

Abstract
Paneth cells at the base of small intestinal crypts of Lieberkühn secrete host defense peptides and proteins, including α-defensins, as mediators of innate immunity. Mouse Paneth cells also express α-defensin-related Defcr-rs genes that code for cysteine-rich sequence 4C (CRS4C) peptides that have a unique CPX triplet repeat motif. In ileitis-prone SAMP1/YitFc mice, Paneth cell levels of CRS4C mRNAs and peptides are induced more than a 1000-fold relative to non-prone strains as early as 4 weeks of age, with the mRNA and peptide levels highest in distal ileum and below detection in duodenum. CRS4C-1 peptides are found exclusively in Paneth cells where they occur only in dense core granules and thus are secreted to function in the intestinal lumen. CRS4C bactericidal peptide activity is membrane-disruptive in that it permeabilizes Escherichia coli and induces rapid microbial cell K+ efflux, but in a manner different from mouse α-defensin cryptdin-4. In in vitro studies, inactive pro-CRS4C-1 is converted to bactericidal CRS4C-1 peptide by matrix metalloproteinase-7 (MMP-7) proteolysis of the precursor proregion at the same residue positions that MMP-7 activates mouse pro-α-defensins. The absence of processed CRS4C in protein extracts of MMP-7-null mouse ileum demonstrates the in vivo requirement for intracellular MMP-7 in pro-CRS4C processing.
Keywords: Cell/Epithelial, Cell/Intracellular Processing, Immunology/Defensins, Immunology/Innate Immunity, Peptides/Biosynthesis, Subcellular Organelles/Endoplasmic Reticulum, Tissue/Organ Systems/Intestine
Introduction
In mammals, antimicrobial protein and peptide genes are expressed by differentiated cell lineages, including epithelial cells and phagocytes, as endogenous mediators of innate immunity (1–4). Of the two major mammalian antimicrobial protein families, cathelicidins and defensins (2, 5), the defensins comprise three subfamilies of cationic peptides, each characterized by a unique tridisulfide array (6, 7). The α-defensins are broad spectrum microbicides that kill via membrane-disruptive mechanisms (8–11) and are abundant in neutrophil azurophilic granules (12) and in dense core secretory granules of Paneth cells, a secretory epithelial cell lineage that is restricted to small intestinal crypts under normal conditions (13–15).
α-Defensins, termed cryptdins (Crps)6 in mice, are synthesized as inactive precursors that must be processed into bactericidal mature forms (16–22). In the α-defensin precursors, anionic proregions in the N-terminal moiety of pro-α-defensin inhibit bactericidal peptide activity by apparent charge neutralization of electropositive amino acids of the α-defensin component (17, 23, 24). Inactive mouse α-defensin precursors are converted to mature bactericidal peptides by matrix metalloproteinase-7 (MMP-7)-mediated proteolysis within the proregion which releases inhibitory acidic amino acids from covalent association with the α-defensin component (17, 19, 23, 25, 26). Mouse pro-Crp activation occurs intracellularly in the Paneth cell-regulated pathway (27, 28).
The two-exon α-defensin genes expressed by small intestinal Paneth cells of rhesus macaque, mouse, and rat undergo extensive duplication events and rapid diversification (29). In the mouse, α-defensin genes duplicated and diversified further to give rise to the Defcr-rs gene subfamily that codes for numerous cysteine-rich sequence 4C (CRS4C) peptides that are unique to mice (30). Defcr-rs and α-defensin (Defcr) genes have similar two-exon structures and normally are expressed only by Paneth cells in mouse small bowel. The first exons of mouse Defcr-rs and Defcr genes have ∼95% nucleotide sequence identity and code for nearly identical proregions (31–33). Despite that extensive identity, however, Defcr-rs gene second exons code for Cys-rich, cationic CRS4C peptides that are not α-defensin paralogs. Instead, they are characterized by seven repeats of a CPX triplet motif that is unique to this defensin peptide subfamily and found only in the mouse (32, 34) (see supplemental Fig. S1). Native CRS4C peptides purified from mouse small intestine exist as disulfide-stabilized homodimers and heterodimers that are antibacterial (33). However, details of their expression patterns, post-translational processing, and mechanisms of action remain obscure.
Here, we report that small intestinal levels of Paneth cell-specific CRS4C mRNAs and peptides are markedly and differentially elevated in the ileum of the SAMP1/YitFc mouse, a strain prone to spontaneous ileitis (35). CRS4C peptides are constituents of Paneth cell dense core granules and are selectively expressed in distal small bowel, and MMP-7 is required to process pro-CRS4C in vivo. MMP-7 can also activate pro-CRS4C-1 in vitro by a mechanism similar to mouse pro-α-defensin processing, converting inactive pro-CRS4C molecules to bactericidal, membrane-disruptive peptides.
EXPERIMENTAL PROCEDURES
Animals and Tissue Preparation
SAMP1/YitFc mice are a substrain derived from SAMP1/Yit mice, originally provided by Professor S. Matsumoto of the Yakult Central Institute for Microbiological Research (Tokyo, Japan) (35), following 20 generations of sibling mating of the colony at the University of Virginia. C57BL/6 mice were purchased from Charles River Breeding Laboratories (Wilmington, MA), and all procedures were performed in compliance with approved protocols of the Institutional Animal Care and Use Committees of the University of California, Irvine and the University of Virginia.
