

Abstract
Iron- and 2-oxoglutarate-dependent dioxygenases are a diverse family of non-heme iron enzymes that catalyze various important oxidations in cells. A key structural motif of these dioxygenases is a facial triad of 2-histidines-1-carboxylate that coordinates the Fe(II) at the catalytic site. Using histone demethylase JMJD1A and DNA repair enzyme ABH2 as examples, we show that this family of dioxygenases is highly sensitive to inhibition by carcinogenic nickel ions. We find that, with iron, the 50% inhibitory concentrations of nickel (IC50 [Ni(II)]) are 25 μm for JMJD1A and 7.5 μm for ABH2. Without iron, JMJD1A is 10 times more sensitive to nickel inhibition with an IC50 [Ni(II)] of 2.5 μm, and approximately one molecule of Ni(II) inhibits one molecule of JMJD1A, suggesting that nickel causes inhibition by replacing the iron. Furthermore, nickel-bound JMJD1A is not reactivated by excessive iron even up to a 2 mm concentration. Using x-ray absorption spectroscopy, we demonstrate that nickel binds to the same site in ABH2 as iron, and replacement of the iron by nickel does not prevent the binding of the cofactor 2-oxoglutarate. Finally, we show that nickel ions target and inhibit JMJD1A in intact cells, and disruption of the iron-binding site decreases binding of nickel ions to ABH2 in intact cells. Together, our results reveal that the members of this dioxygenase family are specific targets for nickel ions in cells. Inhibition of these dioxygenases by nickel is likely to have widespread impacts on cells (e.g. impaired epigenetic programs and DNA repair) and may eventually lead to cancer development.
Keywords: Chromatin/Epigenetics, Chromosomes/Histones, Metals, Metals/Iron, Protein/Iron, X-ray Absorption Spectroscopy, Histone Methylation
Introduction
Nickel compounds are human respiratory carcinogens (1), causing a very high incidence of lung and nasal cancers in nickel refinery workers (2). Over 20 years ago, our group reported that cells phagocytosed particulate nickel compounds, and the dissolution of these particles inside of the cells generated high concentrations of free nickel ions in the cytoplasm and nucleus (3). Using a dye that fluoresces when intracellular nickel ion binds to it, we showed that both soluble and insoluble nickel compounds were able to elevate the levels of nickel ions in the cytoplasmic and nuclear compartments (4). A strong correlation was found between the uptake of particulate nickel compounds by cells and subsequent cell transformation (5), suggesting that intracellular nickel ion concentration is a major determinant of toxicity and carcinogenicity of nickel compounds. Identifying the intracellular targets of nickel ions is therefore crucial to understand the underlying mechanism for the carcinogenic effects of nickel compounds.
Silencing of tumor suppressor gene(s) by epigenetic mechanisms represents one of the potential mechanisms of nickel carcinogenesis. Epigenetic events, which include DNA methylation and histone modifications, are ubiquitously involved in the regulation of gene expression. By using a transgenic cell model with the target gene placed near heterochromatin, we were the first to demonstrate that nickel exposure caused a very high frequency of transgene silencing by increasing DNA methylation and repressive histone marks at the promoter of the silenced transgene (6–8). In animal experiments, injection of particulate nickel compounds (nickel sulfide or nickel subsulfide) into mice induced formation of malignant fibrous histiocytomas and sarcomas, with the p16 and Fhit genes often found to be epigenetically silenced in these cancers (9, 10). Additional studies have demonstrated that nickel exposure caused truncation of histone H2B and H2A as well as global alterations of a variety of histone modifications, such as histone acetylation, methylation, phosphorylation, and ubiquitination (11–20). However, the underlying mechanisms responsible for these nickel-induced epigenetic alterations are poorly understood. In our recent study, we reported that nickel increases the global levels of mono- and di-methylated histone H3 lysine 9 (H3K9me1 and H3K9me2) not by affecting histone methyltransferases but rather by inhibiting a group of unidentified iron- and 2-oxoglutarate-dependent histone demethylases (18).
Since then, five iron- and 2-oxoglutarate-dependent histone H3K9 demethylases, JMJD1A/JHDM2A/KDM3A and JMJD2A-D/KDM4A-D, have been discovered. These enzymes can catalyze the oxidization of methyl groups on histone H3K9, and the resultant hydroxymethyl group is spontaneously lost as formaldehyde to remove one methyl group from the modified lysine (21, 22). JMJD1A demethylates both H3K9me2 and H3K9me1 (23), whereas the JMJD2 family of enzymes target H3K9me3 and H3K9me2 (24–27). JMJD1A functions as a transcriptional co-activator and is involved in multiple biological processes, including androgen receptor signaling, spermatogenesis, smooth muscle cell differentiation, self-renewal of embryonic stem cells, and energy metabolism and weight control (23, 28–31). All these histone H3K9 demethylases belong to a superfamily of iron- and 2-oxoglutarate-dependent dioxygenases, and a key structural motif of this family of dioxygenases is a facial triad of two histidines and one carboxylate that coordinates the Fe(II) ion at the center of the catalytic site (32). Besides histone demethylases, this family also includes hypoxia-inducible factor (HIF) prolyl hydroxylases, factor inhibiting HIF, and DNA repair enzymes ABH2 and ABH3, all of which catalyze various complex oxidations in cells. HIF prolyl hydroxylases 1–3 (PHD1–3) control the degradation of HIF1α through hydroxylating several proline residues located at the oxygen-dependent degradation domain of HIF1α (33); FIH-1 regulates the transcriptional activity of HIF1α through hydroxylating the asparagine residue located at the C-terminal transactivation domain of HIF1α (34); and ABH2 and ABH3 catalyze oxidative demethylation of alkylated DNA bases such as 1-MeA, 3-MeC, 1-MeG, and 3-MeT (35–37).
We have previously reported that HIF prolyl hydroxylase PHD2 was 20 times more sensitive to nickel inhibition when compared with aconitase that binds iron in the form of [4Fe-4S] cluster (38, 39). An interesting question is whether these newly identified histone demethylases and the other members in this Fe(II)- and 2-oxoglutarate-dependent dioxygenase family have similar sensitivity to nickel inhibition as PHD2. We hypothesize that nickel ions inhibit these enzymes by replacing the Fe(II) at the iron-binding motif, resulting in a similar sensitivity of the entire group of enzymes to nickel. In this study, by using JMJD1A and ABH2 as examples, we provide biochemical and structural evidence showing that nickel ions inactivate the Fe(II)- and 2-oxoglutarate-dependent dioxygenases by replacing iron at the catalytic sites. Our study reveals that Fe(II)- and 2-oxoglutarate-dependent dioxygenases are an important group of intracellular targets of human carcinogen nickel compounds.
