

Abstract
α-Synuclein (αS) is a protein involved in the cytopathology and genetics of Parkinson disease and is thought to affect mitochondrial complex I activity. Previous studies have shown that mitochondrial toxins and specifically inhibitors of complex I activity enhance αS pathogenesis. Here we show that αS overexpression specifically inhibits complex I activity in dopaminergic cells and in A53T αS transgenic mouse brains. Importantly, our results indicate that the inhibitory effect on complex I activity is not associated with αS-related pathology. Specifically, complex I activity measured in purified mitochondria from A53T αS transgenic mouse brains was not affected by mouse age; Parkinson disease-like symptoms; levels of αS soluble oligomers; levels of insoluble, lipid-associated αS; or αS intraneuronal depositions in vivo. Likewise, no correlation was found between complex I activity and polyunsaturated fatty acid-induced αS depositions in Lewy body-like inclusions in cultured dopaminergic cells. We further show that the effect of αS on complex I activity is not due to altered mitochondrial protein levels or affected complex I assembly. Based on the results herein, we suggest that αS expression negatively regulates complex I activity as part of its normal, physiological role.
Keywords: Diseases/Aging, Metabolism/Energy, Protein/Folding, Brain, Parkinson Disease, Diseases/Aging, α-Synuclein, Mitochondria, Protein/Misfolding and Aggregation
Introduction
Evidence for the involvement of mitochondrial dysfunction in the pathogenesis of Parkinson disease (PD)2 emerged following the discovery that 1-methyl-4-phenyl-1,2,3,4-tetrahydropyridine (MPTP) causes PD-like symptoms in humans (1) and the subsequent findings that its neurotoxic metabolite, the 1-methyl-4-phenylpyridinium ion, inhibits mitochondrial complex I activity (2). Mitochondrial dysfunction represented by inhibition of complex I activity was described in the brain, skeletal muscle, and platelets of a subset of patients with PD (3–5). The finding that DJ-1, Pink1, HTRA2, LRRK2, and Parkin genes that cause familial PD (6–10) encode mitochondrial proteins has reinforced the link between mitochondrial dysfunction and PD.
α-Synuclein (αS) is a presynaptic protein critically involved in the cytopathology and genetics of PD (reviewed in Refs. 11–13). In PD and the related human synucleinopathies (14), there is a progressive conversion of the normally highly soluble αS protein into insoluble β-sheet rich filamentous assemblies, resulting in its intraneuronal deposition into Lewy bodies and Lewy neurites, the cytopathological hallmarks of this group of disorders (15, 16).
αS partially localizes to mitochondria in neuronal cells (17) and in αS transgenic mice (18), as recently confirmed by others (19–22). The mitochondrial import of αS is energy-dependent and seems to require an outer membrane protein import channel. Once in the mitochondria, αS predominantly associates with the inner mitochondrial membrane, where it can apparently interact with and inhibit complex I activity, resulting in increased reactive oxygen species production (21). This effect is enhanced by the pathogenic A53T αS mutation (21). Importantly, accumulation of αS was also observed in isolated mitochondria from post-mortem PD patients (21).
Here we sought to determine whether αS-related pathology is directly associated with mitochondrial dysfunction, represented by complex I activity. Specifically, we searched for an association between mitochondrial complex I dysfunction and αS-related pathology, represented by either αS-soluble oligomers, αS-insoluble aggregates, PD-like symptoms, or mouse age. We report that αS overexpression in cultured dopaminergic cells or A53T αS tg mouse brains inhibits complex I activity, yet no correlation between αS-related pathology and mitochondrial complex I dysfunction was found. We therefore conclude that αS inhibits complex I activity as part of its normal, physiological function.
MATERIALS AND METHODS
Cells
MN9D, a mesencephalic neuronal cell line with dopaminergic properties (23) was stably transfected with wt human αS cDNA in the pCDNA 3.1 vector. We routinely selected those clones expressing 6–8-fold higher transfected than endogenous αS. Because of the reported tendency of the stable αS clones to lose their expression, we have kept frozen aliquots of specific clones and used the clones 3–8 weeks after thawing (24). To eliminate the possibility of a specific clonal effect, we repeated the experiments in two to four different αS-overexpressing clones. Some of the measurements were repeated in HEK 293 polyclones stably expressing either wt, A53T αS, or the empty vector.
Mice
wt, C3H (Harlan, Israel), and C57BL/J6 (Jackson Laboratories), αS−/− (C57BL/J6, Harlan) (25), or homozygous A53T αS tg (26) (Jackson Laboratories) mice were housed in an animal facility that is fully compliant with the Public Health Service Policy on Humane Care and Use of Laboratory Animals. The overexpression of the transgenic A53T αS was estimated to be 4.6 ± 0.8-fold higher than endogenous αS (26). Similar A53T αS expression levels were detected in the mouse colony we bred (not shown).
Detection of αS Oligomers
αS-overexpressing MN9D cells (5 × 107) or whole mouse brains were fractionated as described (24). Protein samples of high speed supernatant (post 280,000 × g) were incubated at 65 °C for 16–18 h for antigen retrieval (24) before loading on a 10% SDS-PAGE and blotted with syn-1 antibody (BD Biosciences) or LB509 antibody (Zymed Laboratories Inc.).
Brain Fractionation and Chloroform Methanol Extraction
A whole brain of young (4–6 weeks old) or old (12–15 months old) mouse was fractionated as previously described (27), and chloroform/methanol extraction of the lipid-rich fraction was performed as described (27). Briefly, the low speed, lipid-rich fraction floating on top of the sucrose cushion was collected and brought to a final volume of 0.5 ml. Three volumes of chloroform/methanol (2:1) were added. The lipid-rich fraction was extracted for 20 min at room temperature. The extract was centrifuged at 8,000 × g for 10 min, and the interface between the organic (lower) and aqueous (upper) fractions was resuspended in 2× Laemmli buffer, analyzed by Western blotting, and blotted with LB509 antibody.
Isolation of Mitochondria
Fresh tissues of mouse brain or subconfluent cultured cells were used. The cells were washed once in cold PBS prior to homogenization. Alternatively, one mouse brain hemisphere (PBS-perfused) was homogenized in buffer A (320 mm sucrose, 5 mm Tris, 2 mm EGTA, pH 7.4) with 10 strokes of a Teflon Dounce homogenizer. The procedure was performed at 4 °C. The lysate was centrifuged for 3 min at 2000 × g to remove nuclei and cell debris. The supernatants were then transferred to a clean tube and centrifuged for 10 min at 12,000 × g to pellet mitochondria. The crude pellet was resuspended in buffer A with the addition of 0.02% (w/v) digitonin to release trapped mitochondria. The pellet was resuspended and washed two or three times in a large volumes of digitonin-free buffer. Finally, the mitochondria pellet was resuspended in a small volume of buffer A. The mitochondria were either used directly for oxygen consumption or stored in aliquots at −70 °C for enzymatic activities.