For protein extractions, the ileum was removed from mice euthanized by halothane inhalation, and organs were flushed and homogenized on ice in 30% (v/v) acetic acid and incubated overnight at 4 °C with continuous stirring (14, 28, 36, 37). Protein extracts were clarified by centrifugation, diluted 6-fold, dialyzed using SpectraPor3 membranes (Spectrum Labs, Los Angeles, CA) against 5% acetic acid, and lyophilized. Lyophilized protein extracts were concentrated, resuspended in 5% (v/v) acetic acid, and subjected to further purification using preparative acid-urea polyacrylamide gel electrophoresis (AU-PAGE) and reversed phase HPLC (38).
C57BL/6 mouse ileum was prepared for histochemical analysis by immersion in phosphate-buffered formalin fixative. Fixed tissue was processed into paraffin blocks and sectioned by the Histology Laboratory, Department of Pathology and Laboratory Medicine, University of California Irvine Medical Center.
Preparation of Recombinant Pro-CRS4C-1 and CRS4C-1 Peptides
Recombinant pro-CRS4C-1(20–72) and deduced mature CRS4C-1(54–72), corresponding to the Ala53↓Leu54 cleavage of pro-CRS4C-1 by MMP-7 (see supplemental Fig. S4), were expressed in Escherichia coli as N-terminal His6-tagged fusion proteins using pET-28a (Novagen, Madison, WI) as described (17, 39). Pro-CRS4C-1 cDNA (GenBankTM accession number NM_007847) was used as a template to amplify sequences for cloning using forward primer petpro-CRS4cF (5′-GCGCG AATTC ATGGA TTCTA TCCAA AAACA CAGAT-3′) paired with reverse primer petCRS4cR (5′-ATATA TGTCG ACTTA TTTTG GATTG CATTT GCA-3′). For deduced mature CRS4C-1, forward primer pET-LQDAA-CRS4C-F (5′-ATATA TGAAT TCATG CTTCA AGATG CAGCC-3′) and reverse primer petCRS4cR (5′-ATATA TGTCG ACTTA TTTTG GATTG CATTT GCA-3′) were used. The underlined codons in the forward primers denote Met codons introduced to provide a unique CNBr cleavage site within fusion proteins at the N termini of all expressed peptides whose primary structures lack Met (10). PCR was performed using GeneAMP PCR core reagents (Applied Biosystems, Foster City, CA) by incubating the reaction mixture for 94 °C for 5 min followed by 30 cycles of 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s followed by an extension reaction for 7 min at 72 °C. Amplification products were cloned in pCR-2.1 TOPO (Invitrogen), verified by DNA sequencing, subcloned into pET-28a plasmid DNA (Novagen), and transfected into E. coli BL21-CodonPlus (DE3)-RIL (Stratagene, La Jolla, CA) for protein expression.
Recombinant pro-CRS4C-1(20–72), CRS4C-1(59–72), and Crp-5 peptides were expressed in E. coli as N-terminal His6-tagged fusion proteins (1–5). As described above, peptide-coding amplified cDNAs were subcloned into the EcoRI and SalI sites of pET28a (Novagen), transformed into XL-1 Blue cells (Stratagene), and confirmed by DNA sequencing. Recombinant protein expression was induced with 100 μm isopropyl-β-d-1-thiogalactopyranoside for 6 h at 37 °C in Terrific Broth medium (10). Cells were subsequently deposited by centrifugation and lysed by sonication in 6 m guanidine-HCl in 100 mm Tris-HCl (pH 8.0), and the lysates were clarified by centrifugation.
His6-tagged fusion peptides were affinity-purified by selective binding to nickel-nitrilotriacetic acid resin affinity chromatography and eluted with 1 m imidazole, 6 m guanidine-HCl, 100 mm Tris-HCl (pH 5.9) (10, 18). Fusion proteins were exposed to 10 mg/ml CNBr in 80% formic acid for 18 h at 25 °C, diluted with water, and lyophilized. Cleavage products resuspended in 5% (v/v) acetic acid were separated by C18 and C4 reversed phase HPLC with a gradient of acetonitrile and with 0.1% trifluoroacetic acid as an ion-pairing agent. Peptide homogeneity was confirmed by analytical AU-PAGE. The molecular masses were determined by matrix-assisted laser desorption ionization mode mass spectrometry (MALDI-TOF MS) in a Voyager-DE MALDI-TOF spectrometer (PE-Biosystems, Foster City, CA).