EXPERIMENTAL PROCEDURES
Plasmids
pcDNA3-FLAG-JMJD1A and its mutant vector have been previously described (23). pET-28a-ABH2 and ABH2(D173A) bacterial expression vectors were kindly provided by Dr. Timothy R. O'Connor (35). To construct mammalian expression vectors of ABH2 and ABH2(D173A), the coding sequences were excised from pET-28a-ABH2 and ABH2(D173A) vectors and subcloned into the EcoRI and XhoI sites of pcDNA3-FLAG vector.
Purification of FLAG-JMJD1A and His6-ABH2 Recombinant Protein and in Vitro Demethylase Assays
FLAG-tagged JMJD1A was expressed in Sf9 insect cells (Invitrogen) and purified using anti-FLAG (Sigma) affinity chromatography as described previously (23). To detect the demethylase activity of JMJD1A, each in vitro demethylase assay was performed in a reaction buffer containing 50 μm HEPES, pH 7.8, 100 μm FeSO4, 1 mm 2-oxoglutarate, 2 mm ascorbic acid, 5 μg of histones (Roche Applied Sciences), and 1 mm phenylmethylsulfonyl fluoride in a final volume of 25 μl. Varying concentrations of NiCl2 were either added into the reaction buffer prior to the final addition of FLAG-JMJD1A or incubated with FLAG-JMJD1A for 10 min on ice prior to addition of other reaction components. After incubation at 37 °C for 30 min, EDTA was added into the reaction mixtures to a final concentration of 1 mm to terminate the reactions. The remaining levels of H3K9me2 in histones were assessed by immunoblotting.
His6-tagged ABH2 was expressed in Rosetta 2(DE3) bacterial cells (Novagen) and purified by nickel-nitrilotriacetic acid beads (Novagen) as described by Lee et al. (35). The His6 tag was excised from the recombinant proteins using thrombin CleanCleave kit (Sigma) and removed using dialysis. The demethylase activity of ABH2 was measured using an assay essentially the same as described previously by Lee et al. (35). In brief, purified ABH2 was incubated with a 32P-end-labeled single-stranded oligonucleotide (24 nucleotides in length) containing one 3-methylcytosine in its HpaII cutting sequence (synthesized by Midland Certified Reagent Co.). After incubation in a reaction buffer containing 40 μm Fe(II), 1 mm 2-oxoglutarate, and 2 mm ascorbic acid at 37 °C for 30 min, the single-stranded oligonucleotide was purified and annealed with its complementary sequence, which was then subject to digestion with methylation-sensitive restriction enzyme HpaII at 37 °C for 30 min. The reaction products were separated on 20% denaturing polyacrylamide gels. The radioactivity on the gels was detected by autoradiography. The appearance of 7-oligonucleotide fragments indicates the removal of the methyl groups at the 3-N position of cytosines and subsequent cutting of 24-oligonucleotides by HpaII.
The Western blot and autoradiography results were scanned and subject to densitometric analysis using KodakTM 1D 3.52 for Macintosh software or ImageJ software, and values were normalized to the appropriate controls.
X-ray Absorption Spectroscopy
XAS3 data collection and analysis were performed as described previously (40). ABH2 samples (with a metal content of ∼0.3 mm by inductively coupled plasma optical emission spectrometers) in 50 mm HEPES, pH 7.8, and 100 mm NaCl were syringed into polycarbonate sample holders with kapton windows and then frozen in liquid nitrogen. Nickel and iron K-edge XAS data were collected at 10 K using a liquid helium cryostat (Oxford Instruments) on beam line 9-3 at the Stanford Synchrotron Radiation Lightsource. The ring conditions were 3 gigaelectron volts and 80–100 mA. Beamline optics consisted of a Si(220) double-crystal monochromator and two rhodium-coated mirrors for focusing and harmonic rejection. X-ray fluorescence was collected using a 30-element germanium detector (Canberra). Scattering was minimized by placing a set of Soller slits with a Z-1 element filter between the sample chamber and the detector. X-ray absorption near edge spectroscopy (XANES) data were collected from ±200 eV relative to metal edge energies. The energy of each K-edge was calibrated to the first inflection point of the relevant metal foil (7112.5 eV for iron, 8331.6 eV for nickel). Extended x-ray absorption fine structure (EXAFS) data were collected to k = 16 Å−1 (for Ni-ABH2) and k = 14 Å−1 (for Fe-ABH2 and Ni-ABH2 + 2-oxoglutarate) above the edge energy. The data analyzed was truncated at k = 12.5 Å−1 for nickel samples and k = 12 Å−1 for Fe-ABH2 because of poor signal:noise.
XAS data analysis was done using EXAFS123 (41) for XANES analysis and SixPack (42) for EXAFS analysis. Scattering parameters for SixPack fitting were generated using the FEFF 8 software package (43). The SixPack fitting software builds on the ifeffit engine and uses iterative FEFF calculations to fit EXAFS data during model refinement, and it is thus an improvement over previous methods that employ a static set of calculated scattering parameters. For the data shown, five scans were averaged for nickel samples, and 18 scans were averaged for the iron sample, then background-corrected, and normalized by using a three-section cubic spline to fit the base line in the pre-edge and the post-edge regions and then setting the edge jump to 1.0. For XANES analysis, the edge energy reported was taken to be the maximum of the first derivative of the XANES spectrum. For pre-edge XANES analysis, a 75% gaussian and 25% Lorenzian function was used to fit the rise in the fluorescence at the edge. Gaussian peaks were added to the pre-edge fit to account for any peaks in the pre-edge region, and the area of these peaks was taken as a measure of their intensity. In case of EXAFS analysis, the average spectrum was converted to k-space. Least square refinement of fits to the EXAFS data were done over a k range of 2–12.5 for nickel samples and 2–12 for the iron samples. Models involving coordination numbers from 2 to 7 were examined by holding the number of scattering atoms in a shell to integer values. Models involving all possible combinations of nitrogen and sulfur donors were addressed for each coordination number (see supplemental material). The number of histidine imidazole ligands involved in the coordination sphere was estimated by multiple-scattering analysis, as described previously (40). To compare different models to the same data set, ifeffit uses three goodness of fit parameters, χ2 (Equation 1), reduced χ2, and R (Equation 2), where Nidp = number of independent data points; Nϵ2 = number of uncertainties to minimize; Re(fi) = real part of the EXAFS fitting function; Im(fi) = imaginary part of the EXAFS fitting function. Reduced χ2 = χ2/(Nind Nvarys) (where Nvarys = the number of refining parameters) and represents the degrees of freedom in the fit.
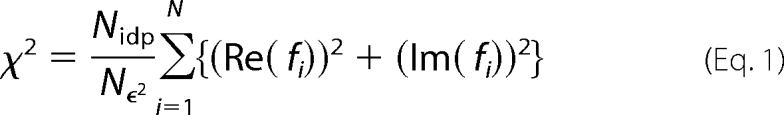 |
Ifeffit also calculates R for the fit, which is given by Equation 2 and is scaled to the magnitude of the data, making it proportional to χ2. To compare different models (fits), the R-factor and reduced χ2 parameters can be assessed to determine which model is the best fit, in which case both parameters should be minimized. Although R will always improve with an increasing number of adjustable parameters, reduced χ2 will go through a minimum and then increase, indicating that the model is over fitting the data.
 |
Best fits were judged by using two goodness of fit parameters, reduced χ2 and R, and the deviation of σ2 from typical values.