Enzymatic and Polarographic Assays
The activity of complex I (rotenone-sensitive NADH:ubiquinone oxidoreductase), complex II (succinate:ubiquinone oxidoreductase), complex I–III (rotenone-sensitive NADH:cytochtome C oxidoreductase), cytochrome c oxidase (COX), and citrate synthase were assayed as previously described (28). Briefly, complex I specific activity was measured by following the decrease in absorbance at 340 nm caused by the oxidation of NADH. A sample (5 μg of protein) of purified mitochondria after osmotic shock in double distilled water was added in buffer containing 25 mm potassium phosphate, pH 7.4, 2 mm KCN, 5 mm MgCl2, 2.5 mg/ml BSA, 2 μm antimycin, 100 μm decyl-ubiquinone, and 0.3 mm K2NADH at 37 °C. The reaction was measured for 3–5 min before adding rotenone (1 μg/ml). The reaction was measured 3 min longer in the presence of rotenone. The nonspecific activity of NADH hydrogenase, represented by rotenone-insensitive activity, was subtracted.
We note that analyses of complex I activity in mitochondria purified from cultured cells was problematic. The results were considered only from batches of purified mitochondria that demonstrated above 50% rotenone sensitivity in the enzymatic assay. Some of the measurements were also performed separately in two labs (Dr. Sharon's lab and the metabolic diseases unit). Notably, rotenone-sensitive NADH-CoQ reductase more accurately represents mitochondrial respiratory complex I as opposed to the rotenone-insensitive outer membrane NADH cytochrome c reductase (29). In mouse brains, purified mitochondria demonstrated high rotenone sensitivity (higher than 90%) and therefore highly repeatable results.
Oxygen Consumption
Oxygen consumption was measured using a Clarck-type electrode in a sealed chamber (Oxygraph; Hansatech) as previously described (30). A fresh protein sample of 15–25 μg of isolated mitochondria (without a digitonin wash) was suspended in Oxymetry buffer (5 mm K2HPO4, 10 mm Tris-HCl, pH 7.4, 100 mm KCl, 5 mm MgCl2, 5 μm EDTA, 90 mm mannitol, 25 mm sucrose). State IV respiration (basal respiration) was determined by the addition of glutamate-malate, and State III respiration (active respiration) was determined by the addition of ADP (0.25 mm).
αS Pathology
Mice, anesthetized with an intraperitoneal overdose injection of sodium pentobarbitone (1 ml/1.5 kg), were perfused with PBS or with PBS-buffered formalin. Following surgical removal of brain and spinal cord, the tissue was kept frozen and used for further biochemical analyses (PBS perfusion) or fixed for another 24 h in the formalin perfusion. Histochemical analysis was performed with formalin-fixed tissue as previously described (31). Briefly, sections of 6 μm were deparaffinized in xylene followed by graded alcohol in descending ethanol concentrations. Endogenous peroxidase activity was inhibited by incubation in methanol/H2O2 (150 ml methanol and 30 ml of 30% H2O2). Antigen retrieval was performed by incubating the slides in 100% formic acid for 5 min followed by extensive washes. The sections were blocked in 2% fetal bovine serum in 0.1 m Tris-HCl, pH 7.6. The sections were then immunostained using anti-αS antibody LB 509 (1:1000) or Syn 303 (1:3000; a gift from Prof. Virginia M.-Y. Lee). Secondary antibody was biotinylated donkey anti-mouse (1:200; Enco Petach Tikvah, Israel), followed by ExtrAvidin (Sigma; 1:100 in blocking solution). Immunoreactivity was visualized with an EnzMet detection kit (Nanoprobes, Yaphank, NY) or diaminobenzidine as chromogen (Zymed Laboratories Inc.).
Pathological Quantitation
We analyzed total αS immunoreactivity. The images were captured with an 60× objective in light microscope, using neutral density filter and calibration of the white color with overlapping spectrum for red, green, and blue. We compared only series of pictures taken together after a particular event of Kohler calibration. The images were quantified with the Image pro plus 6.3 program (Media Cybernetics). The selection of objects was done automatically by color definition. For object specification we used the smoothing options. The selected objects above 2000 pixels were exported to Excel as integrated optical density (area × average density), and background was subtracted from the averaged integrated optical density in each image. The results are presented as 1/integrated optical density. Three brain regions were selected for analyses: cortex, midbrain, and brain stem. In each brain region the total αS immunoreactivity was quantified in three fields for an individual mouse brain. The difference between transgenic mouse brains was compared using Student's t test with correction for unequal (heteroscedastic) variance.
Symptoms
The mice were observed daily for typical clinical signs, including altered gait, kyphosis, ataxia, and wasting. At 7–8 months of age, ∼10–20% of the mice were showing symptoms, including paralysis of hind limbs and freezing for a few seconds. We designated the time of PD symptom appearance when at least two of the indicated symptoms were observed in an individual mouse for at least four consecutive days. Males suffered an acute disease and died within 1 week of symptoms appearance, whereas females suffered milder symptoms that lasted for a longer periods of time. Few mice survived the age of 15 months (5–10% of the colony), with an overall longer life span for the females.
Blue Native Gel
Samples of purified mitochondria were prepared and analyzed by blue native-PAGE as previously described (32). Briefly, frozen aliquots of purified mitochondria were suspended in sample buffer (1 m aminocaproic acid, 50 mm bis-Tris-HCl, and protease inhibitors mixture; Sigma, P8340). The membrane proteins were solubilized with dodecylmaltoside (Sigma) and quantified using Bradford assay (33). Coomassie Brilliant Blue G-250 (Sigma) was added to the samples to a final concentration of 0.36%. The samples were separated on blue native-PAGE (7%) and transferred to polyvinylidene difluoride membrane. The membranes were blocked in 5% nonfat milk-TBST and immunoblotted using anti-complex I (NDUFS3 MS110) or anti-complex V (F1F0 subunit MS501c) mouse monoclonal antibodies (Mitosciences, Eugene, OR).
Statistical Analyses
The nonparametric Kruskal-Wallis analysis of variance test was applied to compare complex I activity between the three age groups. Pair wise comparisons between the wt group and αS tg group at each age were carried out using the nonparametric Mann-Whitney test, with the Bonferroni correction of the significance level for multiple pair wise comparisons (p < = 0.017 was considered statistically significant).