Quantitative Real-time PCR
Levels of Crp and CRS4C mRNAs in mouse small intestinal segments were quantified by real-time PCR analysis using an ABI PRISM SDS7000 sequence detection system (Applied Biosystems). Total RNA was isolated using the RNeasy kit from Qiagen (Valencia, CA) as described by the manufacturer. cDNA was prepared by reverse transcription using random hexamers (1 μg) and 10 ng of total RNA in a final reaction volume of 20 μl containing 200 units of Superscript (Invitrogen). Primers for measuring levels of mRNA were: CRS4C forward primer, 5′-CGCAG CCATG AAGAA ACTTG-3′ and reverse primer, 5′-GAATC AGCCT GGACC TGGAA-3′; Crps, pan-Crp forward primer, 5′-AAGAG ACTAA AACTG AGGAG CAGC-3′ and reverse primer, 5′-GGTGA TCATC AGACC CCAGC ATCAG T-3′, which amplify all mouse Crp mRNA sequences. PCR was performed in triplicate using 10% (v/v) of the first strand synthesis in a total vol of 50 μl that included 25 μl SYBR Green master mix (Applied Biosystems) and a 250 nm final concentration of primers. The amplification of cDNA was performed for 40 cycles by a preset cycling program that included the generation of a melting curve. Thermocycling conditions were: (i) 50 °C for 2 min (activation of AmpErase UNG); (ii) 95 °C for 10 min (activation of AmpliTaq Gold enzyme); (iii) 95 °C for 15 s (denaturation) and 60 °C for 1 min (anneal/extend) for 40 cycles. The ΔCT method was used to quantify relative mRNA levels as described in User Bulletin 2 (Applied Biosystems), using 18 S RNA as the reference and internal standard. A TaqMan primer probe set for 18 S RNA with the Vic/Tamra detection system was used to measure 18 S RNA in replicate samples simultaneously.
The CRS4C primers were based on regions of identity in small intestinal cDNAs or genes from 129/SvJ, C3H/HeJ, C3H/HeN, NMRI/KI, FVB, and BALB/c mice. Those same primers were used to clone and sequence ∼30 CRS4C cDNAs from SAMP1/YitFc ileum, which were identical to reported CRS4C cDNA sequences (32, 33), and CRS4C cDNA cloning frequencies from SAMP1/YitFc mouse ileal RNA were very similar to those from other inbred strains of mice. Thus, the primers are appropriate for quantitation of CRS4C mRNA levels in varied mouse strains, including the SAMP1/YitFc strain.
Preparation of CRS4C-1 and Crp-5 Peptide Antisera
The immune antigens consisted of recombinant CRS4C-1(59–72) and Crp-5 peptide pET-28a fusion proteins expressed in E. coli (1–5), purified by affinity nickel-nitrilotriacetic acid affinity chromatography, dialyzed, and lyophilized. For immunization purposes, a single 7–15-month-old cross-bred goat (35–50 kg) was immunized by subcutaneous injection of 1 mg of Crp-5 or CRS4C-1 fusion proteins emulsified with Complete Freund's Adjuvant (Elmira Biologicals, Iowa City, IA) under conditions approved by the Institutional Animal Care and Use Committee of Elmira Biologicals. Six weeks after initial immunization, goats were boosted by an injection of an additional 1 mg of peptide antigen in the absence of adjuvant. Sera were collected 8–9 days after each antigen boost and tested for peptide specificity by immunoblotting and by Western blots. Immunoglobulin G (IgG) was isolated from antiserum by DEAE Econo-Pac chromatography (Bio-Rad) as recommended by the manufacturer. Peptide specificity of the antibody was determined by assessing immunoreactivity against recombinant proCRS4C-1, mature CRS4C-1, pro-Crp-4, and mature Crp-4 immobilized on nitrocellulose membranes, with the antibody demonstrating reactivity only with the proform and mature form of CRS4C-1 (data not shown).
Immunolocalization of CRS4C Expression in Mouse Small Intestine
For immunohistochemical localization of CRS4C-1 in SAMP1/YitFc mouse, ileal segments were rapidly dissected from SAMP1/YitFc and AKR mice at 10 weeks of age, fixed in Bouin fixative, and embedded in paraffin as described previously (40). Deparaffinized tissue sections were incubated with goat-anti CRS4C-1 (1:5000) and the reaction visualized using horseradish peroxidase-conjugated donkey anti-goat secondary antibody (1:1500, Jackson ImmunoResearch Laboratories) followed by diaminobenzidine precipitation. All sections were counterstained with hematoxylin.
Immunolocalization of CRS4C-1 and Crp-5 in C57Bl/6 mouse small bowel was performed as before (28). Briefly, paraffin sections of formalin-fixed mouse jejunum or ileum were deparaffinized, treated 30 min with 0.3% H2O2, and washed with water and phosphate-buffered saline. Sections were incubated with normal rabbit serum for 30 min, with avidin D blocking solution for 15 min, rinsed with phosphate-buffered saline, and incubated 15 min with biotin blocking solution (Vector Laboratories, Burlingame, CA). Slides were incubated at ambient temperature with 1:1000 dilutions of goat anti-CRS4C-1(59–72) or goat anti-Crp-5 immune antisera or with corresponding preimmune sera, washed with phosphate-buffered saline, incubated 30 min with biotinylated rabbit anti-goat IgG (1:2000), and washed. After a 15-min incubation with Vectastain ABC peroxidase reagent (Vector), slides were washed, flooded with diaminobenzidine, washed, and counterstained before mounting.
For immunogold localization of CRS4C in Paneth cells, C57BL/6 mice were perfused with Hanks' balanced salt solution and then with fixative containing 2% (v/v) formaldehyde and 0.1% (w/v) glutaraldehyde in 0.1 m phosphate buffer (pH 7.4) at 37 °C. The ileum was excised, embedded in gelatin, cut into 100-μm-thick sections, and embedded in Unicryl (Ted Pella, Redding, CA) at −20 °C as described (28, 41). Thin sections were placed on nickel grids and stained on both sides with goat anti-CRS4C-1(59–72) immune sera diluted 1:200 overnight at 4 °C, washed, and then reacted overnight at 4 °C with 1:20 protein G conjugated to 10 nm gold (EMS, Hatfield PA). The sections were then formvar- and carbon-coated, counterstained with uranyl acetate and lead citrate, and evaluated with a JEOL JEM 1200EX II electron microscope at 80 kV (42). Equivalent dilutions of preimmune sera provided negative controls in all experiments.
