transfer processes are ineffective in promoting oxygen activation. As a result, the futile decay of reaction intermediates leads to the release of either O2˙̄ or H2O2. Failed electron and proton transfer can also directly generate RNS by tunneling NOS catalytic cycle toward an unproductive reaction intermediate such as the ferrous heme-nitric oxide complex, whose oxidation can lead to peroxynitrite production (36). The differences in the pKa values of the Nω(H) guanidinium proton of l-Arg and NOHA could modify the characteristics of the proton transfer processes, which might in turn account for the catalytic differences between the first and second steps (37–39) and regulate the nature of NOS catalytic production. However, despite the crucial role of proton transfer in NOS catalysis, little is known about the role of the guanidinium moiety of the NOS substrates, and about the H-bond network surrounding the dioxygen ligand.
SCHEME 1.

Oxidation of l-Arg to NO and citrulline catalyzed by NOS. Step 1, oxidation of l-Arg into Nω-hydroxy-l-arginine consumes two electrons. Step 2, oxidation of NOHA into citrulline and NO requires only one electron.
SCHEME 2.

Proposed mechanism for the first step of the oxidation of l-Arg by NOS and formation of reactive species. After reduction of the native ferric heme, dioxygen binding to the ferrous species leads to a ferrous-dioxygen complex isoelectronic with a ferric-superoxo complex. Autoxidation of this intermediate restores the resting ferric heme with release of superoxide anion (O2˙̄). A rapid electron transfer from the H4B cofactor leads to the build up of a ferric-peroxo complex, the last identified intermediate. A first protonation of this species yields a ferric-hydroperoxo complex. A second protonation of the terminal oxygen atom results in the heterolytic cleavage of the peroxide O–O bond generating the active oxo-ferryl intermediate, responsible for substrate oxidation. By contrast, protonation of the proximal oxygen atom would leads to the regeneration of the ferric heme and to the release of hydrogen peroxide (H2O2).
We have been studying NOS catalytic chemistry for a large series of substrate analogues that were originally designed as alternative, exogenous NO-producing substrates of NOS (40–42). Our results have shown that NOS can catalyze the formation of NO by the oxidation of specific l-Arg analogues, mostly non-amino acid guanidines (42). However, the NOS chemistry seems to vary as a function of the substrate. First, the stability and reactivity of the heme FeII-O2 species depend on the nature of the l-Arg analogue bound at the active site (41, 43). Second, the NADPH/NO ratio was shown to dramatically increase in the presence of these guanidine analogues (40, 43). Finally, the NOS catalytic products were extremely diverse, ranging from the specific and efficient release of NO to the predominant production of various ROS (40, 43). Consequently, the differences in the physicochemical properties of the guanidine analogues appeared to significantly modify and even alter the NOS catalytic mechanism (41, 43). Hence, these analogues represent a good tool to get new information on the NOS molecular mechanism and on the parameters that control the balance between NO and ROS/RNS production.
In this context, we are elucidating the specific role of the guanidinium proton of the NOS substrate in the regulation of the heme distal H-bond network and in the control of NOS chemistry. Specifically, we wish to analyze the role of the interaction between the heme FeII-O2 complex and its distal environment in determining the chemistry of the first and second catalytic steps. For this purpose, we chose a series of l-Arg analogues that exhibit guanidinium groups with different pKa values. Using a combination of vibrational spectroscopies and spectroelectrochemistry, we have examined the effect of these analogues on the structural properties of the heme porphyrin ring, on the heme redox properties, and on the electrostatic properties of the proximal ligand. Focusing on the interaction between the heme FeII-O2 species and its distal environment, we have used the stable mimic species ferrous heme-carbon monoxide (FeII-CO) as an electrostatic probe (44, 45) in combination with resonance Raman (RR) and FTIR spectroscopies to analyze the effects of the analogues on the FeII-CO vibrational modes. Our results lead us to propose a new model for the interaction between the FeII-O2 complex and its distal environment and to assess the role of the surrounding H-bond network in the control of NOS oxidative chemistry.
EXPERIMENTAL PROCEDURES
Chemicals
H4B was obtained from Schircks Laboratory (Jona, Switzerland). Chemicals and reagents of the highest grade commercially available were obtained from Aldrich, Fluka, or Janssen. CO gas was purchased from Messer (Messer France SA, France). The hydrochloride salts of 4,4,4-trifluorobutylguanidine (CF3-(CH2)3-Gua) 1, 4-fluorobutylguanidine (CH2F-(CH2)3-Gua) 2, n-pentylguanidine (CH3-(CH2)4-Gua) 3, cyclopropylguanidine (cyclopropyl-Gua) 4, 4-methoxyphenylguanidine (CH3OPh-Gua) 5, 4-fluorophenylguanidine (FPh-Gua) 6, 4-chlorophenylguanidine (ClPh-Gua) 7, 4-trifluoromethylphenylguanidine (CF3Ph-Gua) 8, and 4-nitrophenylguanidine (NO2Ph-Gua) 9 have been synthesized following general procedures from commercially available amines (46). Their physicochemical characteristics have been described previously (40). See Fig. 1 for structures.
FIGURE 1.

Structure and measured pKa values of all studied l-Arg analogues.
Enzyme Preparation
Mouse inducible NOS oxygenase domain (iNOSoxy) containing a six-histidine tag at its COOH terminus was expressed in Escherichia coli BL21 using the PCWori vector and purified as already described with H4B but without l-Arg (47, 48). It displayed all the spectroscopic properties of the full-length iNOS, and its His6 tag does not modify its reactivity. Its concentration was determined from the visible absorbance at 444 nm of the heme FeII-CO complex using an extinction coefficient of 76 mm−1·cm−1.
pKa Determinations
The direct measure of the pKa in water of a weak acid during its titration by sodium hydroxide is a suitable method to identify pKa up to 11. Identification of the pKa of the arylguanidines was thus achieved by simultaneously monitoring pH and electric conductivity of a solution of 10–20 mm of the guanidinium salt during its titration by 1 m NaOH solution. A small amount of HCl (∼5 mm final) was added to the initial solution to determine more precisely the beginning point of the titration of the guanidinium. The equivalent points of the titration were determined at the intersections of the conductivity straight lines. The pKa value of the guanidine was identified as the pH value at the semi-equivalent point of the titration. In the case of the alkylguanidines, direct measurement could not be achieved. Indeed, their pKa values were expected to be higher than 11, and beyond this value, the glass electrode is no longer reliable due to alkaline error. We thus used the Hammett Correlation (49) method that has been already used for different types of guanidines (50, 51). We fitted the values measured for the arylguanidines (52) and the reference value for l-Arg (53) with the Hammett field parameter σI, and we applied the obtained correlation to the alkylguanidines. We thus obtained extrapolated values for the pKa value of the alkylguanidines.
Spectroelectrochemistry
UV-visible mediated spectroelectrochemical titration of iNOSoxy was performed in a homemade two-compartment bulk electrolysis cell as described elsewhere (54). The working electrode was a gold grid; the auxiliary electrode was a platinum wire, and the reference electrode was a DRIREF-2 Ag/AgCl/KCl 3M (World Precision Instruments, E0 = 0.210 V versus normal hydrogen electrode, T = 20 °C). All potentials are given versus normal hydrogen electrode. The cell was maintained under positive argon pressure during the entire experiment and kept at a constant temperature, 20 °C. Samples for the redox titration experiments were prepared in 100 mm KCl, 100 mm NaPi buffer (pH 7.4) with combinations of l-Arg (5 mm) or substituted guanidines 1–9 (20 mm) and H4B (400 μm). The concentrations of these species were chosen to ensure complete binding to the enzyme. The protein final concentration was 30 μm. Samples were washed by two successive cycles of dilution/centrifugation in this final buffer using a MilliporeTM membrane filter (30-kDa cutoff) at 4 °C. The titration was performed with phenosafranin (5 mm; E0′ = −0.245 V versus normal hydrogen electrode) as mediator. Electrolysis was carried out under stirring and using a homemade potentiostat. Spectral changes of the electrolysis solution were simultaneously monitored on an 8452A diode array spectrophotometer (Hewlett Packard). After each potential drop, the solution was left to equilibrate until two identical UV-visible spectra were recorded. The absorption spectrum at 0 mV was identical to that of ferric iNOSoxy as isolated, exhibiting a Soret band maximum at 395 nm. A −600-mV potential was applied for 20 min to completely reduce the protein and the mediator, and the anaerobicity of the cell was verified by checking the stability of the reduced enzyme and mediator in the absence of any applied potential over a few minutes. Oxidative titration from −500 to 0 mV was then performed before the potential was swept negatively to re-reduce the protein. The potentiometric titration was monitored at 406 nm (phenosafranin isosbestic point), 480 nm (NOS isosbestic point), and 650 nm (no contribution of phenosafranin). The data were analyzed using the Nernst equation (Equation 1),
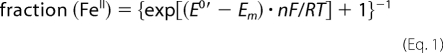 |
where n is the number of electrons, Em is the applied potential, and E0′ is the midpoint potential of interest (F = 96 500 C·mol−1, R = 8.31 J·mol−1·K−1). The number of electrons (n) was found between 0.8 and 1.3 for NOS and between 1.6 and 2.3 for phenosafranin. The obtained midpoint potential for phenosafranin matched the expected value (−245 ± 5 mV), and the difference in the E0′ values obtained for iNOSoxy from the 406 and 650 nm traces was below 10 mV. The reported value for E0′ corresponds to the average of 406 and 650 nm E0′.