RESULTS
Purifications of Mitochondria
Mitochondria were purified from cultured MN9D cells or whole mouse brains and analyzed by Western blotting. The efficacy of the purification methods was determined using specific antibodies against α-tubulin, a cytosolic protein marker, and HSP-60, a mitochondrial protein marker. The fraction obtained, represents purified mitochondria, enriched in HSP-60 and carry only traces of the cytosolic protein, α-tubulin (Fig. 1a).
FIGURE 1.
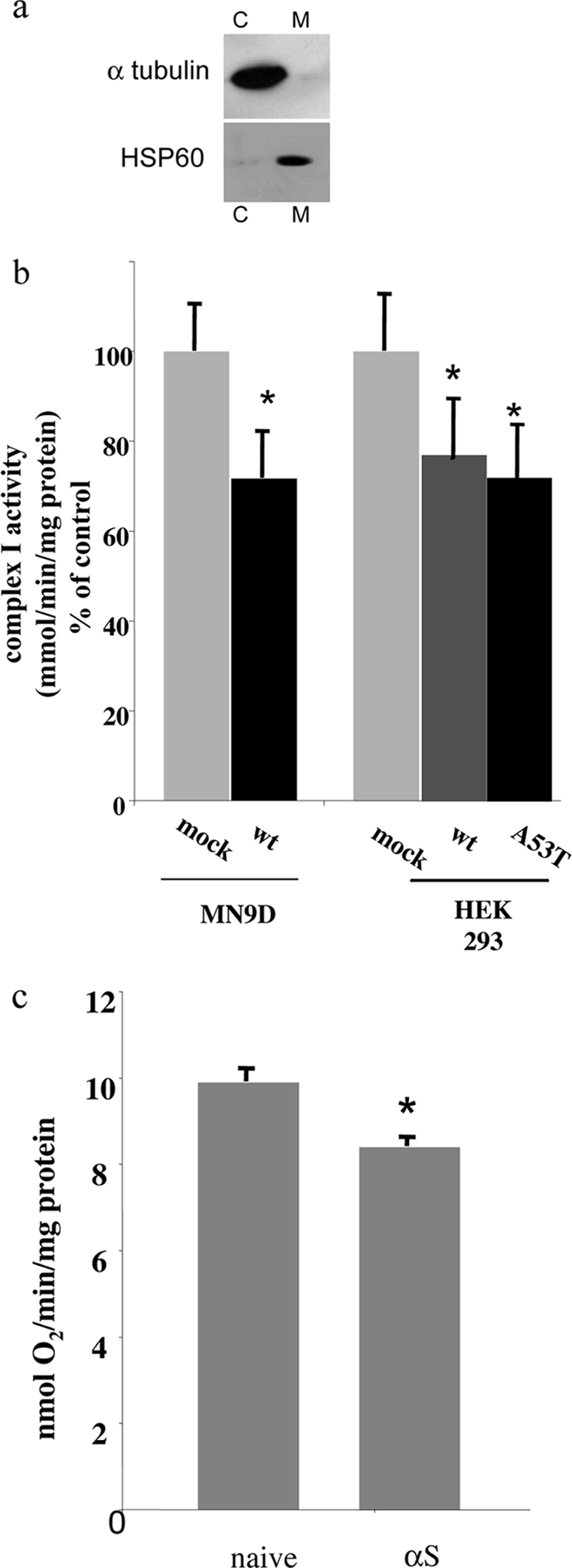
Lower complex I activity in mitochondria purified from αS-overexpressing MN9D dopaminergic cells. a, samples (20 μg protein) of purified mitochondria (M) or cytosol (C) were analyzed by 12% Tris-glycine PAGE, Western blotted, and probed with anti-HSP-60 or anti-α-tubulin antibodies. b, the activity of complex I (rotenone-sensitive NADH:CoQ oxidoreductase) was measured in a sample (5 μg of protein) of purified mitochondria as described under “Materials and Methods.” The results obtained as nmol of oxidized NADH/min/mg of protein are normalized to citrate synthase activity in the same sample and are presented as percentages of mock-transfected cells, designated 100%. The values are the means of n = 4–10 mitochondria preparations ± S.E. *, t test p < 0.02. c, O2 consumption was measured in a sample (100–200 μg of protein) as described under “Materials and Methods” with glutamate-malate (state 4) and ADP (state 3). The values are the means of n = 4 mitochondria preparations ± S.E. *, t test p < 0.05.
αS Overexpression in Cultured Dopaminergic Cells Inhibits Complex I Activity
We initially sought to determine whether αS overexpression affects mitochondrial complex I activity in cultured dopaminergic cells. For this aim, we used stable clones of αS-overexpressing MN9D cells (see methods). Mock-transfected and αS-overexpressing MN9D dopaminergic cells were cultured in parallel; mitochondria were purified; and complex I activity, represented by rotenone-sensitive NADH-CoQ reductase activity, was measured spectrophotometrically (see “Materials and Methods”). Overall, we measured a significant ∼25% lower complex I activity in purified mitochondria from αS-overexpressing cells than mock-transfected cells (Fig. 1b). A similar inhibition in complex I activity was detected in HEK 293 cells stably expressing either wt αS or A53T αS as compared with mock-transfected control cells (Fig. 1b).
To determine whether the αS inhibitory effect is restricted to complex I activity, as previously shown for PD (3–5), we measured complex II, I–III, and complex IV activities in purified mitochondria of mock-transfected and αS-overexpressing MN9D cells. No significant effect on the activity level of these complexes was detected in αS-overexpressing cells as compared with the mock-transfected cells. Furthermore, no significant effect of αS expression on matrix activities, represented by citrate synthase, was detected (not shown). Therefore, αS overexpression in cultured MN9D dopaminergic cells specifically inhibits mitochondrial complex I activity.
To determine whether the inhibitory effect of αS overexpression on complex I activity affects mitochondrial respiration, we measured O2 consumption (see “Materials and Methods”) in naïve and αS-overexpressing cells. A significant reduction of ∼15% in O2 consumption was measured in mitochondria purified from αS-overexpressing than naïve cells (Fig. 1c). Therefore, αS overexpression in MN9D cells resulted in inhibited mitochondria respiration.
The Lower Complex I Activity in A53T-overexpressing Mouse Brains Is Age-independent
To study the effect of αS overexpression on complex I activity in vivo, we purified mitochondria from wt and A53T αS tg mouse brains (26) and measured complex I activity represented by rotenone-sensitive NADH-CoQ reductase. The analysis was performed in young (4–6 weeks old), adult (8–9 months old), and old (12–15 months old) mice (the oldest age obtained for the αS tg mice) (26). Lower complex I activity was measured in purified mitochondria from αS tg than wt mouse brains throughout the tested ages. Importantly, a significant ∼24% lower complex I activity was measured already in young (4–6 weeks) A53T αS than wt mouse brains (Fig. 2). Similarly, a significant ∼22% lower complex I activity was measured in brains of A53T αS ages 8 and 12–15 months. Importantly, no significant differences in complex I activity were detected between the tested ages within the genotypes. Therefore, the inhibitory effect of αS overexpression on complex I activity in mouse brains is independent of mouse age.