In Vitro Activation of Pro-CRS4C-1 by MMP-7
Recombinant pro-Crp-4, Crp-4, pro-CRS4C-1, and CRS4C-1 peptides were digested with MMP-7, and reaction mixtures were analyzed by AU-PAGE and by N-terminal sequencing to determine cleavage sites (18). Peptide samples (1.3 nmol) were incubated with half-molar quantities of human MMP-7 catalytic domain (Chemicon International, Temecula, CA) in buffer containing 10 mm HEPES (pH 7.4), 150 mm NaCl, and 5 mm CaCl2 for 24 h at 37 °C. Reactions were analyzed by AU-PAGE and subjected to N-terminal peptide sequencing. Samples of complete digests were subjected to five cycles of Edman sequencing reactions at the former University of California Irvine Biomedical Protein and Mass Spectrometry Resource Facility on an ABI model 477 system (American Biosystems, Inc., Foster City, CA) configured with on-line phenylthiohydantoin-derivative amino acid analysis as described (18, 28).
Preparative Acid-Urea PAGE
Total soluble proteins extracted from SAMP1/YitFc (35, 40) and MMP-7-null mouse ilea were separated by preparative AU-PAGE using a model 491 PrepCell device (Bio-Rad) as described (38). Lyophilized samples of protein extracts dissolved in 1 ml of 3 m urea in 5% acetic acid (v/v) were electrophoresed continuously on 12.5% AU-polyacrylamide gels for 1 h at 100 V and subsequently 6 h at 250 V. Proteins were eluted from the anodal reservoir with 5% acetic acid at a flow rate of 1 ml/min, and effluent was monitored at A230 nm. Samples of fractions containing resolved proteins were analyzed by analytical AU-PAGE and visualized by staining with Coomassie Blue R-250 after fixation in formalin-containing acetic acid/methanol (43).
Western Blot Analyses
Protein extracts and control peptides resolved by AU-PAGE were transferred to 0.2-μm nitrocellulose membranes using a semidry apparatus, blocked, and incubated with goat anti-CRS4C-1(59–72) immune IgG diluted in Tris-buffered saline/Tween containing 5% (w/v) nonfat milk at room temperature with agitation (27, 28). Washed blots were incubated with peroxidase-conjugated rabbit anti-goat antibody diluted 1:60,000 in 10 mm Tris-HCl, 150 mm NaCl, 0.1% Tween 20 for 1 h, washed, and developed using SuperSignal chemiluminescent substrate (Pierce) with a 30–60-s exposure (27).
Assays of Bactericidal Peptide Activity
To measure bactericidal peptide activities, E. coli ML35 cells suspended in 10 mm PIPES supplemented with 1% (v/v) trypticase soy broth were incubated with recombinant pro-CRS4C-1, CRS4C-1, and CRS4C-1 that had been linearized by reduction and alkylation with iodoacetamide, using Crp-4 as a positive control peptide as described previously (17, 18, 44). Briefly, ∼1 × 106 exponentially growing E. coli ML35 cells were incubated with peptides or MMP-7 digests in 10 mm PIPES (pH 7.4) supplemented with 1% (v/v) trypticase soy broth at 37 °C. After a 60-min incubation, 20 μl of each incubation mixture was diluted 1:2000 with 10 mm PIPES (pH 7.4), and 50 μl of the diluted samples was plated on trypticase soy agar using a Spiral Biotech Autoplate 4000 (Spiral Biotech, Bethesda, MD). Surviving bacteria were quantified as colony-forming units/ml on plates after incubation at 37 °C for 12 h (45).
Assays of Live E. coli ML35 Cell Permeabilization
Exponentially growing E. coli ML35 cells were washed and resuspended in 10 mm PIPES supplemented with 1% (v/v) trypticase soy broth and 2.5 mm 2-ortho-nitrophenyl β-d-galactopyranoside (ONPG). Bacteria were exposed in triplicate to Crp-4, pro-Crp-4, CRS4C-1, and pro-CRS4C-1 in the presence of 2.5 mm ONPG for 2 h at 37 °C. E. coli ML35 is a β-galactosidase constitutive, permease-negative strain that cannot take up ONPG or hydrolyze it to o-nitrophenol (ONP) unless permeabilized by external factors, including defensins (17, 46). β-Galactosidase hydrolysis of ONPG was measured at 405 nm on a 96-well Spectra-Max plate spectrophotometer (Molecular Devices, Inc., Sunnyvale, CA) as described (17, 46).