Resonance Raman Spectroscopy
Samples for the RR experiments were prepared in 100 mm potassium phosphate buffer (pH 7.4) with different combinations of l-arginine (5 mm) or substituted guanidines 1-9 (20 mm), in the presence of H4B (400 μm) and dithiothreitol (3 mm). Samples were conditioned by two successive cycles of dilution/centrifugation in the final buffer using a Millipore membrane filter (30-kDa cutoff) at 4 °C. The binding of the compounds was confirmed by UV-visible absorption spectroscopy via the spin state changes of the Soret absorption band of the ferric heme from 417 nm (low spin) to 395 nm (high spin). Enzyme concentrations for RR studies ranged between 100 and 200 μm. Forty microliters of the anaerobic FeIII iNOSoxy were prepared directly in quartz tubes sealed with airtight rubber septa by alternating 20 cycles of vacuum and argon refilling. Ferrous samples were obtained by reduction of FeIII iNOSoxy with addition of a small volume (∼10 μl) of a sodium dithionite solution (final concentration between 5 and 10 mm) directly into the quartz tube using a gas-tight syringe (Hamilton). FeII-CO samples were obtained by flushing CO inside the quartz tube for 10 min to ensure complete CO saturation of the solution.
The samples were placed into a gas-tight quartz spinning cell to avoid local heating and to prevent photodissociation and degradation of the NOS samples. Excitations at 363.8 and 441.6 nm were obtained with an argon ion laser (Coherent Innova 90) and with a He-Cd laser (Kimmon), respectively. Resonance Raman spectra were recorded at room temperature using a modified single-stage spectrometer (Jobin-Yvon T64000) equipped with a liquid N2-cooled back-thinned CCD detector. Stray scattered light was rejected using a holographic notch filter (Kaiser Optical Systems). Spectra were recorded by the co-addition of 40–240 individual spectra with an exposure time of 10–30 s each (total accumulation time between 20 and 60 min for each spectral window). Three to six successive sets of such spectra were then averaged. Laser power on the sample was kept below 5 milliwatts to avoid photodissociation and photooxidation. To accurately determine small frequency differences, (i) the monochromator was calibrated using the laser excitation wavelength after each sample measurement, and (ii) samples to be directly compared were recorded the same day with the same optical geometry. Spectral precision and accuracy were estimated to be ∼1 cm−1. Base-line corrections were performed using GRAMS 32 software (Galactic Industries). The iNOSoxy RR bands were assigned following previous publications on iNOS and other NOS (44, 55–59).
ATR-FTIR Spectroscopy
iNOSoxy FeII-CO complexes were prepared as described for the RR experiments, except that protein samples were concentrated up to around 600 μm. Oxygen removal was achieved in a sealed cuvette by 20 cycles of alternate vacuum and argon refilling. Small volumes of sodium dithionite solution were added to reach a final dithionite concentration around 10 mm. FeII-CO complexes were then obtained by 5 min of CO flushing inside the cuvette. Room temperature FTIR spectra were recorded using Bruker IFS 66/S Fourier transform infrared spectrometer coupled to a single reflection micro-ATR unit from Pike technologies. Ten microliters of an iNOSoxy FeII-CO sample was placed on the ZnSe crystal surface of the ATR unit. The device was sealed with a gas-tight in-house-built chamber and maintained under a flush of CO for 15 min until the sample was sufficiently dry. Twenty to 30 co-added interferograms were averaged for each spectrum. In some cases, a water vapor spectrum was used for background correction. Base-line correction was achieved using the GRAMS 32 software package. Each curve corresponded to the average of 2–6 individual experiments.
Data Analysis
Identification of spectral components in unresolved Raman and/or FTIR bands was achieved by the combination of Fourier self-deconvolution and second-order derivative analyses of the averaged spectra as follows: valid peaks were identified when both methods resulted in the same frequency values. In the ν3, ν2, νvinyl, and ν12 regions, overlapping bands were reconstructed by fitting (Origin 6.0, OriginLab Corp.) the spectral region to Gaussian functions for which frequencies were unambiguously determined by the above Fourier self-deconvolution and second derivative analyses (GRAMS 32). The determination of the νFe-CO frequencies by RR spectroscopy was made difficult by the existence of several FeII-CO species and the contributions of other porphyrin modes in the 460–570 cm−1 region. Fourier deconvolution and second-order derivative analyses were used to determine the frequencies of spectral components in each complex Fe-CO bands. Using these frequencies, a band-fitting routine was used to construct the band components, assuming ∼10 cm−1 full width at half-maximum bandwidth. Using this method, we found recurring band components centered around 465, 485, 500, and 510 cm−1 for the RR bands we analyzed. The 1900–2000 cm−1 spectral region is uncongested and does not contain contributions from the heme porphyrin, protein amide, or C-H or N-H stretching modes (60). Thus, the determination of νCO mode frequencies in this spectral region is much easier and gives straightforward information about the number and nature of heme pocket conformations. The inverse correlation that exists between the νCO and νFe-CO mode frequencies was used to refine the analysis of the νFe-CO modes in the RR spectra (45).
RESULTS
pKa Variations of l-Arg Guanidine Analogues
We investigated the influence of the Nω(H) guanidinium proton on the NOS oxidative chemistry using a series of alkyl- and aryl guanidine substrates (Fig. 1). The structural and functional characterizations of these analogues have been reported previously (40–42, 61). We have confirmed that all the analogues bind to iNOSoxy 3 (data not shown) with Kd values ranging between 1 and 100 μm (43). The pKa value of each l-Arg analogue was measured using two different methods. The pKa determination of substituted guanidines in water has been already described for phenyl-substituted tetramethylguanidines (62) and monosubstituted guanidines, such as Ph-Gua (50) and NO2Ph-Gua (63). Accordingly, the pKa values of our set of arylguanidines were measured in water by NaOH titration via the simultaneous monitoring of the pH and the electrical conductivity of the guanidinium salt solution (see “Experimental Procedures”). Because such measurements are not possible for substituted guanidines having expected pKa values greater than 11, the pKa values for the alkylguanidines were extrapolated from the correlation equation proposed by Taylor and Wait (63) using the Hammett σI values (52) (see “Experimental Procedures”). We found that arylguanidines exhibit pKa values ranging from 9.3 to 11 (see Fig. 1 and supplemental Table S1), matching those reported in the literature for similar compounds (50, 63). As expected, the values obtained for alkylguanidines were found between 11.8 and 12.6, close to the pKa value of l-Arg (12.48) (supplemental Table S1). This wide range in pKa values suggests that the l-Arg analogues possess distinct proton donating strengths that could favor different H-bonding interactions near the NOS active site.
Effects on Heme Redox Properties
iNOSoxy heme redox midpoint potentials were measured in the presence of H4B and l-Arg or l-Arg analogues (see “Experimental Procedures”). Fig. 2A shows the UV-visible absorption spectra monitored during the spectroelectrochemical titration of iNOSoxy in the presence of FPh-Gua 6. Similar, complete, and reversible reduction-oxidation processes of iNOSoxy were observed in the presence of all tested guanidines (data not shown). The heme iron E0′ midpoint potential and the n values were obtained by fitting the experimental Nernst plot of each spectroelectrochemical titration (Fig. 2B, see “Experimental Procedures”). The heme midpoint potentials of iNOSoxy in the presence of all tested guanidines were found to be between −262 and −270 ± 5 mV. These values are close to the E0′ value measured for iNOSoxy in the presence of l-Arg and to the value reported by Presta et al. (64) (supplemental Table S2). The l-Arg analogues do not significantly change the heme midpoint potential of iNOSoxy heme, which indicates that these compounds do not alter its redox properties. This strongly suggests that the analogues do not significantly alter the coordination chemistry or immediate electrostatic environment of the iNOS heme iron.
FIGURE 2.

Determination of the heme midpoint potentials of FeIII/FeII iNOSoxy in the presence of arylguanidine. Oxidative titration of iNOSoxy in the presence of the arylguanidine FPh-Gua 6 (A) with phenosafranin as redox mediator. Protocol is described under the “Experimental Procedures.” The titration of iNOSoxy was monitored at 406 nm (isosbestic point of phenosafranin) and 650 nm (no absorption of phenosafranin). The evolution of the mediator was checked at 480 nm (isosbestic point of iNOSoxy). The proportions of enzyme oxidized versus the applied potential were monitored at 406 (■) and 650 nm (◇) in the presence of FPh-Gua 6 (B). The solid line displays the theoretical one-electron Nernst plots obtained by simulating each titration.