Isothermal Titration Calorimetry (ITC) Assay
ITC assay was used to study the binding of metals (iron and nickel) to ABH2. A Microcal (model MCS-ITC) instrument was used during this experiment. Apo-ABH2 (1.5 ml of 35 μm protein for the Ni(II) titration and 40 μm protein for the Fe(II) titration) in 50 mm HEPES, 100 mm NaCl, and 2 mm ascorbic acid at pH 7.8 was placed in the sample cell, and metal solution (0.4 mm Ni(OAc)2 or 0.43 mm (NH4)2Fe(SO4)2 in case of iron) was placed in the syringe. The concentration of protein was determined by the BCA protein assay, and the concentration of metals was determined by ICP-OES. Aliquots (9.098 μl) of metal solution were injected 31 times in case of iron and 32 times in case of nickel with 3 min and 20 s between injections. A single-site binding model was used to fit the data in the Microcal interface to the Origin software, allowing the binding constant (ka), enthalpy change (ΔH), and binding stoichiometry (n) to refine freely. Results are shown in Fig. 4 and Table 2. Experimental data are shown in supplemental Fig. S3.
FIGURE 4.

Binding isotherm of apo-ABH2 with iron (top) and nickel (bottom). The continuous line represents a fit of the data to a single-site binding model.
TABLE 2.
ITC analysis of iron or nickel binding to ABH2
| Metal | kd | ΔH | n | ΔG | ΔS |
|---|---|---|---|---|---|
| μm | kcal/mol | kcal/mol | cal/k | ||
| Iron | 4.5 (5) | −10.1 (2) | 1.07 (2) | −7.5 (1) | −8.6 (2) |
| Nickel | 1.7 (4) | −4.2 (3) | 1.04 (4) | −8.1 (2) | 12.9 (9) |
Cell Culture and Transient Transfection
Human embryonic kidney 293T cells were grown in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Invitrogen). The pcDNA3-FLAG-JMJD1A or FLAG-ABH2, the control vectors, or their mutant FLAG-JMJD1A(H1120Y) or FLAG-ABH2(D173A) vectors were transfected into 293T cells using Lipofectamine 2000 (Invitrogen) by following the manufacturer's protocol.
Immunoprecipitation
After transient transfection and nickel ion exposure as specified in the figure legends, about 1 × 107 293T cells were washed with phosphate-buffered saline containing 1 mm EDTA to eliminate nickel ions bound to the cell surface. Nuclear extracts were isolated from cells using the CelLytic Nu-CLEAR extraction kit (Sigma) by following its nondetergent-based protocol. Nuclear extracts were either directly added into the in vitro demethylase assay for demethylase activity measurement or passed through Chelex columns before measurement. To measure the specific demethylase activity of FLAG-tagged JMJD1A, the cell extracts (combining nuclear extracts and cytoplasmic extracts isolated from the same cells) were subjected to immunoprecipitation using anti-FLAG resin (Sigma) with or without the addition of 1 mm EDTA. The anti-FLAG resin was washed three times with wash buffer (80 mm HEPES, pH 7.9, 14 mm MgCl2, 300 mm NaCl, 10% glycerol), and the complex associated with resin was immediately assayed in an in vitro demethylase assay as described above.
63Ni Cell Labeling and Measurement of 63Ni Binding to ABH2 in Intact Cells
About 7 × 106 293T cells were seeded and transfected with pcDNA3-FLAG-ABH2, FLAG empty vector, or its mutant (FLAG-ABH2 [D173A]) vectors. After incubation overnight, media were replaced with 5 ml of Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Four hours after media replacement, cells were exposed to 1 mm nonradioactive NiCl2 supplemented with 0.225 mCi of 63NiCl2 (Eckert & Ziegler, [nonradioactive nickel]/[63Ni] ∼20). After incubation for 20 h, cells were washed with phosphate-buffered saline containing 1 mm EDTA and rinsed twice with phosphate-buffered saline. Cytoplasm and nuclear extract were isolated from cells using the CelLytic Nu-CLEAR extraction kit by following its nondetergent-based protocol. Cytoplasm extract was combined with nuclear extract, and the mixture with about 1.6 × 106 cpm radioactivity was placed in a column and incubated with 100 μl of packed anti-FLAG resin on a rocker at 4 °C for 2 h. The anti-FLAG resin was washed with the wash buffer (80 mm HEPES, pH 7.9, 14 mm MgCl2, and 100 mm NaCl) until radioactivity of the wash solution was within the range of 200–300 cpm/ml. To elute the FLAG-tagged recombinant proteins, the resins were incubated with 500 μl of wash buffer containing 100 μg/ml FLAG peptide (Sigma) for 30 min. Three hundred μl eluate was taken for radioactivity measurement, and the amount of 63Ni associated with FLAG-recombinant protein was calculated by subtracting the radioactivity value of FLAG-peptide eluate with that of the final wash. The amount of FLAG-tagged recombinant protein in the eluates was estimated using Western blot, and the 63Ni-specific activity in FLAG-tagged ABH2 or ABH2 D173A was then calculated (supplemental Fig. S4 and supplemental Table S4). The experiment was performed in triplicate for each vector transfection condition.
Statistical Analysis
The two-tailed Student t test was used to determine the significance of differences between treated sample and control. The difference was considered significant at p < 0.05.
RESULTS
Nickel Ions Inhibit Demethylase Activity of JMJD1A and ABH2 in Vitro
To study whether nickel ions inhibit iron-dependent dioxygenases in vitro, we first expressed recombinant FLAG-tagged JMJD1A in Sf9 insect cells and purified this recombinant protein using anti-FLAG affinity chromatography (supplemental Fig. S1). We measured demethylase activity of FLAG-JMJD1A using an in vitro histone demethylase assay, with varying concentrations of nickel ions in a reaction mixture prior to the final addition of the enzyme. It was found that demethylase activity of recombinant FLAG-JMJD1A was inhibited by nickel ions in a dose-dependent manner (Fig. 1A), with a 50% inhibitory concentration (IC50 [Ni(II)]) of 25 μm in the presence of 100 μm ferrous iron ions (Fig. 1B).
FIGURE 1.

Nickel ion is a potent inhibitor of JMJD1A and ABH2. A, assay of FLAG-JMJD1A demethylase activity in the presence of 100 μm FeSO4 and varying concentrations of NiCl2. The gel was stained with Coomassie Blue to ensure that equal amounts of histones and FLAG-JMJD1A were added into each reaction. B, data quantification of A. Values are means ± S.D. for four replicates in two independent experiments. C, assay of ABH2 demethylase activity in the presence of 40 μm FeSO4 and varying concentrations of NiCl2. Oligo, oligonucleotide. D, data quantification of C.