Potassium Efflux Assays
Exponentially growing E. coli ML35 cells were washed with 10 mm PIPES (pH 7.4) and resuspended in 10 mm PIPES (pH 7.4), 0.1% (v/v) trypticase soy broth. Samples containing 6.25 × 107 colony-forming units of E. coli ML35 cells in a final volume of 250 μl and incubated at 37 °C were used in our experiments. Peptide-mediated potassium efflux from cells was monitored at 10-s intervals after the addition of 7 μm peptide (47) using an MI-442 potassium-selective microelectrode (Microelectrodes, Inc., Bedford, NH) and an MI-409F reference microelectrode (Microelectrodes) both fitted to an Orion SensorLink PCM-700 pH/ISE meter (48) To suppress electrode drift, the tip of the reference electrode was fitted with a salt bridge containing 2 m NaCl in 1% agarose.
Statistical Methods
Data were analyzed by pairwise t tests using the pooled estimate of variance and Bonferroni's correction of the p values for multiple comparisons. Differences were considered significant at p ≤ 0.05.
RESULTS
Dysregulation of Defcr-rs Gene Expression in Ileitis-prone SAMP1/YitFc Mice
At ∼6–8 weeks of age, SAMP1/YitFc mice develop a spontaneous ileitis that resembles many aspects of human Crohn disease (35). Gene expression profiling studies provided evidence that certain Paneth cell-specific mRNAs occur at markedly higher levels in SAMP1/YitFc mice prior to histological evidence of ileal inflammation (data not shown). For example, at 4 weeks of age, transcript levels of the Defcr-rs10 gene, which codes for a CRS4C-4 peptide isoform (32, 33), were already elevated ∼1000-fold in SAMP1/YitFc ileum relative to age-matched AKR and C57BL/6 mice, non-ileitis-prone reference strains in which levels are inherently low (Fig. 1A). Also, Defcr-rs10 gene expression was specific for the ileum, with CRS4C mRNAs occurring at barely detectable levels in jejunum. Using pan-Crp primers that amplify all known mouse α-defensins (see “Experimental Procedures”), a more modest over all increase in Crp gene expression was evident beginning at 4–5 weeks (Fig. 1B), coincident with the time course of increased Paneth cell and intermediate cell numbers that characterizes the SAMP1/YitFc strain (35, 40). In contrast to the elevated Crp gene expression in SAMP1/YitFc ileum, overall expression of Crps remained relatively stable as a function of age in AKR mouse ileum. The time course for CRS4C mRNA accumulation in SAMP1/YitFc ileum diverged markedly from that seen for Crp-coding mRNAs. Increased expression of CRS4C in SAMP1/YitFc ileum began by about 3–4 weeks of age, prior to histologic evidence of inflammation, reaching the highest levels during the inductive phase of inflammation, between 5 and 10 weeks of age (Fig. 1C). Although levels of CRS4C mRNA fell precipitously after inflammation became chronic in SAMP1/YitFc mice, at age >10 weeks (35, 40), they were still elevated compared with the non-ileitis-prone mouse strains.
FIGURE 1.
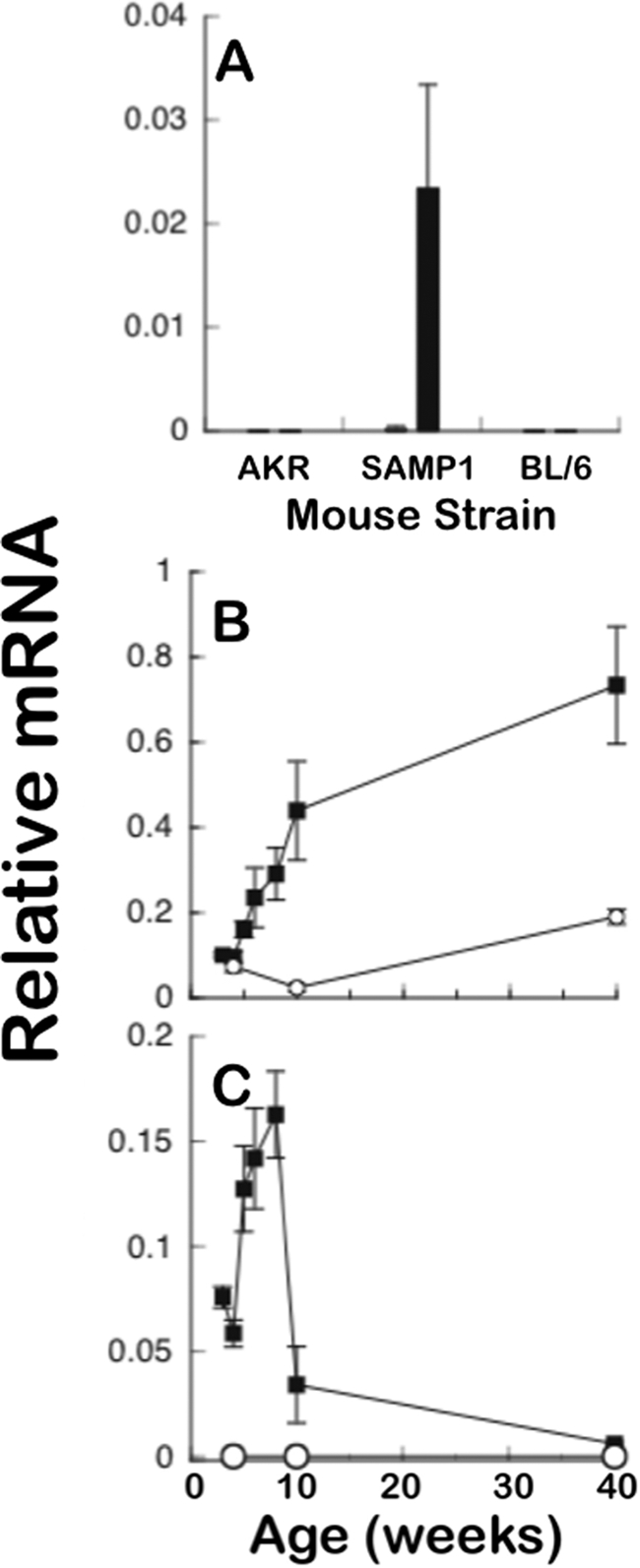
High ileal CRS4C mRNA levels in ileitis-prone SAMP1/YitFc mice. A, CRS4C mRNA levels were measured by quantitative real-time PCR in jejunum and ileum of 10-week-old AKR, SAMP1/YitFc, and C57BL/6 mice relative to 18 S rRNA using sequence-specific primers (see “Experimental Procedures”). Filled bars denote levels in ileum, open bars are levels in jejunum. Data are expressed as mean ± S.D. with n = 4 mice/group. p < 0.001 SAMP1/YitFc ileum versus B6 ileum; p < 0.0001 SAMP1/YitFc ileum versus AKR ileum. B, Paneth cell α-defensin mRNA levels were measured at the ages indicated as given in A. Levels in SAMP1/YitFc (filled squares) and AKR (open circles) mouse ileum were compared using pan-Crp primers Defcrp130 and Defcrm380, which amplify all known mouse Crps. Data are expressed as mean ± S.D. with n = 4–6 mice/group. C, ileal CRS4C mRNA levels were measured as in B. Levels in SAMP1/YitFc (filled squares) and AKR (open circles) mouse ileum were compared using the CRS4C-specific primer set (“Experimental Procedures”). Data are expressed as mean ± S.D. with n = 4–6 mice/group.