FIGURE 2.

The inhibited complex I activity in brains of A53T αS tg mice is independent of mouse age. Complex I activity was measured in purified mitochondria from brains of 1-, 8-, and 12–15-month-old wt and A53T αS tg mice (26) as in Fig. 1b. Relative complex I activity is normalized to wt control for each age group and is plotted as the percentage of activity ± S.D. Significant differences are detected between the two genotypes in the three age groups. No significant differences are detected between age groups in the same genotype (n = 6–8 mice in each age group for A53T αS and wt mice). *, p < 0.017 analysis of variance for multiple pair comparison.
We next sought to compare complex I activity in αS−/− and wt mouse brains. However, no significant effect for αS depletion on complex I activity was detected in the specific mouse line (25).
αS Overexpression Does Not Affect Mitochondrial Assembly
Lower complex I activity could potentially result from affected assembly or fewer mitochondria. We analyzed samples containing 30 μg of protein of purified mitochondrial preparations of wt and A53T αS tg mouse brains by blue native-PAGE followed by Western blotting. The Western blot was reacted with anti-NDUFS3, complex I antibody; anti-COXI, complex IV antibody; or anti-F1F0 ATPase, complex V antibody. No effect on the electrophoretic migration of complex I, IV, or V was detected (Fig. 3a). To determine the total amount of mitochondria, we analyzed, in parallel, samples containing 30 μg of protein of wt and A53T αS whole mouse extracts by SDS-PAGE followed by Western blotting with anti-HSP-60 antibody and found similar levels of HSP-60 immunoreactivity (Fig. 3b). We next normalized the densitometric signal obtained for NDUFS3, COXI, and F1F0 subunits (Fig. 3a) to citrate synthase activity levels, measured in aliquots of the same samples. No differences in the protein levels of the specific subunits were detected (Fig. 3c). We finally normalized HSP-60 immunoreactivity (Fig. 3b) to citrate synthase activity measured for these samples and found a highly similar ratio of the mitochondrial HSP-60 expression levels to matrix protein activity (Fig. 3c).
FIGURE 3.

Complex I or mitochondria assembly are not affected in A53T αS mouse brains. a, samples of 30 μg of protein of purified mitochondria from whole A53T αS or wt mouse brains (8 months of age), processed, and analyzed by 7% blue native-PAGE (see “Materials and Methods”) followed by Western blotting with anti-NDUFS3 antibody (complex I, CI), anti-COX-1 (complex IV, CIV), and anti-F1F0 ATPase (complex V, CV) antibody (Mitosciences). b, protein samples (30 μg) of whole A53T αS or wt mouse brain extracts were analyzed by SDS-PAGE, Western blotted, and probed with anti-HSP60 antibody. c, densitometric quantitation of the specific subunits normalized to citrate synthase activity.
No Correlation between PUFA-induced αS Oligomerization and Mitochondrial Dysfunction in MN9D Dopaminergic Cells
In our recent studies we described aspects of αS and PUFAs association in relevance to αS pathogenesis. Specifically, we have shown that exposure to physiological PUFA (but not MUFA) concentrations, enhances αS oligomerization and aggregation (34), phosphorylation and deposition in cytoplasmic inclusions, in cultured dopaminergic cells (24). We now utilized this experimental system to enhance αS pathogenesis in cultured αS-overexpressing dopaminergic cells. To find out whether enhanced αS pathogenesis associates with mitochondrial complex I dysfunction, we conditioned mock-transfected and αS-overexpressing MN9D cells in serum-free medium (serum being the source for FA in the conditioning media), supplemented with BSA only (serving as a FA carrier at 50 μm) as control or BSA+ FA (at 50 and 250 μm, respectively). The effect of PUFA, α-linolenic acid 18:3, and MUFA, oleic acid 18:1, were tested in parallel, in sister cultures, and compared with BSA-treated sister culture. The cells were conditioned in the specified medium for 16 h and then either processed for mitochondria purification or analyzed for soluble αS oligomerization in the high speed supernatant. Similar to our previous reports (24, 34), 18:1 MUFA did not affect, whereas 18:3 PUFA dramatically induced (approximately ×40), the levels of αS soluble oligomerization (Fig. 4a). Further, 18:3 PUFA but not 18:1 MUFA induced the formation of αS- and ubiquitin-positive cytoplasmic inclusions (Fig. 4b). Nevertheless, complex I activity was not significantly affected by the 18:3 PUFA treatments. Specifically, complex I activity levels were highly similar in 18:1 versus 18:3 treated αS-overexpressing cells (Fig. 4c). Likewise, no effect on O2 consumption was detected with 18:3 PUFA. Therefore, PUFA-enhanced αS oligomerization and inclusion formation in cultured dopaminergic cells is not associated with complex I dysfunction.
FIGURE 4.

18:3 PUFA-induced αS oligomerization and inclusion formation is not associated with complex I inhibition in MN9D dopaminergic cells. a, α-linolenic acid, a 18:3 PUFA but not oleic acid, a 18:1 MUFA of identical carbon chain length, induces the oligomerization of αS. High speed cytosols (15 μg of protein) of human wt αS stably transfected MN9D cells conditioned in standard serum-supplemented medium and then transferred to serum-free medium with or without 18:3 or 18:1 (250 μm) for 16 h. The samples were incubated at 65 °C overnight prior to gel loading and blotting with H3C antibody. b, αS-overexpressing cells were conditioned in standard serum-supplemented medium and then transferred to serum-free medium supplemented with BSA + 18:3 or 18:1 (50 and 250 μm for BSA and FA, respectively) for 18 h. The cells were then processed for ICC (24) using antibodies against αS (LB509; Zymed Laboratories Inc.) and ubiquitin (Dako), followed by Alexa 488 and Cy5, respectively. The bars represents 10 μm. c, complex I activity as described for b. M.W, molecular weight.