We next studied effect of nickel on another iron-dependent dioxygenase, ABH2. His6-tagged ABH2 was expressed in bacterial cells and purified using nickel-nitrilotriacetic acid affinity chromatography (supplemental Fig. S1). After removing the His6 tag from the recombinant protein (supplemental Fig. S1), we measured the effect of nickel ions on the demethylase activity of ABH2 using an assay developed by Lee et al. (35). In this assay, a synthesized single-stranded oligodeoxyribonucleotide containing one 3-methylcytosine (3-MeC) in the HpaII restriction enzyme cleavage site (3-MeCCGG) is subject to demethylation catalyzed by ABH2. After demethylation, this oligodeoxyribonucleotide is annealed to another oligodeoxyribonucleotide with complementary sequence, and the annealed DNA is digested with HpaII. Because HpaII can only digest the DNA with unmodified CCGG sequence but not the one with 3-MeCCGG, the amount of cleaved DNA by HpaII can be used to measure the demethylase activity of ABH2. Similar to the results with JMJD1A, we found that the demethylase activity of ABH2 was inhibited by nickel ions in a dose-dependent manner (Fig. 1C), with a 50% inhibiting concentration (IC50 [Ni(II)]) of 7.5 μm in the presence of 40 μm ferrous iron ions (Fig. 1D). In our previous study, we have reported that aconitase, a Krebs cycle enzyme where iron is bound in a form of [4Fe-4S] cluster, has a 50% inhibiting concentration (IC50 [Ni(II)]) of 500 μm (39). Compared with aconitase, both JMJD1A and ABH2 are considerably more sensitive to nickel inhibition.
JMJD1A Is More Sensitive to Nickel Inhibition in the Absence of Iron
Based on our hypothesis that nickel ions inhibit Fe(II)- and 2-oxoglutarate-dependent dioxygenases by replacing the ferrous iron at the iron-binding site of these enzymes, JMJD1A should be more sensitive to nickel ion inhibition in the absence of ferrous iron. Recombinant JMJD1A protein was incubated with nickel ions prior to addition of ferrous iron ions and other essential cofactors for demethylase activity measurement. As expected, under these conditions, JMJD1A was 10 times more sensitive to nickel inhibition with an IC50 [Ni(II)] = 2.5 μm (Fig. 2, A and B). Interestingly, JMJD1A demethylase activity was completely inhibited by nickel ions when the molar ratio of Ni(II)/JMJD1A approached 1, indicating that one molecule of nickel ion inhibits one molecule of JMJD1A (Fig. 2B). More impressively, inhibition of JMJD1A by nickel ions could not be reversed by addition of excessive iron ions even up to 2 mm (Fig. 2C), suggesting a higher binding affinity of Ni(II) than Fe(II) for the enzyme. We also studied whether the demethylase activity of JMJD1A can be inhibited by other metal ions. JMJD1A was first incubated with 5 μm of various metal ions and subsequently assayed for demethylase activity. With a molar ratio of metal/enzyme = 1.6, we found that Co(II) ions completely inhibited JMJD1A demethylase activity, whereas Cu(II) caused a lower degree of inhibition and Mn(II), As(III), and Cr(VI) did not have any inhibitory effect (supplemental Fig. S2).
FIGURE 2.

Nickel ions inhibit JMJD1A by competing with iron ions. A, FLAG-JMJD1A was incubated with varying concentrations of NiCl2 on ice for 10 min, and its demethylase activity was subsequently measured using in vitro demethylase assay. The results of one typical experiment from three independent experiments are shown here. B, data quantification of A and comparison with Fig. 1A. Values are means ± S.D. for four replicates in two independent experiments. C, inhibition of JMJD1A by nickel ions could not be reversed by addition of excessive iron ions. Purified FLAG-JMJD1A was preincubated with 5 μm NiCl2 and was then assayed for its demethylase activity in the presence of varying concentrations of ferrous iron ions. The results of one typical experiment from two independent experiments are shown here.
Nickel Replaces the Ferrous Iron at the Iron-binding Site of ABH2
To gain more insight into how nickel inhibits the iron-dependent dioxygenases, we utilized XAS to study the nickel binding to ABH2. XAS is one of the most powerful techniques available for studying the structural details of metal ion binding to proteins. This technique yields information about the coordination number, ligand environment, and metric details regarding the metal-binding site(s). In XAS, comparison of shifts in metal K-edge energies may reveal redox-active metal sites; the analysis of extended x-ray absorption fine structure (EXAFS) provides information regarding the types and number of ligands bound to a metal ion and metric details for the metal ion site; and the analysis of x-ray absorption near edge structure (XANES) provides information about the coordination number of a metal ion and the geometry of the metal site (44, 45). Because XAS analysis requires a large amount of purified protein that can only be obtained by using a bacterial expression system and because we had no success in expressing JMJD1A in bacteria due to its large molecular weight, ABH2 was chosen for this analysis.
XAS studies were performed to investigate the structures of nickel and iron bound to ABH2. XANES data are summarized in Fig. 3A and Table 1. Comparison of the XANES data for Ni-ABH2 and Ni-ABH2 + 2-oxoglutarate (Fig. 3A) shows a structural change occurs in the nickel site when 2-oxoglutarate is added, consistent with binding of this cofactor to nickel. Both nickel and iron have vacancies in 3d manifold, and peaks associated with 1s → 3d transitions are observed in the pre-edge XANES for all three samples (Fig. 3A). The peak areas will depend on the coordination number and geometry of the metal site. From comparison of the peak areas of the observed 1s → 3d transitions (Table 1) with samples of known coordination numbers and geometries (44, 45), both iron and nickel (in presence and absence of 2-oxoglutarate) have a coordination number of five, which is in agreement with the previous work with Fe(II)-AlkB (46). In the case of the nickel samples, small shoulders associated with peaks involving 1-s → 4-pz transitions are observed and consistent with a geometry that is closer to square pyramidal than trigonal bipyramidal (44).
FIGURE 3.

XAS analysis of nickel binding to ABH2. A, K-edge XANES spectra for Fe-ABH2 (red), Ni-ABH2 (blue), and Ni-ABH2 + 2-oxoglutarate (green). Insets, expansions of the pre-edge XANES region showing peaks associated with 1s → 3d electronic transitions. B, unfiltered, k3-weighted EXAFS spectra (colored lines, red = Fe-ABH2, blue = Ni-ABH2 and green = Ni-ABH2 + 2-oxoglutarate) and best fits from Table 1 (black lines). FT, Fourier transform. Left, k-space spectra and fits. Right, FT-data and fits.
TABLE 1.