Effects on Heme Fe-S Proximal Bond
The characteristics of the bond between the proximal cysteine ligand and the NOS heme iron atom are expected to directly influence the reactivity of the distal oxygen ligand of the NOS heme FeII-O2 species. We investigated the effect of l-Arg analogues on the proximal side of iNOSoxy heme by analyzing iNOSoxy FeIII-S(Cys) vibration mode. This stretching mode can be examined by RR spectroscopy by exploiting the S → FeIII charge transfer band of iNOSoxy FeIII high spin species. This mode is indeed preferentially enhanced upon excitation at 363.8 nm but is absent from the RR spectrum when iNOSoxy is in the low spin FeIII state (65, 66). The RR spectra of FeIII high spin iNOSoxy recorded in the presence of H4B and l-Arg or l-Arg analogues exhibits a band attributable to the νFe-S mode around 338 cm−1 that disappears in the absence of any cofactor and substrate and l-Arg (i.e. when iNOSoxy is in the FeIII low spin state). This frequency matches the value obtained in the presence of l-Arg (supplemental Fig. S1A) (66) and indicates that the binding of our l-Arg analogues does not induce any sizeable modification of the Fe-S(Cys) bond strength relative to l-Arg binding. This conclusion is consistent with the above spectroelectrochemical results.
Effects on Heme Structure
Resonance Raman spectroscopy was used to determine the effects of the bound l-Arg analogues on the conformation of the iNOSoxy porphyrin ring in the resting ferric state (laser excitation at 363.8 nm) and for the FeII-CO complex (laser excitation at 441.6 nm, see “Experimental Procedures”). The high frequency region (1300–1700 cm−1) of the RR spectrum of iNOSoxy exhibits core-size sensitive porphyrin modes that reflect the oxidation, spin, and coordination states of the heme iron. The RR spectra of ferric iNOSoxy prepared in the presence of saturating amounts of H4B and of l-Arg analogues are extremely similar to those obtained in the presence of H4B and l-Arg (supplemental Fig. S1B). Diagnostic heme porphyrin modes were observed around 1372 cm−1 (ν4), 1487 cm−1 (ν3), 1562 cm−1 (ν2), and 1625 cm−1 (νvinyl) (supplemental Table S3), which are all characteristic of a sole population of iNOSoxy in the FeIII five-coordinated (5c) high spin state (56, 58, 65–67). In the same way, the RR spectra of iNOSoxy FeII-CO complexes prepared in the presence of saturating amounts of H4B and of l-Arg analogues are similar to those recorded in the presence of H4B and l-Arg (supplemental Fig. S2) (55–57, 59, 66, 67). Minor bands at 689, 1390, 1424, and 1601 cm−1 arise from minor ferrous heme species resulting from partial photodissociation of CO (supplemental Table S4) (55, 68). All of these results suggest that l-Arg and l-Arg analogues exhibit similar (and negligible) interactions with iNOS heme and that the analogues do not sterically perturb the heme conformation and environment.
Effects on Heme Distal Environment
We also used the RR spectra of iNOSoxy FeII-CO complexes (supplemental Fig. S2) to analyze more specifically the direct effects of the pKa values of bound analogues on the CO ligand coordination and electrostatic environment. Based on previous reports (55, 59, 69), the RR bands at 480–510 cm−1 are attributable to the νFe-CO stretching modes. As described previously for NOS (69), in the absence of both substrate and cofactor, νFe-CO RR bands appear broad (∼50 cm−1 full width at half-maximum) and unresolved, centered at ∼490 cm−1 (supplemental Fig. S2A). This reflects the contributions of several distinct νFe-CO modes attributed to different heme distal pocket conformations resulting in multiple populations of slightly different νFe-CO modes. The binding of l-Arg or NOHA is believed to constrain the heme pocket to one of these conformations (59), leading to a more homogeneous population of νFe-CO modes, which results in a narrower RR band. Indeed, upon l-Arg or NOHA binding, the νFe-CO RR band significantly narrowed (∼18 cm−1 full width at half-maximum for l-Arg) and shifted up in frequency to 510 and 500 cm−1, respectively (supplemental Fig. S2A). However, the RR spectra in the presence of our series of l-Arg analogues showed clear differences as a function of the l-Arg analogue bound to the active site (supplemental Fig. S2A).
To analyze more precisely the interaction between the FeII-CO complex and the l-Arg analogues, we fitted the 425–550 cm−1 spectral region of all RR spectra to a multigaussian function (see “Experimental Procedures”) (41). All νFe-CO bands could be simulated by three to four bands centered at approximately 465, 485, 500, and 510 cm−1. The guanidines studied here could be classified into three families according to their νFe-CO band patterns (Fig. 3 and Table 1). Family 1 (Fig. 3A) includes alkylguanidines that result in iNOSoxy FeII-CO RR spectra similar to that of iNOSoxy with l-Arg as substrate, exhibiting a prominent component at ∼510 cm−1 and weaker contributions around 485 and 500 cm−1. Family 2 includes some arylguanidines, such as PPh-Gua (6), ClPh-Gua (7), and CF3Ph-Gua (8), for which the FeII-CO RR spectra are similar to that of iNOSoxy obtained with NOHA as the bound substrate, exhibiting a main contribution at 497 cm−1 and minor contributions around 485 and 510 cm−1 (Fig. 3B). Family 3 (Fig. 3C) includes other arylguanidines (CH3OPh-Gua (5) and NO2Ph-Gua (9)) that lead to RR spectra similar to that obtained in the absence of substrate (55, 69).
FIGURE 3.

Analysis of the νFe-CO modes of iNOSoxy FeII-CO complex in the presence of l-Arg analogues. The resonance Raman spectra of FeII-CO complexes in the presence of the combination of H4B and l-Arg analogues (supplemental Fig. S2A) were obtained using an excitation wavelength at 441.6 nm. Experiments were achieved in the presence of H4B, except for the −/− experiment that was achieved in the absence of both l-Arg analogues and H4B. The 425–550-cm−1 spectral regions were fitted to multigaussian function. Full protocol is described under “Experimental Procedures.”
TABLE 1.
Analysis of νFe-CO, δFe-CO, and νFe-CO vibration modes of various iNOSoxy FeII-CO complexes based on the resonance Raman and ATR-FTIR spectra of Figs. 3 and 4 and supplemental Fig. S2.
Multigaussian simulation of RR and FTIR spectra was achieved as described under “Experimental Procedures.” Frequencies are expressed in cm−1. Bold values correspond to the predominant conformation. nNOS, neuronal NOS; saNOS, bacterial NOS-like protein from Staphylococcus aureus.
| Protein | Compound | νFe-CO, spectral deconvolution (width, a % intensity) | δFe-C-O | νC-O | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| iNOSoxy | l-Arg | 485 (8, 5) | 501 (15, 39) | 513 (13, 56) | 566 | 1903 | b | |
| 3 | 480 (18, 17) | 494 (14, 22) | 507 (14, 61) | 567 | 1907 | b | ||
| 1 | 478 (18, 20) | 497 (16, 35) | 509 (13, 45) | 566 | 1905 | b | ||
| 4 | 490 (15, 14) | 503 (12, 35) | 511 (12, 50) | 565 | 1911 | b | ||
| NOHA | 485 (17, 20) | 499 (16, 66) | 514 (10, 14) | 563 | b | |||
| 6 | 483 (22, 38) | 497 (16, 43) | 511 (17, 19) | 563 | 1915 | b | ||
| 7 | 479 (17, 36) | 497 (15, 50) | 513 (12, 15) | 565 | 1915 | b | ||
| 8 | 479 (12, 27) | 495 (17, 50) | 511 (14, 24) | 561 | b | |||
| None | 460 (16, 13) | 475 (16, 34) | 492 (17, 40) | 507 (14, 13) | 559 | 1949–1963 | 55, 70 b | |
| 5 | 465 (12, 15) | 479 (15, 33) | 495 (14, 36) | 510 (14, 15) | 562 | 1918 | b | |
| 9 | 465 (17, 20) | 480 (17, 39) | 499 (17, 40) | 514 (10, 7) | 563 | 1921 | b | |
| nNOSoxy | l-Arg | 489 (26, 52) | 502 (12, 28) | 514 (12, 20) | 565 | 1929 | 59,69 | |
| NOHA | 490 (26, 56) | 501 (12, 30) | 514 (12, 14) | 563 | 1928 | 59, 69 | ||
| −/− | 489 (26, 80) | 501 (12, 14) | 514 (12, 6) | 562 | 1936 | 59, 69 | ||
| iNOSoxy | l-Arg | 482 (26, 11) | 503 (26, 34) | 512 (15, 55) | 567 | 1906 | 55, 59, 69, 70 | |
| NOHA | 481 (26, 31) | 500 (26, 53) | 513 (28, 16) | 59, 69 | ||||
| −/− | 482 (26, 67) | 502 (26, 33) | 560 | 1943–1951 | 55, 59, 69, 70 | |||
| saNOS | l-Arg | 567 | 1917 | 56 | ||||
| −/− | 560 | 1930–1949 | 56 | |||||
| P450 | Camphor | 560 | 68 | |||||
| 558 | 68 | |||||||
a Data are from this work.
b Width is in cm−1.