No Correlation between Levels of αS-soluble Oligomers and Complex I Activity in A53T αS Mouse Brains
We have recently shown evidence indicating that soluble αS oligomers are cytotoxic and precede the formation of αS-positive intracellular inclusions (24). To test whether αS oligomerization associates with mitochondrial complex I dysfunction in vivo, we sacrificed a cohort of 12 adult, 8–9-month-old A53T αS tg mice, including symptomatic and asymptomatic mice. One brain hemisphere was processed for αS oligomers detection in high speed supernatant (Fig. 5, a and b), and mitochondria were purified from the second hemisphere and analyzed for complex I activity (Fig. 5c). For each mouse brain the results for levels of αS oligomers were aligned versus complex I activity and the appearance of symptoms. We then statistically analyzed the results of mice 1–12 using nonparametric analysis of variance and found no correlation between αS oligomers levels and complex I activity (Fig. 5d) or symptom appearance (see “Materials and Methods”). Similarly, no correlation between mouse endogenous αS oligomerization and complex I activity was found in wt mouse brains (Fig. 5e).
FIGURE 5.

αS oligomers do not associate with inhibited complex I activity. The brains of a cohort of 12 A53T αS tg mice 8–9 months old (Symptomatic and asymptomatic) were divided in hemispheres. a, αS soluble oligomers in high speed cytosols (15 μg protein) of A53T αS tg mice. The samples were treated at 65 °C overnight prior to gel loading and blotting with LB509 antibody. b, densitometry of Western blot in a, presented as the ratio of αS oligomers (migrating higher than 35 kDa)/monomer (migrating as 17 kDa) and normalized to actin. c, complex I activity measured in purified mitochondria from the respective mouse brain hemisphere. The activity of complex I (rotenone-sensitive NADH:CoQ oxidoreductase) was measured as described for b. d, no correlation between human A53T αS oligomerization and complex I activity (n = 12). e, no correlation between endogenous mouse αS and complex I activity (n = 9).
No Correlation between αS Pathology and Complex I Activity in Vivo
Finally, we sought to determine whether mitochondrial complex I activity correlates with αS pathology in brains of old A53T αS tg mice. The detection of αS pathology was performed in mouse brains perfused with formaldehyde, whereas mitochondria were purified from mouse brains perfused with PBS. For this reason, an individual αS tg mouse brain was tested either for pathology or for complex I activity. αS pathology in A53T αS mouse brains was analyzed by immunohistochemistry in three brain regions: cerebellum, brain stem, and substantia nigra (Fig. 6, b–d). αS pathology was indistinguishable from total αS protein levels and involved enhanced total αS protein levels (35, 36) including Lewy neurites. We quantified the total αS signal obtained by immunohistochemistry in the brain stem, where αS pathology was previously reported for this transgenic mouse line (26). Comparing the total αS signal by immunohistochemistry in the five old (12–15 months) and six young (4–6 weeks) A53T αS mouse brains, we found a significant ∼2-fold higher αS signal in the old mouse brains than in the young mouse brains (p < 0.05, t test with correction for unequal variance) (Fig. 6e), indicating pathogenic accumulations of αS in the old A53T tg mouse brains.
FIGURE 6.

αS pathology is not associated with inhibited complex I activity. Representative images of immunohistochemistry analysis of A53T αS tg mouse brains aged 12–15 months. Formalin-fixed tissue immunostained with anti-αS antibody (Syn 303) and visualized with diaminobenzidine as chromogen (see “Materials and Methods”). a–d, Nontransgenic (a), transgenic brain stem (b), transgenic substantia nigra (c), and transgenic cerebellum (d). αS staining appears throughout the cell body with some Lewy neurites and Lewy bodies (arrowheads). e, quantitation of total αS immunoreactivity (including pathogenic and normal expression) in the brain stem of young (4–6 weeks) and old (12–15 months) A53T αS mouse brains. The values are the means of n = 5–6 brains and 3–4 images for each mouse brain ± S.D. *, t test, p < 0.05. f, insoluble, lipid associated-αS in young (4–6 weeks old) and old (12–15 months old) A53TαS tg mouse brains. Chloroform/methanol extraction of a sample of the lipid-rich low speed fraction (50 μl of total 500 μl of suspension) of young and old A53T αS mouse brains. The interface between the organic (lower part) and aqueous (upper part) fractions was resuspended in 2× Laemmli buffer, analyzed by 10% Tris-glycine PAGE, and blotted with LB509. g, quantitation of αS immunoreactivity in f presented as the ratio between the high molecular weight immunoreactive species to monomer in the same lane.
We then compared the levels of insoluble αS immunoreactivity associated with the lipid-rich, low speed fraction (27) extracted from one hemisphere of young or old αS tg mouse PBS-perfused brains. Importantly, this high molecular weight, insoluble αS signal was associated with the pathogenic αS signal in the synucleinopathies PD and DLB (34). The lipid-rich fraction was extracted by chloroform methanol (2:1), and the interface between the aqueous and organic phases was analyzed by Western blot and probed with LB509 antibody (Fig. 6f). The result indicates a ∼7–9-fold higher ratio of high molecular weight αS to monomer signal in the old compared with the young mouse brains (Fig. 6g).
Importantly, although αS pathology was significantly higher in the old versus young αS tg mouse brains, complex I activity did not differ between these age groups (Fig. 2). We therefore conclude that age-dependent accumulations of αS pathology do not associate with mitochondrial complex I inhibition in the A53T αS mouse brains.
DISCUSSION
In this study we aimed at assessing whether αS pathology, i.e. accumulation of soluble oligomers, insoluble aggregates, or inclusion formation, is directly associated with mitochondrial dysfunction. We report that either human wt αS overexpression in cultured dopaminergic cells or transgenic overexpression of A53T αS in mouse brains inhibits mitochondrial complex I activity. Our results suggest that the inhibitory effect of αS overexpression on complex I activity is not associated with levels of αS-related pathology in cultured dopaminergic cells nor in mouse brains. Specifically, enhanced αS oligomerization and inclusion formation, induced by 18:3 PUFA treatment, did not significantly affect complex I activity in cultured αS-overexpressing MN9D dopaminergic cells. Further, no correlation between complex I activity and either age of αS tg mice, levels of αS oligomers, or levels of insoluble αS was found. Based on the results presented herein, we conclude that αS expression and not its pathology associates with inhibited mitochondrial complex I activity.
Recent studies have reported that αS expression induces oxidative stress and impairs complex I activity, as part of its deleterious effects attributed to mitochondrial dysfunction in PD pathogenesis (21, 37). Transgenic overexpression of A53T αS was shown to inhibit complex IV in mouse brains (18). Attempts to correlate αS toxicity and mitochondrial dysfunction have recently been made using in vivo and in vitro models. Specifically, it was shown that aggregated, but not nonaggregated, αS associates with isolated rat brain mitochondria and induces calcium-dependent opening of the mitochondrial permeability transition pore and cytochrome c release (22). In this regard, it was suggested that aggregated αS forms amyloid pores capable of puncturing cell membranes leading to the release of cytochrome c from the mitochondria (38–40). However, other studies have suggested that αS expression may have a constitutive role in maintaining or regulating mitochondrial respiratory chain complexes. Specifically, using αS siRNA transfections there was a low but significant decrease in NADH-cytochrome c reductase levels, representing complex I–III activity (21), and lower NADH-cytochrome c reductase activity was detected in αS null mouse brains (41). In line with the functional link between αS expression and mitochondrial activities, αS null mice are resistant to the mitochondrial toxins MPTP, malonate, and 3-nitropropionic acid (42, 43). In this regard, the results presented herein, describing the absence of any correlation between αS pathology and mitochondrial complex I activity, suggest that the inhibitory effect of αS expression on complex I activity results from a regulatory, physiological, effect of αS rather than a pathogenic effect.