XAS analysis
SPY is square pyramidal, and αKG is α-ketoglutarate; CN is coordination number; Im is imidazole; and E is energy.
| ABH2 sample | XANES analysis |
EXAFS analysis |
||||||
|---|---|---|---|---|---|---|---|---|
| Edge E | 1s → 3d peak area (×102 eV) | CN/geometry | Shell | r | σ2 (×10−2 eV) | ΔE0 | R | |
| eV | Å | eV | ||||||
| Iron | 7125.9 (2) | 9 (1) | 5 | 5N (2 Im) | 2.02 (1) | 7 (1) | −9 (2) | 0.027 |
| 3C | 3.09 (6) | 1 (5) | ||||||
| Nickel | 8344.7 (2) | 5 (1) | 5/SPY | 5N (2 Im) | 2.08 (1) | 4.7 (5) | −4 (1) | 0.023 |
| Nickel + αKG | 8345.3 (2) | 5.4 (8) | 5/SPY | 5N (2 Im) | 2.103 (6) | 6 (1) | −6 (1) | 0.011 |
| 1C | 2.46 (3) | 5 (3) | ||||||
EXAFS data show that the best fit for Fe-ABH2 consists of five oxygen/nitrogen donor ligands of which two are histidine ligands (Table 1 and Fig. 3B), consistent with the structures of other resting non-heme iron dioxygenase enzymes (47). When nickel binds to ABH2, it is bound in a site that is indistinguishable from the iron-binding site, consisting of five oxygen/nitrogen donor ligands of which two are histidines. When 2-oxoglutarate is added to Ni-ABH2, the EXAFS spectrum is altered (features between r = 2–3 Å are greatly reduced in intensity), consistent with a change in the coordination sphere of the nickel center arising from binding of the cofactor to the nickel site and with the crystal structure of ABH2 bound to double strand DNA with Mn(II) and 2-oxoglutarate (48). Taken together, these results demonstrate that nickel binds to the Fe(II)-binding site of ABH2 and does not prevent the binding of the cofactor 2-oxoglutarate.
Nickel Ions Bind to ABH2 More Tightly than Iron Ions
We next examined whether nickel ions bind to ABH2 more tightly than iron. The affinity of iron and nickel toward the active site of ABH2 was measured using ITC. In both cases, the binding stoichiometry is approximately one metal per apo-ABH2. ITC experiments show that the binding affinity (in terms of kd) of Fe(II) for ABH2 is 4.5 μm, which is comparable with the reported kd values of Fe(II) for other mononuclear non-heme iron dioxygenases, such as TfdA (kd of 7.45 μm measured by fluorescence) and Dke1 (kd of 5 μm for site I). It was found that the binding affinity of nickel to ABH2 is 1.7 μm, indicating nickel binds to ABH2 2.65 times more tightly than iron. We also see a larger change in enthalpy for the Fe(II) experiment than that for Ni(II) (Fig. 4), which becomes important during the calculation of entropy change (ΔS). The data in Table 2 show that the binding of Ni(II) to ABH2 is entropically favored but that of Fe(II) is not entropically favored.
Nickel Ions Bind to and Inhibit JMJD1A in Cells
We next asked whether nickel ions inhibit the activity of dioxygenases in cells by directly binding to the enzymes. To this end, we overexpressed FLAG-JMJD1A in human embryonic kidney 293T cells and assessed the levels of H3K9me2 in endogenous chromatin following nickel treatment. Overexpression of FLAG-JMJD1A significantly decreased the global level of H3K9me2 in the chromatin of cells, whereas this effect was almost completely reversed by nickel treatment of intact cells without affecting the expression of recombinant demethylase (Fig. 5).
FIGURE 5.
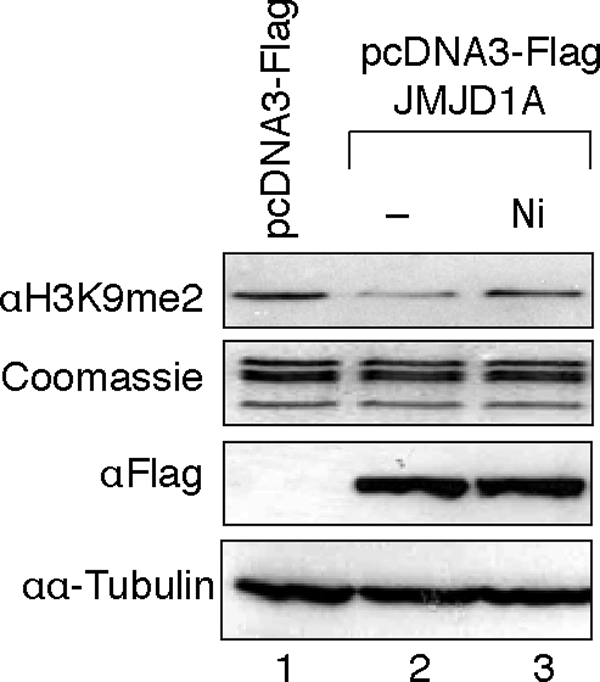
Nickel ions inhibit demethylase activity of JMJD1A in cells. 293T cells were transiently transfected with FLAG-JMJD1A expression vectors and were then exposed to 1 mm NiCl2 for 24 h. Histones were extracted and used for H3K9me2 detection by immunoblotting. The levels of overexpressed FLAG-JMJD1A were detected by immunoblotting using anti-FLAG antibody. The results of one typical experiment from three independent experiments are shown here.
To further study whether inactivation of the overexpressed JMJD1A was due to direct binding of nickel ions to the enzyme in cells, we isolated nuclear extracts from the nickel ion-treated 293T cells with overexpression of FLAG-JMJD1A and measured their demethylase activity in vitro. To prevent cell extracts from being contaminated by extracellular nickel ions during isolation, cells were carefully washed with phosphate-buffered saline containing 1 mm EDTA twice before lysis. Compared with the cells transfected with the empty vectors, a higher H3K9 demethylase activity was observed in the nuclear extract isolated from the cells with overexpression of FLAG-JMJD1A (Fig. 6A). In the cells exposed to nickel ions, a lower demethylase activity was detected in the nuclear extract, and this inhibitory effect was completely reversed by passage of the nuclear extracts through Chelex (a metal chelator) columns (Fig. 6A). To measure the specific demethylase activity of FLAG-JMJD1A, we performed immunoprecipitation (IP) using anti-FLAG antibody with or without addition of EDTA into the IP buffer. The immunoprecipitated FLAG-JMJD1A in the nickel-treated cells retained some residual demethylase activity (Fig. 6B, lane 4), which is likely due to the dissociation of the nickel ions from FLAG-JMJD1A during the IP. Nevertheless, compared with the IP sample without addition of EDTA, a significantly higher FLAG-JMJD1A demethylase activity was detected in the same sample with EDTA (Fig. 6B, lane 4 versus 8). Similar to the results in Fig. 6B, an inhibition of JMJD1A demethylase activity was observed in cells pretreated with a much lower concentration of nickel ions prior to FLAG-JMJD1A overexpression, and this inhibition could be reversed by addition of EDTA during IP (Fig. 6C, lane 4 versus 3). Because of a lower expression level of exogenous FLAG-JMJD1A in the cells pretreated with nickel ions, a lower demethylase activity was detected in the IP samples from the nickel-treated cells as compared with the untreated cells even when EDTA was present in the IP buffer (Fig. 6B, lane 4 versus 2). Probably due to the instability of mutant protein, a much lower expression level of recombinant protein was found in cells transfected with FLAG-JMJD1A(H1120Y) expression vectors. Taken together, these results demonstrate that nickel inactivates JMJD1A in intact cells by direct binding to this demethylase enzyme.