Abundant CRS4C Peptides in SAMP1/YitFc Mouse Ileum
The unusual pattern of CRS4C expression seen in SAMP1/YitFc mice coupled with the distinct behavior of expression of the Defcr-rs genes relative to the Crp (Defcr) α-defensin gene expression prompted us to investigate the levels and pattern of CRS4C protein distribution in the intestine of the SAMP1/YitFc mouse and additional inbred strains. By immunohistochemical detection, the anti-CRS4C-1 antibody reacted with Paneth cells in the small bowel of 10-week-old SAMP1/YitFc mice (Fig. 2B). When staining was performed at low antibody concentration, the intensity of the ileal Paneth cell immunostaining of SAMP1/YitFc mouse (Fig. 2B) was much more intense than the staining of Paneth cells of AKR mice, a non-ileitis-prone strain (Fig. 2A). Also, in Western blots performed on three individual AKR and SAMP1/YitFc mice, CRS4C-1 protein was not detected immunohistochemically in the proximal intestine of either strain and was at undetectable levels in AKR mouse ileum under those conditions (see supplemental Fig. S2). Consistent with the results of mRNA quantitation (Fig. 1A), CRS4C protein accumulates to markedly higher levels in the distal small bowel.
FIGURE 2.

Temporal and regional CRS4C peptide levels in SAMP1/YitFc mouse small intestine. A and B, immunohistochemical staining of crypts of the non-ileitis-prone AKR (A) and SAMP1/YitFc (B) mouse strains using anti-CRS4C-1 antiserum (see “Experimental Procedures”). Arrows indicate strongly immunopositive SAMP1/YitFc Paneth cells in B and AKR Paneth cells in A that exhibit markedly lower levels of immunoreactivity. CRS4C peptides are products of Paneth cells in the intestinal crypts of both mice. C, age-related appearance and regional distribution of CRS4C peptide accumulation determined for the SAMP1/YitFc mouse by Western blotting. Samples (0.50 mg) of total organ protein-extracted jejunum (J) and ileum (I) of individual 4- or 10-week-old SAMP1/YitFc mice were separated by AU-PAGE as noted and blotted onto a nitrocellulose membrane. The blot was subjected to Western blot analysis using anti-CRS4C-1 IgG diluted 1:5 (see “Experimental Procedures”). Recombinant pro-Crp-4 (leftmost lane) was a negative control peptide, and recombinant CRS4C-1 and pro-CRS4C-1 provided positive controls as shown by their strong immunoreactivity. Immunopositive bands that co-migrate with recombinant CRS4C-1, indicated by the lower right arrow and the curly brace, are readily detected in ileum of both 4- and 10-week-old SAMP1/YitFc mice, with the intensity of the signal markedly increased by 10 weeks of age.
CRS4C-1 and cross-reactive peptide isoforms were immunolocalized exclusively to Paneth cells in the C57BL/6 mouse small intestine (supplemental Fig. S3). However, a much higher antiserum concentration than that used for SAMP1/YitFc immunolocalization was required to detect CRS4C-1 in C57BL/6 Paneth cells. Under these conditions, anti-CRS4C-1 reacted with Paneth cells in C57BL/6 mouse small intestinal crypts (supplemental Fig. S3, A and B), with an intensity and specificity similar to that of antisera for Crp-5 (supplemental Fig. S3, C and D), consistent with in situ hybridization of CRS4C-1 to mouse Paneth cells (32) and as reported for Crp-1, Crp-4, and the pro-Crp proregion (14, 28, 49). The CRS4C peptide family consists of many isoforms with conservative amino acid substitutions (32, 33, 50), and the anti-CRS4C-1 serum is likely to detect those variants as well as the original peptide antigen.