Complex I inhibitors and therefore mitochondrial dysfunction enhance αS pathology in mouse models. Specifically, chronic MPTP treatment was shown to enhance the formation of endogenous αS- and ubiquitin-positive inclusions in wt mouse brains (44) and mitochondrial degeneration in wt αS tg mouse brains (45). Similarly, rotenone, a classical inhibitor of complex I, was shown to induce the formation of αS-positive inclusions (46–48). Neuronal αS pathology and mitochondrial degeneration were enhanced in A53T but not wt αS tg mice chronically treated with the combination of two pesticides, paraquat and maneb. Importantly, the combination of the two pesticides was needed to detect the deleterious effects (49). Interestingly, paraquat alone was reported to induce αS expression levels and the formation of intraneuronal inclusions in A53T αS tg mice; however, it did not induce nigral degeneration (50). These studies consistently suggest that mitochondrial dysfunction may enhance αS pathology leading to PD and therefore suggest that mitochondrial dysfunction occurs prior to αS pathology and Lewy body formation. Interestingly, it was suggested that the enhanced αS pathogenesis by complex I inhibitors may be protective (50, 51).
αS has a role in deleterious mechanisms leading to neuronal cell death. However, the precise mechanism activating cell death is unknown. A key event in αS-related toxicity is its tendency toward misfolding and aggregation, resulting in its deposition in neuronal cytoplasmic inclusions, the Lewy bodies (recently reviewed in Ref. 52). Various cellular mechanisms were shown to mediate αS toxicity, including inhibition of the proteasome and the cellular quality control, disruption of axonal transport, synaptic transmission, and mitochondrial dysfunction and enhancement of oxidative stress (reviewed in Ref. 11). Mitochondria play a central role in the regulation of apoptotic cell death, and indeed there is considerable evidence suggesting that apoptosis might account for cell death in PD (53). Enhanced expression of apoptotic markers, including expression of p53, Bcl-2-associated X protein, caspase-3, and nuclear translocation of glyceraldehydes phosphate dehydrogenase, has been detected in PD (53–55).
It was recently suggested that αS toxicity involves aspects of membrane trafficking, possibly in endocytosis or in the secretory pathway. Specifically, αS expression inhibits endoplasmic reticulum to Golgi trafficking, resulting in cytotoxicity that was prevented by Rab1 expression (56). It was further shown that αS expression affected vesicle docking or fusion to the Golgi apparatus after an efficient budding from the endoplasmic reticulum (57). In PC12 cells, αS overexpression inhibited evoked catecholamine release and increased the “docked” vesicle pool (58). Other studies have suggested an indirect role for αS in promoting the assembly of the SNARE complex (59, 60). SNAREs catalyze the fusion of vesicles with their target membranes to enable the release of cargo from the vesicle (reviewed in Refs. 61 and 62). Collectively, growing evidence implicates αS in membrane trafficking, including endocytosis and exocytosis. We have recently shown that αS specifically affects synaptic vesicle recycling by activating clathrin-mediated endocytosis. Based on our findings we suggested that αS affects synaptic function and strength (63). Importantly, we found a correlation between the αS activity and levels of αS soluble oligomers. Specifically, the more αS oligomers, the more endocytic activity (63).
Membrane trafficking is also implicated in mitochondrial dynamics, involving fusion and fission events. PD toxins, such as 6-hydroxydopamine, rotenone, and the 1-methyl-4-phenylpyridinium ion induce mitochondrial fission and cell death (64–66). The role of mitochondrial dynamics in PD was recently emphasized by the finding that PD-related genes PINK-1 and parkin regulate mitochondrial fission/fusion events (67). It is therefore of interest to find out whether αS oligomers or αS cytotoxicity is involved in mitochondrial dynamics.
The ongoing debate concerning the identification of the toxic αS forms, whether it is the soluble αS oligomers, insoluble aggregates, or Lewy bodies, relies on analogies and evidence from other neurodegenerative diseases such as Alzheimer and Huntington diseases (recently reviewed in Ref. 68). A major difficulty in identifying the toxic αS form is the absence of clear definite information regarding the normal function of αS or the initial deleterious mechanism(s) activated in PD by αS. Additionally, aging and αS pathology were recently shown to amplify αS expression and toxicity (36), adding an additional aspect of αS complexity. The results presented herein provide a negative clue as to the pathogenic effects of αS by denying a deleterious role for αS toxic forms in complex I activity.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 NS051318.
- PD
- Parkinson disease
- αS
- α-synuclein
- tg
- transgenic
- wt
- wild type
- FA
- fatty acid
- PUFA
- polyunsaturated fatty acids
- MUFA
- monounsaturated fatty acids
- MPTP
- 1-methyl-4-phenyl-1,2,3,4-tetrahydropyridine
- PBS
- phosphate-buffered saline
- COX
- cytochrome c oxidase
- BSA
- bovine serum albumin
- bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- NDUFS3
- NADH dehydrogenase [ubiquinone] iron-sulfur protein 3
- SNARE
- soluble NSF attachment protein receptors.