ATR-FTIR characterization of the iNOSoxy FeII-CO complexes for the same combination of H4B and l-Arg analogues led to a similar analysis. Although two νCO mode bands were observed at 1949 and 1963 cm−1 in the absence of substrate and H4B, the addition of H4B and l-Arg analogues led to a simple IR spectrum with a single narrow νCO band whose frequency varied between 1905 and 1921 cm−1 (Fig. 4). This effect, similar to the one observed upon the addition of substrate l-Arg and cofactor H4B (Fig. 4 and Table 1) (55–57, 59, 70), corresponds to a transition from a multiconformation state to a state where a single conformation predominates. As observed previously, three classes of behaviors could be observed. Binding of alkylguanidines (family 1) led to a conformation with a νCO frequency between 1903 and 1911 cm−1, similar to that observed upon l-Arg binding (1903 cm−1). Binding of the arylguanidines from the second family yielded spectra similar to those for iNOSoxy binding NOHA with a νCO frequency around 1915 cm−1. Finally, the IR spectra obtained in the presence of arylguanidines from the third family were characterized by a νCO frequency between 1918 and 1921 cm−1, close to those observed for iNOSoxy in the absence of l-Arg or NOHA (55, 70).
FIGURE 4.

Effects of l-Arg analogues binding on the νC-O stretching frequencies of the FeII-CO complexes of iNOSoxy as measured by ATR-FTIR. All the experiments were achieved in the presence of H4B as described under “Experimental Procedures.”
Thus, our RR and ATR-FTIR results are completely consistent and suggest that substrate analogues can be classified into the following three groups on the basis of their effect on CO coordination. (i) The first group of guanidines (family 1) leads to a distal pocket conformation of the iNOSoxy FeII-CO complex similar to that observed in the presence of l-Arg; these spectra (νFe-CO = ∼510 cm−1 and νC-O = ∼1907 cm−1) indicate strong electrostatic or H-bond interactions on the CO ligand (41). (ii) The second group (family 2) exhibits a spectral profile reminiscent to that observed in the presence of NOHA; these spectra (νFe-CO = ∼500 cm−1 and νC-O = ∼1915 cm−1) indicate weaker electrostatic or H-bond interactions at the level of the CO ligand. (iii) Finally, no significant modification of the structure of the FeII-CO complexes can be observed in the presence of arylguanidines from family 3; these spectra resemble those of iNOSoxy in the absence of bound substrate and indicate several weak and very weak electrostatic or H-bond interactions on CO that are probably not interacting specifically with the analogue.
DISCUSSION
The mechanistic difference between the first and second steps of NOS catalytic activity (Scheme 1) is commonly explained by the difference in pKa values of the guanidinium proton of l-Arg and that of NOHA (37–39). However, until now, no clear picture of specific proton and electron transfer events has emerged to clarify this aspect of the NOS mechanism (24, 25, 30, 32). On the other hand, we and others have shown that the nature of the guanidine substrate can significantly modify the oxidation chemistry, along with the resulting reaction intermediates and products (41, 42, 71, 72). These considerations led us to address, in this work, the actual role of the guanidinium proton in the mechanism of NOS by analyzing the interactions between the heme active site and the guanidinium moiety of l-Arg analogues that display a significant range of pKa values. Using the stable FeII-CO species as an isoelectronic mimic, we report here experimental data that are addressing for the first time the effect of NOS substrate guanidinium proton pKa on the crucial intermediate NOS-FeII-O2.
Both oxidation steps in the NOS mechanism require the binding of O2 to the heme in its ferrous state to form the heme FeII-O2 intermediate, precursor to the oxidizing species. The FeII-CO complex is a useful mimic of the unstable NOS FeII-O2 intermediate, and numerous investigations have shown the effects of substrate binding on the geometry of the FeII-C-O moiety for NOS (41, 55–57, 59, 69, 70) and cytochromes P450 (44, 68, 73–76). In addition to bond angles, the Fe-CO vibrational modes are very sensitive to the electrostatic and polar properties of the heme distal pocket because of changes in the back donation from the iron dπ* orbital to the empty π* CO orbital (44, 45). Changes in the electrostatic distal environment have direct consequences on the νC-O and νFe-CO stretching frequencies (44): higher νFe-CO and lower νC-O frequencies will reflect a greater net positive charge in the vicinity of the O-terminal ligand of the FeII-CO moiety. Thus, the analysis of these two vibrational modes allows the characterization of these electrostatic and steric effects on the FeII ligand as well as providing a correlation between the guanidinium proton pKa values and the structure of the FeII-CO complexes. This information will reflect any electrostatic and/or steric effects the l-Arg analogues will have on the structure, stability, and reactivity of the FeII-O2 moiety.
New Model for FeII-CO Coordination
To date, NOS-heme-FeII-CO complexes have been described as exhibiting two distinct conformations named “open” and “closed” with respect to their heme distal pocket. Substrate binding was believed to suppress the open conformation and to modify the structure of the closed one. However, this representation is vague, and it is unable to explain the chemical basis behind the significant differences observed between the three NOS isoforms (55, 69) and between the substrates l-Arg and NOHA (59, 69). Based on the observations of Li et al. (69), our results lead us to propose a new and more detailed model (see below) involving an equilibrium between at least three distinct conformational families that corresponds to specific interactions between the FeII-CO complex and its heme distal environment. The strongest electrostatic or H-bond interaction on the CO ligand is seen when alkylguanidines of family 1 (including l-Arg) are bound at the substrate site; this leads to the predominance of conformation 1 for which νFe-CO and νC-O frequencies are observed at around 510 and 1903–1911 cm−1, respectively. A weaker electrostatic or H-bond interaction at CO is observed in the presence of bound NOHA and some arylguanidines (family 2, Table 1), resulting in conformation 2 (νFe-CO and νC-O frequencies around 500 and 1915 cm−1, respectively). In the case of some arylguanidines (family 3, Table 1) the interaction was found to be weak to very weak with a conformational structure close to the one observed in the absence of substrate (conformation 3, νFe-CO and νC-O frequencies around 475–500 and 1960–1920 cm−1, respectively; see Table 1), suggesting that analogues of family 3 are not significantly interacting with the CO ligand.
Various Causes for the Changes in FeII-CO Coordination
The changes induced by bound guanidines on the electrostatic, polar, or H-bonding interactions between the heme distal pocket and the CO distal ligand could originate from several causes. The similar binding geometries of l-Arg and NOHA (37, 38), as well as crystallographic structural analyses of NOS in the presence of hydroxyguanidines (77, 78), led us to exclude that differences in the positioning of the guanidinium moiety or significant steric effects could account for a νFe-CO frequency shift of up to 30 cm−1 (41). The RR spectra of FeIII and FeII-CO iNOSoxy complexes presented here did not show any significant differences in the frequencies of core-size sensitive porphyrin modes, suggesting very similar heme conformations and heme-protein interactions for the series of bound l-Arg analogues. In addition, our data do not indicate any modification of the νFe-S frequency in response to l-Arg analogues binding, indicating that the Fe-Cys bond strength remains constant and that the differences in the FeII-CO coordination do not arise from a change in the “Push” effect exerted by the proximal cysteine ligand (44, 79, 80). Furthermore, our spectroelectrochemical titrations did not reveal any significant variation of the heme redox midpoint potentials upon the binding of the l-Arg analogues that could account for the variations of the properties of the FeII-O2 complexes (81, 82). Thus, our complete set of results strongly suggests that the differences observed in the coordination of iNOSoxy FeII-CO moiety do not arise from changes in the heme conformation or the proximal Fe-S bond but rather arise from changes in electrostatic (H-bonding) effects on the CO ligand, in the distal pocket, near the guanidinium group of the bound analogues.
Interestingly, we observed a striking correlation between the FeII-CO vibrational frequencies and the pKa value of the Nω(H) guanidinium proton of the l-Arg analogues (Fig. 5). Conformation 1 (high νFe-CO and low νC-O frequencies) was mostly found in the presence of substrates that exhibit the highest pKa values (l-Arg and alkylguanidines), whereas low pKa guanidines seemed to favor conformations 2 and 3 that are characterized by lower νFe-CO frequencies (Fig. 5). The changes in iNOSoxy FeII-CO coordination thus appear to be determined by the pKa of the guanidine moiety. The pKa values directly reflect the polarization of the N–H bond and thus the strength of the H-bond donated by the guanidinium moiety and/or its ability to exert electrostatic effects. As the guanidinium pKa value decreases, the N–H bond becomes more polarized and the proton more acidic. As a consequence, a guanidinium group of lower pKa value should lead to a stronger, direct H-bond interaction with the FeII-CO complex. However, we report here the opposite effect: this H-bond interaction is weak in the presence of low pKa arylguanidines, and its strength increases with the pKa value of the substrate guanidinium. This counter-intuitive result implies that the effects observed in the presence of our analogues are not linked to a direct interaction between the guanidinium and the CO ligand and suggests that other intervening molecules are involved with the distal ligand in mediating the influence of the guanidinium group.