The induction of ileal CRS4C peptide levels and the marked accumulation of CRS4C peptides in SAMP1/YitFc ileum reflected increased levels of CRS4C mRNA and preceded the onset of ileitis. Nominal ≤10-kDa peptides were prepared from protein extracts of ileum and jejunum from 4- and 10-week-old SAMP1/YitFc mice using Centricon 10 centrifugal filtration units (Millipore). The ≤10-kDa peptide fractions, along with recombinant CRS4C and pro-CRS4C peptide controls, were subjected to AU-PAGE Western blot analysis with CRS4C antiserum (Fig. 2C). The resolved CRS4C peptide isoforms are abundant in SAMP1/YitFc mouse ileum (Fig. 2C, lanes I) at both ages examined and contrasts with the low levels detected in jejunum (Fig. 2C, lanes J). CRS4C peptide immunoreactivity in the SAMP1/YitFc mouse ileum was present at 4 weeks of age and continued to increase during the development of the acute phase of ileitis, consistent with the increased expression of CRS4C mRNA observed at this time (Fig. 1C).
CRS4C-1 in Paneth Cell Dense Core Secretory Granules
CRS4C peptides are constituents of mouse Paneth cell secretory granules as shown by subcellular location in mouse jejunum using a gold-conjugated second antibody. Anti-CRS4C-1 antibody reacted specifically with Paneth cell dense core granules (Fig. 3, A, B, and D) with negligible background staining with preimmune serum (Fig. 3C). The electron-lucent halos of Paneth cell granules, which are rich in O-linked GalNAc glycoconjugates (51), were not immunoreactive (Fig. 3C). Staining was highly specific for the electron-dense region of secretory granules (Fig. 3D), where peptide constituents are concentrated in the regulated pathway. Thus, CRS4C peptides co-localize with mouse enteric α-defensins (28) and, along with the varied host defense molecules secreted by Paneth cells (13, 52), are released into the small intestinal lumen to function in that environment. The localization of CRS4C and α-defensins in Paneth cell secretory granules and the extensive proregion sequence identity of their precursors (32, 33, 50) suggested that CRS4C and Crp proforms are processed by a similar MMP-7-mediated mechanism (27, 28).
FIGURE 3.

CRS4C protein localization to dense core secretory granules of mouse Paneth cells. Sections of C57/BL6 mouse small intestinal tissue were incubated with preimmune (C) and anti-CRS4C-1 antisera and anti-goat IgG secondary antibody conjugated with 10-nm gold particles (A, B, and D), as described under “Experimental Procedures.” Magnification, ×20,000 (A). Scale bars, 200 nm (all panels). Electron micrographs show that the anti-CRS4C antibody reacts specifically with the electron-dense regions of Paneth cell secretory granules (arrows).
In Vitro Pro-CRS4C-1 Activation by MMP-7
To test whether MMP-7 mediates in vitro activation of pro-CRS4C-1, recombinant pro-CRS4C-1 and CRS4C-1 peptides were exposed to MMP-7 and compared with the well characterized MMP-7 conversion products of pro-Crp-4 (17, 18, 23, 27). In vitro MMP-7 digestion products of pro-CRS4C-1 and pro-Crp-4 were analyzed by AU-PAGE (43, 53), by direct N-terminal sequencing of reaction mixtures, and for bactericidal peptide activity (18). AU-PAGE analyses showed that the single major product of pro-CRS4C-1 digestion with MMP-7 co-migrated with recombinant CRS4C-1 (Fig. 4A, upper arrow). Similarly, a single major product resulted from pro-Crp-4 exposure to MMP-7, and it co-migrated with mature Crp-4 as shown before (17, 18, 23, 28).
FIGURE 4.

In vitro enzymatic activation of pro-CRS4C-1 bactericidal activity by MMP-7. A, equimolar samples of recombinant Crp-4, pro-Crp-4, deduced mature CRS4C-1, and pro-CRS4C-1 were incubated overnight at 37 °C with (+) or without (−) 0.5-mol equivalents of MMP-7 and subjected to analytical AU-PAGE. Note that MMP-7cleaves pro-Crp-4 as well as pro-CRS4C. Pro-CRS4C peptides are processed by MMP-7. B, exponentially growing E. coli ML35 were exposed to mature CRS4C-1 (○) and pro-CRS4C-1 (●) as well as mature CRS4C-1 (▿) and pro-CRS4C-1 (▾) that were exposed to MMP-7 for 1 h at 37 °C. Surviving bacteria were quantified as colony-forming units (CFU)/ml (see “Experimental Procedures”). The displayed result is representative of three independent experiments. MMP-7 converts the inactive CRS4C precursor to a bactericidal peptide activity level equivalent to that of mature CRS4C.