FIGURE 6.

Nickel ions directly bind to JMJD1A in cells to cause inhibition. A, assay of demethylase activity in the nuclear extracts of nickel ion-exposed cells. 293T cells were treated under the same conditions as described in Fig. 5. The nuclear extracts were isolated, and a portion of these extracts was passed through Chelex columns to eliminate any metals present in the extracts. In vitro histone demethylase assay was performed to measure histone demethylase activity of the nuclear extracts. The levels of FLAG-JMJD1A present in the nuclear extracts were assessed by immunoblotting using anti-FLAG antibody. B, assay of specific JMJD1A demethylase activity in the nickel ion-exposed cells. 293T cells were treated under the same condition as described in A. Cytoplasmic and nuclear extracts were isolated and combined. The combined lysates were subject to immunoprecipitation (I.P.) using anti-FLAG resin with or without addition of 1 mm EDTA into IP buffer. In vitro histone demethylase assay was performed to measure histone demethylase activity present in the immunoprecipitates. The same membrane was stained for histone H3 to assess the amount of histones loaded into the gel. The levels of immunoprecipitated FLAG-JMJD1A were assessed by immunoblotting using anti-FLAG antibody. C, assay of specific JMJD1A demethylase activity in cells pretreated with nickel ions. 293T cells were exposed to 150 μm NiCl2 for 3 days and were then transfected with FLAG-JMJD1A or FLAG-JMJD1A H1120Y expression vectors. Two days after the transfection, cell extracts were isolated and were subject to IP as described in B. In vitro histone demethylase assay was performed as in B. The results of one typical experiment from at least two independent experiments are shown in A–C.
Nickel Ion Binds to the Catalytic Site of ABH2 in Cells
The findings described above suggest that nickel ions inactivate the iron-dependent dioxygenases in cells by directly binding to their iron-binding sites. To prove it, we sought to measure the amount of nickel binding to a wild type iron-dependent dioxygenase or a point mutant with a disrupted iron-binding site in intact cells exposed to nickel. Because the expression level of FLAG-JMJD1A(H1120Y) point mutant is much lower than FLAG-JMJD1A wild type in cells (Fig. 6C), we used ABH2 for these experiments. Fig. 7A shows that, in 293 cells, the expression level of FLAG-ABH2(D173A), a point mutant with the aspartic acid residue in the 2-His-1-carboxylate motif replaced by alanine, was about 80% of the FLAG-ABH2 level. To measure the binding of nickel to ABH2 in cells, we labeled 293T cells with 63NiCl2 24 h after transfection with FLAG, FLAG-ABH2, or FLAG-ABH2(D173A) expression vectors. FLAG-tagged recombinant protein was purified using anti-FLAG resin column and eluted with FLAG peptide. Fig. 7B shows that the radioactivity associated with FLAG-ABH2 eluates was double that of FLAG-ABH2(D173A) samples. After normalizing the 63Ni radioactivity using the amount of FLAG-ABH2 or ABH2 D173A protein present in the eluates (raw data and calculations are provided in supplemental Fig. S4 and supplemental Table S4), the 63Ni-specific radioactivity associated with FLAG-ABH2 was still significantly higher than that of ABH2(D173A) (Fig. 7C). These results demonstrated that nickel binds to the iron-binding site of ABH2 in cells.
FIGURE 7.

Nickel ions bind to the iron-binding site of ABH2 in cells. A, measurement of FLAG-ABH2 and ABH2(D173A) expression levels in the nickel-treated 293T cells. 293T cells were transiently transfected with FLAG-ABH2 and FLAG-ABH2(D173A) expression vectors and then treated with 1 mm NiCl2 that contained 0.22 mCi of 63NiCl2. Expression of FLAG-ABH2 or ABH2(D173A) in cell lysates was measured by Western blot using anti-FLAG antibody. The intensity of bands was quantified using ImageJ software and marked below the graph. The quantification results were graphed on the right. B, cell lysates collected in A were subject to IP with anti-FLAG resin. The FLAG-tagged recombinant proteins were eluted with FLAG peptide, and their associated radioactivity was measured. C, 63Ni-specific radioactivity associated with FLAG-ABH2 or ABH2(D173A) was calculated. The experiment was conducted in triplicate, and values are means ± S.D. for triplicates. The difference in 63Ni-specific radioactivity between FLAG-ABH2 and FLAG-ABH2(D173A) samples is statistically significant because a two-tailed Student t test analysis gives a p value of 0.044.
DISCUSSION
The family of iron- and 2-oxoglutarate-dependent dioxygenases uses the 2-His-1-carboxylate motif to bind the cofactor ferrous iron ion. In this study, we demonstrate that two distinct members of this enzyme family, histone demethylase JMJD1A and DNA repair enzyme ABH2, are sensitive to nickel ion inhibition. The IC50 [Ni(II)] values for FLAG-JMJD1A (about 25 μm in the presence of 100 μm Fe(II)) and ABH2 (about 7.5 μm in the presence of 40 μm Fe(II)) are comparable with that of PHD2 (about 22 μm in the presence of 100 μm Fe(II)) (38). Presumably, this is because all three of these dioxygenases use the same structural motif (His-Asp-His) to bind ferrous iron at their active sites, and Ni(II) competes with Fe(II) and replaces it at the iron-binding site. In support of this hypothesis, our results showed that the IC50 [Ni(II)] value for FLAG-JMJD1A decreased 10-fold to 2.5 μm when the recombinant demethylase protein was incubated with nickel prior to addition of iron, and one molecule of nickel ion stoichiometrically caused inhibition of one molecule of JMJD1A demethylase. This condition likely reflects how nickel inhibits these demethylases in cells. In cells, the free-available iron level is maintained within a relatively narrow range of concentrations (estimated to be 0.8 μm) to minimize generation of harmful reactive oxygen species (49), whereas no known intracellular molecule is capable of “chelating” nickel ions. Without being effectively competed by iron ions, nickel ions are more likely to be placed at the iron-binding sites of these iron- and 2-oxoglutarate-dependent dioxygenases and cause permanent inhibition of these enzymes.