FIGURE 5.

Correlation between the νC-O (upper figure) and νFe-CO (lower figure) stretching frequencies of the FeII-CO complexes of iNOSoxy and the pKa of the free guanidines in solution.
Structural Model of Interaction between FeII-CO and NOS Substrate
The crystallographic structures of NOS-heme-FeII-CO (39) and ferrous heme-nitric oxide complex (39, 83), in the presence of l-Arg and NOHA, suggest the existence of an H-bond between the l-Arg-guanidinium and the distal ligand (39, 83). Additionally, in the presence of l-Arg, all crystallographic structures reveal the presence of a structural water molecule involved in H-bonding interactions with both the ligand (CO or NO) and the guanidinium moiety of l-Arg (Fig. 6) (39, 83). In the presence of NOHA, the crystal structure shows this water molecule is shifted away from the distal ligand and closer to the guanidinium, eventually resulting in the loss of the H-bond between the water and the distal CO/NO ligand (83).
FIGURE 6.

Crystallographic structure of the active site of iNOSoxy highlighting the H-bonds network in the NOS FeII-CO complex. This structure was obtained from the crystallographic structure of neuronal nitric-oxide synthase (nNOS) FeII-CO complex (Protein Data Bank code 2G6M (39), numbering for iNOSoxy) obtained in the presence of l-Arg (yellow) and H4B (data not shown). Important H-bonds are shown with dashed gray lines. A conserved water molecule (in red) plays a key role in the H-bond network between the guanidinium of l-Arg and the heme-bound CO.
In light of the above structural data, our results allow us to specify the respective roles of the water molecule and of the substrate in the tuning of the structural and electronic properties of the FeII-CO complex (Scheme 3). When l-Arg and other guanidinium substrates of high pKa (family 1) values are bound to iNOSoxy, the CO distal ligand is H-bonded to both the water molecule and the guanidinium, leading to a strong “double” H-bond interaction between CO and the heme distal pocket resulting in conformation 1 (Scheme 3A). Because a stronger H-bond is expected between CO and a guanidinium moiety with lower pKa values, the net decrease in interaction observed in the presence of low pKa guanidines (family 2, including NOHA) can only arise from the weakening (and/or the loss) of the other H-bond, i.e. that between CO and the water molecule. The stronger H-bond between the water and the guanidinium (83) could displace and/or reorient the water molecule and disrupt its H-bond with the CO resulting in an H-bonding situation similar to that of NOS with bound NOHA (see above and Ref. 83). The ensuing change in the distal H-bond network, where the net H-bond interaction on the CO ligand is weaker, gives rise to conformation 2 (Scheme 3B). For high pKa guanidines, the guanidinium would be engaged in fewer interactions leading to a predominant conformation (such as conformations 1 and 2). In contrast, a bound analogue of lower pKa (such as some arylguanidines, family 3) would favor the formation of vaguely defined conformations characterized by very weak and nonspecific interactions with the distal ligand (conformation 3).
SCHEME 3.

Schematic view of the proposed active-site H-bond networks in the iNOS FeII-CO complexes in the presence of l-Arg analogues from family 1 (A) and family 2 (B).
Implications on NOS Mechanism of the Oxygen Activation
Our results show that the pKa value of the exchangeable protons of the guanidine moiety influences the H-bond network at the NOS active site and can modify the electrostatic environment of the FeII-CO complex. The iNOS FeII-CO complex is an isoelectronic mimic of the crucial NOS FeII-O2 intermediate (44, 45), and indeed NOS FeII-O2 complex exhibits the same sensitivity to the nature of the guanidines (80). For instance, the NOS FeII-O2 complex exhibits a weaker O–O bond in the presence of l-Arg than in the presence of NOHA (νO-O = 1132–35 versus 1323–26 cm−1) (69, 80, 84) indicating stronger interactions on the O2 ligand for the l-Arg binding case. According to our above analysis of the H-bond interactions with the (CO) distal ligand, the binding of high pKa guanidines, such as l-Arg, would result in the guanidinium and the structural water molecule strongly interacting with the oxygen ligand of the FeII-O2 moiety (see Scheme 3A). For low pKa-bound guanidine substrates such as NOHA, there would be an increase in H-bond interaction between the guanidinium and the water molecule, which will in turn weaken the interaction between the water molecule and the FeII-O2 oxygen ligand (see Scheme 3B).
The properties of the substrate guanidinium therefore seem to influence the structural and electronic properties of the FeII-O2 complex. This should be crucial for NOS catalytic efficiency that primarily relies on the kinetic balance between futile FeII-O2 autoxidation and FeII-O2 activation (26, 85). Actually, we observed a good correlation between the pKa values of our series of l-Arg analogues and the rates of FeII-O2 autoxidation (Table 2) (41). The lowest autoxidation rates are observed for NOS bound with high pKa substrate analogues (family 1) that promote a strong H-bond between the active site water molecule and the terminal oxygen of the heme-bound O2. This strong double H-bond situation (Scheme 3A) would stabilize the FeII-O2 complex, prevent dissociation, and decrease the FeII-O2 autoxidation rate allowing time for electron transfer from the reductase domain to FeII-O2 to give the ferric peroxo (FeIII-OO−) intermediate (i.e. oxygen activation), and ultimately favoring efficient NO production (Table 2). In contrast, the loss of such a strong H-bond would decrease the stability of the FeII-O2 complex; this is observed with guanidines of families 2 and 3 for which the autoxidation rates increase by 100-fold (Table 2). Thus, the decrease of the guanidinium pKa value can be related to the decrease in NO production by the uncoupling of electron transfer and the associated release of high amounts of ROS (Table 2) (40, 42, 86).
TABLE 2.
Relationship between the pKa of the guanidine, the autoxidation process, the uncoupling ratio, and the iNOS-catalyzed production of NO from the guanidines and the corresponding N-hydroxyguanidines
| Compound | pKa | Autoxidation rate a | Production of NO (% l-Arg) b | Production of NO (% NOHA) c |
|---|---|---|---|---|
| s−1 | ||||
| l-Arg | 12.48 | 0.2 ± 0.04 | 100 | 100 |
| 3 | 12.6 | 1.9 ± 0.4 | 11 ± 2 | 32 ± 5 |
| 1 | 11.8 | 1.6 ± 0.5 | 35 ± 2 | 95 ± 8 |
| 4 | 11.8 | <0.5 | 10 ± 3 | |
| 6 | 10.8 | 12 ± 3 | <0.5 | 41 ± 6 |
| 7 | 10.3 | 7.3 ± 1.1 | <0.5 | 13 ± 3 |
| 8 | 10.0 | <0.5 | 0.5 ± 0.2 | |
| 5 | 11.0 | 19 ± 5 | <0.5 | 6 ± 2 |
| 9 | 9.3 | <0.5 | 2 ± 1 | |
a Autoxidation rates correspond to the decay rate of iNOSoxy FeII-O2 measured by stopped-flow in the presence of H2B and l-Arg analogues. Values are from Ref. 41.
b Formation of NO by oxidation of some guanidines; values are relative to the production of NO observed using l-Arg as substrate. Values are from Refs. 41, 42.
c Formation of NO by oxidation of the corresponding N-hydroxyguanidines; values are relative to the production of NO observed using NOHA as substrate. Values are from Ref. 42.
Additionally, our data can help understanding the NOS mechanism of oxygen activation. The current picture of the NOS molecular mechanism remains conflicting. The first step of NOS chemistry, Arg → NOHA, is generally believed to follow a typical P450-like mono-oxygenation reaction based on the double protonation of the FeIII-OO− peroxo intermediate, the heterolytic cleavage of the peroxo O–O bond leading to the formation of a Compound I species, followed by a typical P450-like “radical rebound” mechanism (Scheme 2). The second step, NOHA → citrulline + NO, is supposed to involve the direct reaction of the (hydro)peroxo species on the NOHA hydroxyguanidinium moiety, followed by the rearrangement of the resulting tetrahedral complex and the release of citrulline and NO (26, 29). These models are supported by crystallographic, spectroscopic, and cryogenic experimental evidences (39, 69, 80, 83, 87), but recently serious questions have been raised (30, 32). In the absence of the definitive isolation, characterization, and identification of key reaction intermediates, an unequivocal NOS mechanism cannot be advanced and therefore several alternative mechanisms have been proposed for each catalytic step. These proposals differ in the number and/or the source of transferred protons 4 and thus in the nature of the resulting oxidative species, e.g. a FeIII-peroxo complex (30) or a Compound II intermediate (32) for l-Arg oxidation, a FeII-O2 complex (30, 88), or an oxoferryl complex (31) for NOHA oxidation.