REFERENCES
- 1.Langston J. W., Ballard P. A., Jr. (1983) N. Engl. J. Med. 309, 310. [DOI] [PubMed] [Google Scholar]
- 2.Nicklas W. J., Vyas I., Heikkila R. E. (1985) Life Sci. 36, 2503–2508 [DOI] [PubMed] [Google Scholar]
- 3.Keeney P. M., Xie J., Capaldi R. A., Bennett J. P., Jr. (2006) J. Neurosci. 26, 5256–5264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mizuno Y., Ohta S., Tanaka M., Takamiya S., Suzuki K., Sato T., Oya H., Ozawa T., Kagawa Y. (1989) Biochem. Biophys. Res. Commun. 163, 1450–1455 [DOI] [PubMed] [Google Scholar]
- 5.Schapira A. H., Cooper J. M., Dexter D., Jenner P., Clark J. B., Marsden C. D. (1989) Lancet 333, 1269. [DOI] [PubMed] [Google Scholar]
- 6.Martins L. M., Morrison A., Klupsch K., Fedele V., Moisoi N., Teismann P., Abuin A., Grau E., Geppert M., Livi G. P., Creasy C. L., Martin A., Hargreaves I., Heales S. J., Okada H., Brandner S., Schulz J. B., Mak T., Downward J. (2004) Mol. Cell. Biol. 24, 9848–9862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang L., Shimoji M., Thomas B., Moore D. J., Yu S. W., Marupudi N. I., Torp R., Torgner I. A., Ottersen O. P., Dawson T. M., Dawson V. L. (2005) Hum. Mol. Genet. 14, 2063–2073 [DOI] [PubMed] [Google Scholar]
- 8.Darios F., Corti O., Lücking C. B., Hampe C., Muriel M. P., Abbas N., Gu W. J., Hirsch E. C., Rooney T., Ruberg M., Brice A. (2003) Hum. Mol. Genet. 12, 517–526 [DOI] [PubMed] [Google Scholar]
- 9.Clark I. E., Dodson M. W., Jiang C., Cao J. H., Huh J. R., Seol J. H., Yoo S. J., Hay B. A., Guo M. (2006) Nature 441, 1162–1166 [DOI] [PubMed] [Google Scholar]
- 10.Canet-Avilés R. M., Wilson M. A., Miller D. W., Ahmad R., McLendon C., Bandyopadhyay S., Baptista M. J., Ringe D., Petsko G. A., Cookson M. R. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 9103–9108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee V. M., Trojanowski J. Q. (2006) Neuron 52, 33–38 [DOI] [PubMed] [Google Scholar]
- 12.Moore D. J., West A. B., Dawson V. L., Dawson T. M. (2005) Annu. Rev. Neurosci 28, 57–87 [DOI] [PubMed] [Google Scholar]
- 13.Hardy J., Cai H., Cookson M. R., Gwinn-Hardy K., Singleton A. (2006) Ann. Neurol. 60, 389–398 [DOI] [PubMed] [Google Scholar]
- 14.Duda J. E., Lee V. M., Trojanowski J. Q. (2000) J. Neurosci. Res. 61, 121–127 [DOI] [PubMed] [Google Scholar]
- 15.Spillantini M. G., Schmidt M. L., Lee V. M., Trojanowski J. Q., Jakes R., Goedert M. (1997) Nature 388, 839–840 [DOI] [PubMed] [Google Scholar]
- 16.Goedert M. (2001) Nat. Rev. Neurosci. 2, 492–501 [DOI] [PubMed] [Google Scholar]
- 17.Perez R. G., Waymire J. C., Lin E., Liu J. J., Guo F., Zigmond M. J. (2002) J. Neurosci. 22, 3090–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin L. J., Pan Y., Price A. C., Sterling W., Copeland N. G., Jenkins N. A., Price D. L., Lee M. K. (2006) J. Neurosci. 26, 41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang L., Zhang C., Zhu Y., Cai Q., Chan P., Uéda K., Yu S., Yang H. (2008) Brain Res. 1244, 40–52 [DOI] [PubMed] [Google Scholar]
- 20.Shavali S., Brown-Borg H. M., Ebadi M., Porter J. (2008) Neurosci. Lett. 439, 125–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Devi L., Raghavendran V., Prabhu B. M., Avadhani N. G., Anandatheerthavarada H. K. (2008) J. Biol. Chem. 283, 9089–9100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parihar M. S., Parihar A., Fujita M., Hashimoto M., Ghafourifar P. (2008) Cell Mol. Life Sci. 65, 1272–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi H. K., Won L. A., Kontur P. J., Hammond D. N., Fox A. P., Wainer B. H., Hoffmann P. C., Heller A. (1991) Brain Res. 552, 67–76 [DOI] [PubMed] [Google Scholar]
- 24.Assayag K., Yakunin E., Loeb V., Selkoe D. J., Sharon R. (2007) Am.5 J. Pathol. 171, 2000–2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Specht C. G., Schoepfer R. (2001) BMC Neurosci. 2, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giasson B. I., Duda J. E., Quinn S. M., Zhang B., Trojanowski J. Q., Lee V. M. (2002) Neuron 34, 521–533 [DOI] [PubMed] [Google Scholar]
- 27.Sharon R., Goldberg M. S., Bar-Josef I., Betensky R. A., Shen J., Selkoe D. J. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 9110–9115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saada A., Bar-Meir M., Belaiche C., Miller C., Elpeleg O. (2004) Anal. Biochem. 335, 66–72 [DOI] [PubMed] [Google Scholar]
- 29.Ragan C. I., Wilson M. T., Darley-Usmar V. M., Lowe P. N. (1987) Subfractionation of Mitochondria and Isolation of the Proteins of Oxidative Phosphorylation, pp. 80–82, IRL Press, Washington, D.C. [Google Scholar]
- 30.Barth P. G., Scholte H. R., Berden J. A., Van der Klei-Van Moorsel J. M., Luyt-Houwen I. E., Van 't Veer-Korthof E. T., Van der Harten J. J., Sobotka-Plojhar M. A. (1983) J. Neurol. Sci. 62, 327–355 [DOI] [PubMed] [Google Scholar]
- 31.Beach T. G., White C. L., Hamilton R. L., Duda J. E., Iwatsubo T., Dickson D. W., Leverenz J. B., Roncaroli F., Buttini M., Hladik C. L., Sue L. I., Noorigian J. V., Adler C. H. (2008) Acta Neuropathol. 116, 277–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schägger H. (2001) Methods Cell Biol. 65, 231–244 [DOI] [PubMed] [Google Scholar]
- 33.Bradford M. M. (1976) Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 34.Sharon R., Bar-Joseph I., Frosch M. P., Walsh D. M., Hamilton J. A., Selkoe D. J. (2003) Neuron 37, 583–595 [DOI] [PubMed] [Google Scholar]
- 35.Li W., Lesuisse C., Xu Y., Troncoso J. C., Price D. L., Lee M. K. (2004) J. Neurosci. 24, 7400–7409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chu Y., Kordower J. H. (2007) Neurobiol. Dis. 25, 134–149 [DOI] [PubMed] [Google Scholar]