Both XANES and EXAFS data suggest that the catalytic inactivity of Ni-ABH2 is not because it does not bind the cofactor but presumably arises from an inability of the nickel site to activate oxygen. Although a coordination position is available for oxygen binding, the nickel center is not easily oxidized. By using factor-inhibiting HIF as a model, Topol et al. (50) performed a theoretical calculation of the energy required to route from the reaction components to a high spin metal oxide intermediate with Fe(II), Ni(II), or Co(II) in the active site. They found that substitution of the Fe(II) by Ni(II) or Co(II) significantly increased the reaction energy barrier and thus deterred the oxidization reaction (50). In the only known nickel-containing dioxygenase, Ni-ARD, the nickel is not redox-active and serves only to coordinate the substrate and facilitate ligand oxidation by O2 (51).
Previously, we calculated the binding constant of various divalent metals to two imidazole ligands that were used to model the two histidines in the His-Asp-His triad (38). The logarithm of binding strength of Ni(II) coordination to two imidazole molecules is approximately 3 orders of magnitude greater than that for Fe(II) to bind to the same two molecules (38). However, in this study, the results of ITC analysis show that the binding of nickel to ABH2 is only about three times stronger than iron. This discrepancy is probably caused by some factors, such as the amino acid environment surrounding the iron-binding histidines, which were not taken into consideration in the original model. Nevertheless, our data still indicate that the binding of nickel to ABH2 is tighter than iron, which may explain why even 2 mm Fe(II) could not reactivate JMJD1A that had been incubated with 5 μm Ni(II).
Several hypotheses have been proposed to explain how nickel ions may inhibit the function of iron- and 2-oxoglutarate- dependent dioxygenases in cells. It has been suggested that interference with intracellular iron homeostasis, oxidation/depletion of ascorbic acid that is also an essential cofactor for most of the dioxygenases in this family, and direct binding to the enzyme may collectively contribute to the ability of nickel ions to inhibit iron- and 2-oxoglutarate-dependent dioxygenases (39, 52, 53). Because interference with intracellular iron homeostasis and oxidation/depletion of ascorbic acid are readily correctable after removal of nickel ions, these dioxygenases would be easily reactivated. In contrast, direct binding of nickel ion to these enzymes would cause permanent inactivation (Fig. 2C). Our results demonstrated that nickel ions inactivate JMJD1A by directly binding to this enzyme in cells, suggesting that the inhibitory effects of nickel on these dioxygenases may be persistent even after discontinuation of exposure. Because higher concentration of intracellular nickel ions causes a greater degree of inhibition of iron-dependent demethylases, it agrees with the hypothesis that the intracellular nickel ion concentration is a major determinant of toxicity and carcinogenicity of nickel compounds. Given that the iron- and 2-oxoglutarate-dependent family of dioxygenases have multiple functions in cells, inhibition of these enzymes by nickel is likely to have widespread impact on cells and could eventually lead to development of cancer. Inhibition of the histone H3K9 demethylases, for example, could increase repressive histone mark H3K9me2 in the promoters of susceptible tumor suppressor genes and result in silencing of gene expression.
In summary, our study demonstrates that iron-and 2-oxoglutarate dioxygenases are direct targets of carcinogenic nickel compounds in cells. Future study should be directed toward understanding how inactivation of these dioxygenases is involved in nickel carcinogenesis.
Supplementary Material
Acknowledgment
We thank Dr. Timothy R. O'Connor for providing the His6-tagged ABH2 and ABH2(D173A) bacterial expression vectors.
This work was supported, in whole or in part, by National Institutes of Health Grants ES000260, ES014454, and ES005512 from NIEHS and Grant CA16087 from NCI.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–4 and Tables S1–S4.
- XAS
- x-ray absorption spectroscopy
- EXAFS
- extended x-ray absorption fine structure
- ITC
- isothermal titration calorimetry
- IP
- immunoprecipitation
- XANES
- x-ray absorption near edge spectroscopy.
REFERENCES
- 1.International Agency for Research on Cancer (1990) Monographs on the Evaluation of Carcinogenic Risks to Humans: Chromium, Nickel and Welding, pp. 1–648, Lyon, France: [PMC free article] [PubMed] [Google Scholar]
- 2.Grandjean P., Andersen O., Nielsen G. D. (1988) Am. J. Ind. Med. 13, 193–209 [DOI] [PubMed] [Google Scholar]
- 3.Costa M., Simmons-Hansen J., Bedrossian C. W., Bonura J., Caprioli R. M. (1981) Cancer Res. 41, 2868–2876 [PubMed] [Google Scholar]
- 4.Ke Q., Davidson T., Kluz T., Oller A., Costa M. (2007) Toxicol. Appl. Pharmacol. 219, 18–23 [DOI] [PubMed] [Google Scholar]
- 5.Costa M., Abbracchio M. P., Simmons-Hansen J. (1981) Toxicol. Appl. Pharmacol. 60, 313–323 [DOI] [PubMed] [Google Scholar]
- 6.Yan Y., Kluz T., Zhang P., Chen H. B., Costa M. (2003) Toxicol. Appl. Pharmacol. 190, 272–277 [DOI] [PubMed] [Google Scholar]
- 7.Sutherland J. E., Peng W., Zhang Q., Costa M. (2001) Mutat. Res. 479, 225–233 [DOI] [PubMed] [Google Scholar]
- 8.Lee Y. W., Klein C. B., Kargacin B., Salnikow K., Kitahara J., Dowjat K., Zhitkovich A., Christie N. T., Costa M. (1995) Mol. Cell. Biol. 15, 2547–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Govindarajan B., Klafter R., Miller M. S., Mansur C., Mizesko M., Bai X., LaMontagne K., Jr., Arbiser J. L. (2002) Mol. Med. 8, 1–8 [PMC free article] [PubMed] [Google Scholar]
- 10.Kowara R., Salnikow K., Diwan B. A., Bare R. M., Waalkes M. P., Kasprzak K. S. (2004) Mol. Cell. Biochem. 255, 195–202 [DOI] [PubMed] [Google Scholar]