Our analysis of the structural and electrostatic influences of the guanidinium pKa, combined with the characterization of NOS-catalyzed oxidation of the series analogues studied (Table 2), leads us to propose a model for the mechanism of FeII-O2 activation (Scheme 4) that completes the ones proposed by Rousseau and co-workers (69) and Poulos and co-workers (39). High pKa alkylguanidines (including l-Arg) that favor the formation of the second H-bond between the distal water molecule and the terminal oxygen atom of the FeIII-peroxo complex prove to be good substrates of iNOS despite slightly higher autoxidation rates (Table 2). Reciprocally, no formation of NO was detected for the bound low pKa arylguanidine analogues for which the above-mentioned H-bond is not present (Table 2). This strongly supports the involvement of two protonation events leading to the formation of a Compound I as the oxidative species for l-Arg hydroxylation (Scheme 4, step 1). However, whereas low pKa arylguanidines failed to become hydroxylated, the corresponding N-hydroxyguanidines still led to significant production of NO (Table 2) (61, 86, 89). This suggests that the FeIII-peroxo complex might be sufficient to achieve NOHA oxidation (29); without the participation of an additional water molecule, low pKa substrates, such as NOHA, will stabilize the FeIII-peroxo complex and favor its direct reaction on the guanidinium moiety (Scheme 4, step 2).
SCHEME 4.

Molecular mechanisms proposed for both steps of NOS catalysis. High pKa alkylguanidines would favor the formation of an H-bond between the distal water molecule and the terminal oxygen atom of the FeIII-peroxo complex. Heterolytic cleavage of the O–O bond would yield a high valent Por⨥-FeIV=O intermediate as the substrate oxidant (step 1). Step 2, low pKa substrates such as NOHA would stabilize the FeIII-peroxo intermediate and favor its direct reaction on the hydroxyguanidinium moiety.
Our analysis can help to comprehend the parameters that control NOS biological activities. We showed that the guanidinium pKa determines the stability of the FeII-O2 complex and the properties of proton transfer onto the ferric-peroxide complex. As such, it is a key element of the efficiency of NOS-catalyzed oxygen activation and NO production. Changes in the guanidinium pKa value will lead to the autoxidation of the FeII-O2 complex, and thus to superoxide production or to a futile proton transfer, leading to the release of hydrogen peroxide. The production of ROS will have catastrophic downstream consequences on NOS production such as the self-inhibiting oxidation of BH4 (90–92) or the production of the highly toxic peroxynitrite from the reaction of NO with superoxide (93–95). Consequently, the pKa properties of the substrate guanidinium appear crucial in the control of the balance between NO and ROS/RNS production. This balance, which is increasingly considered as the major determinant of NOS biological impact (5, 96, 97), is observed in NO-related pathological settings such as neurodegenerative diseases: whereas NO is believed to exert physiological and neuroprotective roles, ROS and peroxynitrite are mostly responsible for NOS-related neurotoxicity (98–101). The understanding of the parameters that control the NO/ROS balance is thus essential for therapeutic strategies (102, 103).
In this context, the selective inhibition of NOS isoforms has long been a major challenge for the biomedical community, leading to the discovery of potent and selective NOS inhibitors (104–107). The importance of H-bonds and pKa has been evoked to explain the inhibitory power of compounds such as substituted guanidines, iso-thioureas, or amidines (108–110). Although the effect of NOS inhibitors on ROS/RNS production is crucial to determine the actual biological impact of these compounds, this effect has been poorly investigated, and only a limited number of inhibitors that fully block all NOS catalytic productions are presently identified (111, 112). In this context, our model can analyze the effects of known or potential inhibitors on the stability of NOS reaction intermediates and thus on the potential production of ROS-peroxynitrite. As the distal H-bond network proved to be specific for each NOS isoform (55, 66, 113), our model could also provide a rationale for the selectivity and efficiency of the various classes of inhibitors. This functional approach could help with predicting the actual biological impact of tested compounds and designing selective inhibitors that block ROS and RNS production.
This approach might also be valuable for the design of alternative substrates of NOS. This study highlights the requirement for the presence of a high pKa guanidinium to prevent FeII-O2 autoxidation and favor a productive proton transfer sequence. In this regard, the introduction of electron-donating groups close to the guanidinium moiety might enhance the efficiency of l-Arg-based NO donors. As an example, such substitution on the aromatic ring of aryl-guanidines could increase their pKa values and allow their transformation into NO. Thus, the modulation of guanidinium high pKa might be a useful tool for the design of new l-Arg analogues with improved NO formation efficiency.
Conclusions
By using analogues of l-Arg, we were able to reveal a direct influence of the guanidinium proton pKa on H-bond interactions between the distal ligand, the substrate, and a crucial neighboring water molecule. Our results suggest that by modifying the structure of the distal H-bond network the substrate guanidinium can finely tune NOS oxidative chemistry, in particular the nature of NOS reaction intermediate and thus the specificity of each catalytic step. Additionally, we propose that the properties of the guanidinium moiety determine the stability of the FeII-O2 complex and the efficiency of the proton transfer processes. Our study establishes a direct correlation between the ROS/NO production and the guanidinium pKa, which supports a major role for the substrate guanidinium in the regulation of the NOS catalytic production. In this regard, by allowing the design of efficient and selective NOS inhibitors or substrates, our model could be a promising tool to understand and control the oxidative stress that consists of NOS-catalyzed ROS production, BH4 depletion, and peroxynitrite formation, which is the hallmark of several pathologies such as atherosclerosis and neurodegenerative diseases.
Supplementary Material

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2 and Tables S1–S4.
The binding of l-Arg analogues was confirmed by the 420 → 395-nm spectral shift of the Soret UV-visible absorption band, characteristic of a ferric heme iron low spin → high spin transition.
J. Santolini, submitted for publication.
- NO
- nitric oxide
- FeII-O2
- ferrous heme-oxy complex
- FeII-CO
- ferrous heme-carbon monoxide complex
- FTIR
- Fourier-transformed infra-red spectroscopy
- H4B
- tetrahydrobiopterin, (6R)-5,6,7,8-tetrahydro-l-biopterin
- iNOSoxy
- inducible NOS oxygenase domain
- NOS
- nitric-oxide synthase
- NOHA
- Nω-hydroxy-l-arginine
- iNOS
- inducible nitric oxide synthase
- ROS
- reactive oxygen species
- RNS
- reactive nitrogen species
- RR
- resonance Raman spectroscopy
- ATR
- attenuated total reflectance.