- 37.Hsu L. J., Sagara Y., Arroyo A., Rockenstein E., Sisk A., Mallory M., Wong J., Takenouchi T., Hashimoto M., Masliah E. (2000) Am. J. Pathol. 157, 401–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maries E., Dass B., Collier T. J., Kordower J. H., Steece-Collier K. (2003) Nat. Rev. Neurosci. 4, 727–738 [DOI] [PubMed] [Google Scholar]
- 39.Volles M. J., Lansbury P. T., Jr. (2002) Biochemistry 41, 4595–4602 [DOI] [PubMed] [Google Scholar]
- 40.Lashuel H. A., Petre B. M., Wall J., Simon M., Nowak R. J., Walz T., Lansbury P. T., Jr. (2002) J. Mol. Biol. 322, 1089–1102 [DOI] [PubMed] [Google Scholar]
- 41.Ellis C. E., Murphy E. J., Mitchell D. C., Golovko M. Y., Scaglia F., Barceló-Coblijn G. C., Nussbaum R. L. (2005) Mol. Cell. Biol. 25, 10190–10201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dauer W., Kholodilov N., Vila M., Trillat A. C., Goodchild R., Larsen K. E., Staal R., Tieu K., Schmitz Y., Yuan C. A., Rocha M., Jackson-Lewis V., Hersch S., Sulzer D., Przedborski S., Burke R., Hen R. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 14524–14529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klivenyi P., Siwek D., Gardian G., Yang L., Starkov A., Cleren C., Ferrante R. J., Kowall N. W., Abeliovich A., Beal M. F. (2006) Neurobiol. Dis. 21, 541–548 [DOI] [PubMed] [Google Scholar]
- 44.Fornai F., Schlüter O. M., Lenzi P., Gesi M., Ruffoli R., Ferrucci M., Lazzeri G., Busceti C. L., Pontarelli F., Battaglia G., Pellegrini A., Nicoletti F., Ruggieri S., Paparelli A., Südhof T. C. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 3413–3418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song D. D., Shults C. W., Sisk A., Rockenstein E., Masliah E. (2004) Exp. Neurol. 186, 158–172 [DOI] [PubMed] [Google Scholar]
- 46.Sherer T. B., Betarbet R., Testa C. M., Seo B. B., Richardson J. R., Kim J. H., Miller G. W., Yagi T., Matsuno-Yagi A., Greenamyre J. T. (2003) J. Neurosci. 23, 10756–10764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sherer T. B., Kim J. H., Betarbet R., Greenamyre J. T. (2003) Exp. Neurol. 179, 9–16 [DOI] [PubMed] [Google Scholar]
- 48.Betarbet R., Sherer T. B., MacKenzie G., Garcia-Osuna M., Panov A. V., Greenamyre J. T. (2000) Nat. Neurosci. 3, 1301–1306 [DOI] [PubMed] [Google Scholar]
- 49.Norris E. H., Uryu K., Leight S., Giasson B. I., Trojanowski J. Q., Lee V. M. (2007) Am. J. Pathol. 170, 658–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Manning-Bog A. B., McCormack A. L., Purisai M. G., Bolin L. M., Di Monte D. A. (2003) J. Neurosci. 23, 3095–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Manning-Bog A. B., McCormack A. L., Li J., Uversky V. N., Fink A. L., Di Monte D. A. (2002) J. Biol. Chem. 277, 1641–1644 [DOI] [PubMed] [Google Scholar]
- 52.Gupta A., Dawson V. L., Dawson T. M. (2008) Ann. Neurol. 64, (Suppl. 2) S3–S15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hirsch E. C., Hunot S., Faucheux B., Agid Y., Mizuno Y., Mochizuki H., Tatton W. G., Tatton N., Olanow W. C. (1999) Mov. Disord. 14, 383–385 [DOI] [PubMed] [Google Scholar]
- 54.Nair V. D., McNaught K. S., González-Maeso J., Sealfon S. C., Olanow C. W. (2006) J. Biol. Chem. 281, 39550–39560 [DOI] [PubMed] [Google Scholar]
- 55.Tatton W. G., Chalmers-Redman R., Brown D., Tatton N. (2003) Ann. Neurol. 53, (Suppl. 3) S61–S72 [DOI] [PubMed] [Google Scholar]
- 56.Cooper A. A., Gitler A. D., Cashikar A., Haynes C. M., Hill K. J., Bhullar B., Liu K., Xu K., Strathearn K. E., Liu F., Cao S., Caldwell K. A., Caldwell G. A., Marsischky G., Kolodner R. D., Labaer J., Rochet J. C., Bonini N. M., Lindquist S. (2006) Science 313, 324–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gitler A. D., Bevis B. J., Shorter J., Strathearn K. E., Hamamichi S., Su L. J., Caldwell K. A., Caldwell G. A., Rochet J. C., McCaffery J. M., Barlowe C., Lindquist S. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 145–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Larsen K. E., Schmitz Y., Troyer M. D., Mosharov E., Dietrich P., Quazi A. Z., Savalle M., Nemani V., Chaudhry F. A., Edwards R. H., Stefanis L., Sulzer D. (2006) J. Neurosci. 26, 11915–11922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chandra S., Gallardo G., Fernández-Chacón R., Schlüter O. M., Südhof T. C. (2005) Cell 123, 383–396 [DOI] [PubMed] [Google Scholar]
- 60.Chandra S., Fornai F., Kwon H. B., Yazdani U., Atasoy D., Liu X., Hammer R. E., Battaglia G., German D. C., Castillo P. E., Südhof T. C. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 14966–14971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ungar D., Hughson F. M. (2003) Annu. Rev. Cell Dev. Biol. 19, 493–517 [DOI] [PubMed] [Google Scholar]
- 62.Sudhof T. C. (2004) Annu. Rev. Neurosci. 27, 509–547 [DOI] [PubMed] [Google Scholar]
- 63.Ben Gedalya T., Loeb V., Israeli E., Altschuler Y., Selkoe D. J., Sharon R. (2009) Traffic 10, 218–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barsoum M. J., Yuan H., Gerencser A. A., Liot G., Kushnareva Y., Gräber S., Kovacs I., Lee W. D., Waggoner J., Cui J., White A. D., Bossy B., Martinou J. C., Youle R. J., Lipton S. A., Ellisman M. H., Perkins G. A., Bossy-Wetzel E. (2006) EMBO J. 25, 3900–3911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meuer K., Suppanz I. E., Lingor P., Planchamp V., Göricke B., Fichtner L., Braus G. H., Dietz G. P., Jakobs S., Bähr M., Weishaupt J. H. (2007) Cell Death Differ. 14, 651–661 [DOI] [PubMed] [Google Scholar]
- 66.Gomez-Lazaro M., Bonekamp N. A., Galindo M. F., Jordán J., Schrader M. (2008) Free Radic. Biol. Med. 44, 1960–1969 [DOI] [PubMed] [Google Scholar]
- 67.Vila M., Ramonet D., Perier C. (2008) J. Neurochem. 107, 317–328 [DOI] [PubMed] [Google Scholar]
- 68.Winklhofer K. F., Tatzelt J., Haass C. (2008) EMBO J. 27, 336–349 [DOI] [PMC free article] [PubMed] [Google Scholar]