- 11.Bal W., Liang R., Lukszo J., Lee S. H., Dizdaroglu M., Kasprzak K. S. (2000) Chem. Res. Toxicol. 13, 616–624 [DOI] [PubMed] [Google Scholar]
- 12.Kang J., Zhang Y., Chen J., Chen H., Lin C., Wang Q., Ou Y. (2003) Toxicol. Sci. 74, 279–286 [DOI] [PubMed] [Google Scholar]
- 13.Karaczyn A. A., Golebiowski F., Kasprzak K. S. (2005) Chem. Res. Toxicol. 18, 1934–1942 [DOI] [PubMed] [Google Scholar]
- 14.Karaczyn A. A., Golebiowski F., Kasprzak K. S. (2006) Exp. Cell Res. 312, 3252–3259 [DOI] [PubMed] [Google Scholar]
- 15.Ke Q., Davidson T., Chen H., Kluz T., Costa M. (2006) Carcinogenesis 27, 1481–1488 [DOI] [PubMed] [Google Scholar]
- 16.Ke Q., Ellen T. P., Costa M. (2008) Toxicol. Appl. Pharmacol. 228, 190–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ke Q., Li Q., Ellen T. P., Sun H., Costa M. (2008) Carcinogenesis 29, 1276–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen H., Ke Q., Kluz T., Yan Y., Costa M. (2006) Mol. Cell. Biol. 26, 3728–3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou X., Li Q., Arita A., Sun H., Costa M. (2009) Toxicol. Appl. Pharmacol. 236, 78–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Golebiowski F., Kasprzak K. S. (2005) Mol. Cell. Biochem. 279, 133–139 [DOI] [PubMed] [Google Scholar]
- 21.Shi Y., Whetstine J. R. (2007) Mol. Cell 25, 1–14 [DOI] [PubMed] [Google Scholar]
- 22.Klose R. J., Zhang Y. (2007) Nat. Rev. Mol. Cell Biol. 8, 307–318 [DOI] [PubMed] [Google Scholar]
- 23.Yamane K., Toumazou C., Tsukada Y., Erdjument-Bromage H., Tempst P., Wong J., Zhang Y. (2006) Cell 125, 483–495 [DOI] [PubMed] [Google Scholar]
- 24.Cloos P. A., Christensen J., Agger K., Maiolica A., Rappsilber J., Antal T., Hansen K. H., Helin K. (2006) Nature 442, 307–311 [DOI] [PubMed] [Google Scholar]
- 25.Fodor B. D., Kubicek S., Yonezawa M., O'Sullivan R. J., Sengupta R., Perez-Burgos L., Opravil S., Mechtler K., Schotta G., Jenuwein T. (2006) Genes Dev. 20, 1557–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klose R. J., Yamane K., Bae Y., Zhang D., Erdjument-Bromage H., Tempst P., Wong J., Zhang Y. (2006) Nature 442, 312–316 [DOI] [PubMed] [Google Scholar]
- 27.Whetstine J. R., Nottke A., Lan F., Huarte M., Smolikov S., Chen Z., Spooner E., Li E., Zhang G., Colaiacovo M., Shi Y. (2006) Cell 125, 467–481 [DOI] [PubMed] [Google Scholar]
- 28.Tateishi K., Okada Y., Kallin E. M., Zhang Y. (2009) Nature 458, 757–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Okada Y., Scott G., Ray M. K., Mishina Y., Zhang Y. (2007) Nature 450, 119–123 [DOI] [PubMed] [Google Scholar]
- 30.Loh Y. H., Zhang W., Chen X., George J., Ng H. H. (2007) Genes Dev. 21, 2545–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lockman K., Taylor J. M., Mack C. P. (2007) Circ. Res. 101, e115–e123 [DOI] [PubMed] [Google Scholar]
- 32.Straganz G. D., Nidetzky B. (2006) ChemBioChem 7, 1536–1548 [DOI] [PubMed] [Google Scholar]
- 33.Bruick R. K., McKnight S. L. (2001) Science 294, 1337–1340 [DOI] [PubMed] [Google Scholar]
- 34.Lando D., Peet D. J., Whelan D. A., Gorman J. J., Whitelaw M. L. (2002) Science 295, 858–861 [DOI] [PubMed] [Google Scholar]
- 35.Lee D. H., Jin S. G., Cai S., Chen Y., Pfeifer G. P., O'Connor T. R. (2005) J. Biol. Chem. 280, 39448–39459 [DOI] [PubMed] [Google Scholar]
- 36.Duncan T., Trewick S. C., Koivisto P., Bates P. A., Lindahl T., Sedgwick B. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 16660–16665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aas P. A., Otterlei M., Falnes P. O., Vågbø C. B., Skorpen F., Akbari M., Sundheim O., Bjørås M., Slupphaug G., Seeberg E., Krokan H. E. (2003) Nature 421, 859–863 [DOI] [PubMed] [Google Scholar]
- 38.Davidson T. L., Chen H., Di Toro D. M., D'Angelo G., Costa M. (2006) Mol. Carcinog. 45, 479–489 [DOI] [PubMed] [Google Scholar]
- 39.Chen H., Davidson T., Singleton S., Garrick M. D., Costa M. (2005) Toxicol. Appl. Pharmacol. 206, 275–287 [DOI] [PubMed] [Google Scholar]
- 40.Leitch S., Bradley M. J., Rowe J. L., Chivers P. T., Maroney M. J. (2007) J. Am. Chem. Soc. 129, 5085–5095 [DOI] [PubMed] [Google Scholar]
- 41.Padden K. M., Krebs J. F., MacBeth C. E., Scarrow R. C., Borovik A. S. (2001) J. Am. Chem. Soc. 123, 1072–1079 [DOI] [PubMed] [Google Scholar]
- 42.Webb S. M. (2005) Physica. Scripta. T. 115, 1011–1014 [Google Scholar]
- 43.Ankudinov A. L., Ravel B., Rehr J. J., Conradson S. D. (1998) Physical Rev. B 58, 7565–7576 [Google Scholar]
- 44.Colpas G. J., Maroney M. J., Bagyinka C., Kumar M., Willis W. S., Suib S. L., Baidya N., Mascharak P. K. (1991) Inorg. Chem. 30, 920–928 [Google Scholar]
- 45.Randall C. R., Shu L. J., Chiou Y. M., Hagen K. S., Ito M., Kitajima N., Lachicotte R. J., Zang Y., Que L. (1995) Inorg. Chem. 34, 1036–1039 [Google Scholar]
- 46.Mishina Y., Chen L. X., He C. (2004) J. Am. Chem. Soc. 126, 16930–16936 [DOI] [PubMed] [Google Scholar]
- 47.Que L., Jr., Ho R. Y. (1996) Chem. Rev. 96, 2607–2624 [DOI] [PubMed] [Google Scholar]
- 48.Yang C. G., Yi C., Duguid E. M., Sullivan C. T., Jian X., Rice P. A., He C. (2008) Nature 452, 961–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Konijn A. M., Glickstein H., Vaisman B., Meyron-Holtz E. G., Slotki I. N., Cabantchik Z. I. (1999) Blood 94, 2128–2134 [PubMed] [Google Scholar]
- 50.Topol I. A., Nemukhin A. V., Salnikow K., Cachau R. E., Abashkin Y. G., Kasprzak K. S., Burt S. K. (2006) J. Phys. Chem. A 110, 4223–4228 [DOI] [PubMed] [Google Scholar]
- 51.Ju T., Goldsmith R. B., Chai S. C., Maroney M. J., Pochapsky S. S., Pochapsky T. C. (2006) J. Mol. Biol. 363, 823–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davidson T., Chen H., Garrick M. D., D'Angelo G., Costa M. (2005) Mol. Cell. Biochem. 279, 157–162 [DOI] [PubMed] [Google Scholar]
- 53.Karaczyn A., Ivanov S., Reynolds M., Zhitkovich A., Kasprzak K. S., Salnikow K. (2006) J. Cell. Biochem. 97, 1025–1035 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.