REFERENCES
- 1. Sessa W. C. (2004) J. Cell Sci. 117, 2427–2429 [DOI] [PubMed] [Google Scholar]
- 2. Mungrue I. N., Bredt D. S. (2004) J. Cell Sci. 117, 2627–2629 [DOI] [PubMed] [Google Scholar]
- 3. Bredt D. S. (2003) J. Cell Sci. 116, 9–15 [DOI] [PubMed] [Google Scholar]
- 4. Zhang L., Dawson V. L., Dawson T. M. (2006) Pharmacol. Ther. 109, 33–41 [DOI] [PubMed] [Google Scholar]
- 5. Pacher P., Beckman J. S., Liaudet L. (2007) Physiol. Rev. 87, 315–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Malinski T. (2007) J. Alzheimers Dis. 11, 207–218 [DOI] [PubMed] [Google Scholar]
- 7. Förstermann U., Münzel T. (2006) Circulation 113, 1708–1714 [DOI] [PubMed] [Google Scholar]
- 8. Pollock J. S., Förstermann U., Mitchell J. A., Warner T. D., Schmidt H. H., Nakane M., Murad F. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 10480–10484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stuehr D. J., Cho H. J., Kwon N. S., Weise M. F., Nathan C. F. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 7773–7777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bredt D. S., Hwang P. M., Glatt C. E., Lowenstein C., Reed R. R., Snyder S. H. (1991) Nature 351, 714–718 [DOI] [PubMed] [Google Scholar]
- 11. Xie Q. W., Cho H. J., Calaycay J., Mumford R. A., Swiderek K. M., Lee T. D., Ding A., Troso T., Nathan C. (1992) Science 256, 225–228 [DOI] [PubMed] [Google Scholar]
- 12. Sessa W. C., Harrison J. K., Barber C. M., Zeng D., Durieux M. E., D'Angelo D. D., Lynch K. R., Peach M. J. (1992) J. Biol. Chem. 267, 15274–15276 [PubMed] [Google Scholar]
- 13. Stuehr D. J. (1999) Biochim. Biophys. Acta 1411, 217–230 [DOI] [PubMed] [Google Scholar]
- 14. Marletta M. A., Hurshman A. R., Rusche K. M. (1998) Curr. Opin. Chem. Biol. 2, 656–663 [DOI] [PubMed] [Google Scholar]
- 15. Knowles R. G., Moncada S. (1994) Biochem. J. 298, 249–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ghosh D. K., Salerno J. C. (2003) Front. Biosci. 8, d193–209 [DOI] [PubMed] [Google Scholar]
- 17. Alderton W. K., Cooper C. E., Knowles R. G. (2001) Biochem. J. 357, 593–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roman L. J., Martásek P., Masters B. S. (2002) Chem. Rev. 102, 1179–1190 [DOI] [PubMed] [Google Scholar]
- 19. Garcin E. D., Bruns C. M., Lloyd S. J., Hosfield D. J., Tiso M., Gachhui R., Stuehr D. J., Tainer J. A., Getzoff E. D. (2004) J. Biol. Chem. 279, 37918–37927 [DOI] [PubMed] [Google Scholar]
- 20. Panda K., Haque M. M., Garcin-Hosfield E. D., Durra D., Getzoff E. D., Stuehr D. J. (2006) J. Biol. Chem. 281, 36819–36827 [DOI] [PubMed] [Google Scholar]
- 21. Abu-Soud H. M., Stuehr D. J. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 10769–10772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stuehr D. J., Abu-Soud H. M., Rousseau D. L., Feldman P. L., Wang J. (1995) Adv. Pharmacol. 34, 207–213 [DOI] [PubMed] [Google Scholar]
- 23. Stuehr D. J., Kwon N. S., Nathan C. F., Griffith O. W., Feldman P. L., Wiseman J. (1991) J. Biol. Chem. 266, 6259–6263 [PubMed] [Google Scholar]
- 24. Li H., Poulos T. L. (2005) J. Inorg. Biochem. 99, 293–305 [DOI] [PubMed] [Google Scholar]
- 25. Gorren A. C., Mayer B. (2007) Biochim. Biophys. Acta 1770, 432–445 [DOI] [PubMed] [Google Scholar]
- 26. Stuehr D. J., Santolini J., Wang Z. Q., Wei C. C., Adak S. (2004) J. Biol. Chem. 279, 36167–36170 [DOI] [PubMed] [Google Scholar]
- 27. Wei C. C., Wang Z. Q., Hemann C., Hille R., Stuehr D. J. (2003) J. Biol. Chem. 278, 46668–46673 [DOI] [PubMed] [Google Scholar]
- 28. Wei C. C., Wang Z. Q., Tejero J., Yang Y. P., Hemann C., Hille R., Stuehr D. J. (2008) J. Biol. Chem. 283, 11734–11742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Woodward J. J., Chang M. M., Martin N. I., Marletta M. A. (2009) J. Am. Chem. Soc. 131, 297–305 [DOI] [PubMed] [Google Scholar]
- 30. Zhu Y., Silverman R. B. (2008) Biochemistry 47, 2231–2243 [DOI] [PubMed] [Google Scholar]
- 31. Robinet J. J., Cho K. B., Gauld J. W. (2008) J. Am. Chem. Soc. 130, 3328–3334 [DOI] [PubMed] [Google Scholar]
- 32. de Visser S. P., Tan L. S. (2008) J. Am. Chem. Soc. 130, 12961–12974 [DOI] [PubMed] [Google Scholar]
- 33. Cho K. B., Carvajal M. A., Shaik S. (2009) J. Phys. Chem. B 113, 336–346 [DOI] [PubMed] [Google Scholar]
- 34. Cho K. B., Derat E., Shaik S. (2007) J. Am. Chem. Soc. 129, 3182–3188 [DOI] [PubMed] [Google Scholar]
- 35. Cho K. B., Gauld J. W. (2005) J. Phys. Chem. B 109, 23706–23714 [DOI] [PubMed] [Google Scholar]
- 36. Tejero J., Santolini J., Stuehr D. J. (2009) FEBS J. 276, 4505–4514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Crane B. R., Arvai A. S., Ghosh D. K., Wu C., Getzoff E. D., Stuehr D. J., Tainer J. A. (1998) Science 279, 2121–2126 [DOI] [PubMed] [Google Scholar]
- 38. Crane B. R., Arvai A. S., Ghosh S., Getzoff E. D., Stuehr D. J., Tainer J. A. (2000) Biochemistry 39, 4608–4621 [DOI] [PubMed] [Google Scholar]
- 39. Li H., Igarashi J., Jamal J., Yang W., Poulos T. L. (2006) J. Biol. Inorg. Chem. 11, 753–768 [DOI] [PubMed] [Google Scholar]
- 40. Dijols S., Boucher J. L., Lepoivre M., Lefevre-Groboillot D., Moreau M., Frapart Y., Rekka E., Meade A. L., Stuehr D. J., Mansuy D. (2002) Biochemistry 41, 9286–9292 [DOI] [PubMed] [Google Scholar]
- 41. Moreau M., Boucher J. L., Mattioli T. A., Stuehr D. J., Mansuy D., Santolini J. (2006) Biochemistry 45, 3988–3999 [DOI] [PubMed] [Google Scholar]
- 42. Mansuy D., Boucher J. L. (2004) Free Radic. Biol. Med. 37, 1105–1121 [DOI] [PubMed] [Google Scholar]
- 43. Moreau M. (2005) Etude du Mécanisme d'Oxydation des Guanidines par les NO-Synthases, Recherche de Nouveaux Précurseurs de NO. Thèse de Chimie, Université de Paris Descartes, Paris [Google Scholar]
- 44. Spiro T. G., Wasbotten I. H. (2005) J. Inorg. Biochem. 99, 34–44 [DOI] [PubMed] [Google Scholar]
- 45. Ray G. B., Li X. Y., Ibers J. A., Sessler J. L., Spiro T. G. (1994) J. Am. Chem. Soc. 116, 162–176 [Google Scholar]
- 46. Bernatowicz M. S., Wu Y., Matsueda G. R. (1993) Tetrahedron Lett. 34, 3389–3392 [Google Scholar]
- 47. Ghosh D. K., Crane B. R., Ghosh S., Wolan D., Gachhui R., Crooks C., Presta A., Tainer J. A., Getzoff E. D., Stuehr D. J. (1999) EMBO J. 18, 6260–6270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stuehr D. J., Ikeda-Saito M. (1992) J. Biol. Chem. 267, 20547–20550 [PubMed] [Google Scholar]
- 49. Hammett L. P. (1937) J. Am. Chem. Soc. 59, 96–103 [Google Scholar]
- 50. Charton M. (1965) J. Org. Chem. 30, 969–973 [Google Scholar]
- 51. Yamamoto Y., Kojima S. (1991) in The Chemistry of Amidines and Imidate (Patai S., Rappoport Z. eds) pp. 485–526, John Wiley & Sons, Inc., New York [Google Scholar]
- 52. Hansch C., Leo A., Taft R. W. (1991) Chem. Rev. 91, 165–195 [Google Scholar]
- 53. Perrin D. D. (1972) Dissociation Constants of Organic Bases in Aqueous Solution, Butterworths, London [Google Scholar]
- 54. Balland V., Hureau C., Cusano A. M., Liu Y., Tron T., Limoges B. (2008) Chemistry 14, 7186–7192 [DOI] [PubMed] [Google Scholar]
- 55. Fan B., Wang J., Stuehr D. J., Rousseau D. L. (1997) Biochemistry 36, 12660–12665 [DOI] [PubMed] [Google Scholar]
- 56. Chartier F. J., Couture M. (2004) Biophys J. 87, 1939–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li D., Stuehr D. J., Yeh S. R., Rousseau D. L. (2004) J. Biol. Chem. 279, 26489–26499 [DOI] [PubMed] [Google Scholar]
- 58. Wang J., Stuehr D. J., Rousseau D. L. (1995) Biochemistry 34, 7080–7087 [DOI] [PubMed] [Google Scholar]
- 59. Wang J., Stuehr D. J., Rousseau D. L. (1997) Biochemistry 36, 4595–4606 [DOI] [PubMed] [Google Scholar]
- 60. Ingledew W. J., Smith S. M., Salerno J. C., Rich P. R. (2002) Biochemistry 41, 8377–8384 [DOI] [PubMed] [Google Scholar]
- 61. Lefèvre-Groboillot D., Boucher J. L., Stuehr D. J., Mansuy D. (2005) FEBS J. 272, 3172–3183 [DOI] [PubMed] [Google Scholar]
- 62. Leffek K. T., Pruszynski P., Thanapaalasingham K. (1989) Can. J. Chem-Rev. Can. Chim. 67, 590–595 [Google Scholar]
- 63. Taylor P. J., Wait A. R. (1986) J. Chem. Soc. Perkin Trans. 2, 1765–1770 [Google Scholar]
- 64. Presta A., Weber-Main A. M., Stankovich M. T., Stuehr D. J. (1998) J. Am. Chem. Soc. 120, 9460–9465 [Google Scholar]
- 65. Schelvis J. P., Berka V., Babcock G. T., Tsai A. L. (2002) Biochemistry 41, 5695–5701 [DOI] [PubMed] [Google Scholar]
- 66. Santolini J., Roman M., Stuehr D. J., Mattioli T. A. (2006) Biochemistry 45, 1480–1489 [DOI] [PubMed] [Google Scholar]
- 67. Rodríguez-Crespo I., Moënne-Loccoz P., Loehr T. M., Ortiz de Montellano P. R. (1997) Biochemistry 36, 8530–8538 [DOI] [PubMed] [Google Scholar]
- 68. Wells A. V., Li P., Champion P. M., Martinis S. A., Sligar S. G. (1992) Biochemistry 31, 4384–4393 [DOI] [PubMed] [Google Scholar]
- 69. Li D., Kabir M., Stuehr D. J., Rousseau D. L., Yeh S. R. (2007) J. Am. Chem. Soc. 129, 6943–6951 [DOI] [PubMed] [Google Scholar]
- 70. Jung C., Stuehr D. J., Ghosh D. K. (2000) Biochemistry 39, 10163–10171 [DOI] [PubMed] [Google Scholar]
- 71. Martin N. I., Woodward J. J., Winter M. B., Beeson W. T., Marletta M. A. (2007) J. Am. Chem. Soc. 129, 12563–12570 [DOI] [PubMed] [Google Scholar]
- 72. Luzzi S. D., Marletta M. A. (2005) Bioorg. Med. Chem. Lett. 15, 3934–3941 [DOI] [PubMed] [Google Scholar]
- 73. Deng T. J., Proniewicz L. M., Kincaid J. R., Yeom H., Macdonald I. D., Sligar S. G. (1999) Biochemistry 38, 13699–13706 [DOI] [PubMed] [Google Scholar]
- 74. Tsubaki M., Hiwatashi A., Ichikawa Y. (1986) Biochemistry 25, 3563–3569 [DOI] [PubMed] [Google Scholar]
- 75. Jung C., Hoa G. H., Schröder K. L., Simon M., Doucet J. P. (1992) Biochemistry 31, 12855–12862 [DOI] [PubMed] [Google Scholar]
- 76. Jung C., Schulze H., Deprez E. (1996) Biochemistry 35, 15088–15094 [DOI] [PubMed] [Google Scholar]
- 77. Raman C. S., Li H., Martásek P., Southan G., Masters B. S., Poulos T. L. (2001) Biochemistry 40, 13448–13455 [DOI] [PubMed] [Google Scholar]
- 78. Li H., Shimizu H., Flinspach M., Jamal J., Yang W., Xian M., Cai T., Wen E. Z., Jia Q., Wang P. G., Poulos T. L. (2002) Biochemistry 41, 13868–13875 [DOI] [PubMed] [Google Scholar]
- 79. Tosha T., Kagawa N., Ohta T., Yoshioka S., Waterman M. R., Kitagawa T. (2006) Biochemistry 45, 5631–5640 [DOI] [PubMed] [Google Scholar]
- 80. Chartier F. J., Couture M. (2007) J. Biol. Chem. 282, 20877–20886 [DOI] [PubMed] [Google Scholar]
- 81. Ost T. W., Miles C. S., Munro A. W., Murdoch J., Reid G. A., Chapman S. K. (2001) Biochemistry 40, 13421–13429 [DOI] [PubMed] [Google Scholar]
- 82. Adak S., Wang Q., Stuehr D. J. (2000) J. Biol. Chem. 275, 17434–17439 [DOI] [PubMed] [Google Scholar]
- 83. Pant K., Crane B. R. (2006) Biochemistry 45, 2537–2544 [DOI] [PubMed] [Google Scholar]
- 84. Chartier F. J., Blais S. P., Couture M. (2006) J. Biol. Chem. 281, 9953–9962 [DOI] [PubMed] [Google Scholar]
- 85. Wei C. C., Wang Z. Q., Meade A. L., McDonald J. F., Stuehr D. J. (2002) J. Inorg. Biochem. 91, 618–624 [DOI] [PubMed] [Google Scholar]
- 86. Renodon-Cornière A., Dijols S., Perollier C., Lefevre-Groboillot D., Boucher J. L., Attias R., Sari M. A., Stuehr D., Mansuy D. (2002) J. Med. Chem. 45, 944–954 [DOI] [PubMed] [Google Scholar]
- 87. Davydov R., Ledbetter-Rogers A., Martásek P., Larukhin M., Sono M., Dawson J. H., Masters B. S., Hoffman B. M. (2002) Biochemistry 41, 10375–10381 [DOI] [PubMed] [Google Scholar]
- 88. Huang H., Hah J. M., Silverman R. B. (2001) J. Am. Chem. Soc. 123, 2674–2676 [DOI] [PubMed] [Google Scholar]
- 89. Renodon-Cornière A., Boucher J. L., Dijols S., Stuehr D. J., Mansuy D. (1999) Biochemistry 38, 4663–4668 [DOI] [PubMed] [Google Scholar]
- 90. Vásquez-Vivar J. (2009) Free Radic. Biol. Med. 47, 1108–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Stuehr D., Pou S., Rosen G. M. (2001) J. Biol. Chem. 276, 14533–14536 [DOI] [PubMed] [Google Scholar]
- 92. Schulz E., Jansen T., Wenzel P., Daiber A., Münzel T. (2008) Antioxid. Redox. Signal. 10, 1115–1126 [DOI] [PubMed] [Google Scholar]
- 93. Förstermann U. (2006) Biol. Chem. 387, 1521–1533 [DOI] [PubMed] [Google Scholar]
- 94. Patel R. P., McAndrew J., Sellak H., White C. R., Jo H., Freeman B. A., Darley-Usmar V. M. (1999) Biochim. Biophys. Acta 1411, 385–400 [DOI] [PubMed] [Google Scholar]
- 95. Radi R., Peluffo G., Alvarez M. N., Naviliat M., Cayota A. (2001) Free Radic. Biol. Med. 30, 463–488 [DOI] [PubMed] [Google Scholar]
- 96. Pall M. L. (2007) Med. Hypotheses 69, 821–825 [DOI] [PubMed] [Google Scholar]
- 97. Liaudet L., Soriano F. G., Szabó C. (2000) Crit. Care Med. 28, Suppl. 4, N37–N52 [DOI] [PubMed] [Google Scholar]
- 98. Lipton S. A. (1999) Cell Death Differ. 6, 943–951 [DOI] [PubMed] [Google Scholar]
- 99. Reynolds M. R., Berry R. W., Binder L. I. (2007) Biochemistry 46, 7325–7336 [DOI] [PubMed] [Google Scholar]
- 100. Guix F. X., Uribesalgo I., Coma M., Muñoz F. J. (2005) Prog. Neurobiol. 76, 126–152 [DOI] [PubMed] [Google Scholar]
- 101. Dawson V. L., Dawson T. M. (1996) Neurochem. Int. 29, 97–110 [DOI] [PubMed] [Google Scholar]
- 102. Szabo C., Ischiropoulos H., Radi R. (2007) Nat. Rev. 6, 662–680 [DOI] [PubMed] [Google Scholar]
- 103. Trujillo M., Ferrer-Sueta G., Radi R. (2008) Antioxid. Redox. Signal. 10, 1607–1620 [DOI] [PubMed] [Google Scholar]
- 104. Garvey E. P., Oplinger J. A., Furfine E. S., Kiff R. J., Laszlo F., Whittle B. J., Knowles R. G. (1997) J. Biol. Chem. 272, 4959–4963 [DOI] [PubMed] [Google Scholar]
- 105. Collins J. L., Shearer B. G., Oplinger J. A., Lee S., Garvey E. P., Salter M., Duffy C., Burnette T. C., Furfine E. S. (1998) J. Med. Chem. 41, 2858–2871 [DOI] [PubMed] [Google Scholar]
- 106. Silverman R. B. (2009) Acc. Chem. Res. 42, 439–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Garcin E. D., Arvai A. S., Rosenfeld R. J., Kroeger M. D., Crane B. R., Andersson G., Andrews G., Hamley P. J., Mallinder P. R., Nicholls D. J., St-Gallay S. A., Tinker A. C., Gensmantel N. P., Mete A., Cheshire D. R., Connolly S., Stuehr D. J., Aberg A., Wallace A. V., Tainer J. A., Getzoff E. D. (2008) Nat. Chem. Biol. 4, 700–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Babu B. R., Frey C., Griffith O. W. (1999) J. Biol. Chem. 274, 25218–25226 [DOI] [PubMed] [Google Scholar]
- 109. Li H., Raman C. S., Martásek P., Král V., Masters B. S., Poulos T. L. (2000) J. Inorg. Biochem. 81, 133–139 [DOI] [PubMed] [Google Scholar]
- 110. Igarashi J., Li H., Jamal J., Ji H., Fang J., Lawton G. R., Silverman R. B., Poulos T. L. (2009) J. Med. Chem. 52, 2060–2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Sennequier N., Stuehr D. J. (1996) Biochemistry 35, 5883–5892 [DOI] [PubMed] [Google Scholar]
- 112. Abu-Soud H. M., Feldman P. L., Clark P., Stuehr D. J. (1994) J. Biol. Chem. 269, 32318–32326 [PubMed] [Google Scholar]
- 113. Rousseau D. L., Li D., Couture M., Yeh S. R. (2005) J. Inorg. Biochem. 99, 306–323 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.