To identify the major sites of proteolysis, pro-CRS4C-1 was incubated with MMP-7, and the N termini of reaction products were determined by five cycles of Edman sequencing reactions (18). Three N-terminal sequences, VSFGD…, LQDAA…, and LGWGR… were the most prevalent in the digests (see supplemental Fig. S4). These N termini correspond to proregion cleavage events at Ser43↓Val44, Ala53↓Leu54 and Ala58↓Leu59 in the precursor, the same sites cleaved in pro-Crp-4 (17, 18) (see supplemental Fig. S4). Because the N termini of mouse enteric α-defensins correspond to the Ala58↓Leu59 cleavage site (14, 27, 28), and the predominant N terminus of native, purified CRS4C peptides (33) corresponds to the product of Ala53↓Leu54 cleavage, we speculate that MMP-7 processing of pro-CRS4C molecules may be less efficient than pro-α-defensin processing in vivo.
MMP-7 Proteolysis Activates Bactericidal Peptide Activity
Generally, pro-α-defensins lack bactericidal activity until cleaved by their convertases (17, 19, 22, 23, 54). To test whether MMP-7 proteolysis is an activating conversion step, we assayed the bactericidal peptide activity of pro-CRS4C-1(20–72) and CRS4C-1(54–72) before and after incubation with MMP-7. E. coli ML35 cell survival was unaffected by incubation with untreated pro-CRS4C-1 (Fig. 4B), indicating that pro-CRS4C-1, as all pro-α-defensins, lacks bactericidal activity. However, digestion mixtures consisting of pro-CRS4C-1 incubated with MMP-7 for 1 h at 37 °C reduced E. coli cell survival by 99.9%, as observed for mature CRS4C-1 in the presence or absence of MMP-7 (Fig. 4B). Fig. 4B is representative of three independent experiments performed on different days. CRS4C-1(54–72) has slightly greater activity in the presence of MMP-7 than in its absence, suggesting that the LQDAA N terminus of CRS4C-1(54–72) is somewhat attenuated relative to the final CRS4C(59–72) product of MMP-7 cleavage which terminates in LGWGR (see supplemental Fig. S4). Thus, removal of the proregion from covalent association with the CRS4C-1(54–72) component converts the precursor to an active form, even though the three proregion digestion fragments remain in the assay mixture. Because the predominant form of CRS4C isolated from mouse small intestine had LQDAA at the N terminus (33), that peptide was prepared and used to investigate the mechanisms of action of this unusual peptide. These in vitro experiments show the ability of MMP-7 to process pro-CRS4C-1 to the mature deduced CRS4C-1 peptide and demonstrate that the mature CRS4C-1 peptide is resistant to proteolysis by this enzyme. Also, MMP-7 converts the inactive CRS4C-1 precursor to a bactericidal peptide that is equivalent in activity to mature CRS4C.
MMP-7 Processing of CRS4C Precursors in Vivo
To determine whether the in vitro activation by MMP-7 is predictive of in vivo pro-CRS4C conversion, the status of CRS4C activation was characterized in protein isolated from intact mouse small bowel. Protein was extracted from the ilea of SAMP1/YitFc (35, 40) and MMP-7-null (28) mice (see “Experimental Procedures”) and separated by preparative AU-PAGE (38), and the levels of processed CRS4C-1 were evaluated by Western blot analyses with CRS4C-1 antiserum. Processed Paneth cell α-defensins were abundant in AU-PAGE separations of SAMP1/YitFc protein extracts as judged by their co-migration with Crp-4 (Fig. 5A, boxed region, 7th–11th lanes). As expected, mature α-defensins were not detected in MMP-7-null ileal protein extracts as judged by staining with Coomassie Blue (Fig. 5B) (27). AU-PAGE Western blot analyses (55) of fractionated SAMP1/YitFc mouse ileal proteins (Fig. 5A) showed robust immunoreactivity of mature CRS4C peptides that eluted from preparative AU-PAGE slightly later than α-defensins and co-migrated with recombinant CRS4C-1 (Fig. 5C, boxed region, 15th–20th lanes). Consistent with the requirement for MMP-7 in α-defensin activation (27), mature CRS4C-1 peptides were not detected in MMP-7-null mouse extracts (Fig. 5D). Ileal proteins from MMP-7-null mice contained apparent CRS4C precursors that eluted from preparative AU-PAGE with lower mobility (fractions 35 and later) as judged by their immunoreactivity and co-migration with recombinant pro-CRS4C-1 (data not shown). Collectively, the results of these in vitro and in vivo experiments implicate MMP-7 as the in vivo activating convertase for both pro-α-defensins and the related CRS4C precursors in mouse Paneth cells.
FIGURE 5.

Analysis of in vivo processing of native CRS4C peptides. Acid extracts of ileal organs from wild-type SAMP1/YitFc mice (A and C) and MMP-7 gene knock-out mice (B and D) were separated by preparative AU-PAGE and analyzed in analytical AU-polyacrylamide gels by Coomassie Blue staining (A and B) and Western blotting using anti-CRS4C-1 IgG (1:15) (C and D). Peptide controls included pro-Crp-4 (PC4), CRS4C-1 (4C), and pro-CRS4C-1 (P4C), and fraction numbers are denoted at the tops of the gels. Processed mouse enteric α-defensins (A, boxed region) and immunoreactive, processed CRS4C (C, boxed region) are found in wild-type mice but absent in MMP-7-null mice. MMP-7 is required for native CRS4C processing in vivo.
Mechanisms of CRS4C-1 Bacterial Cell Killing
Disulfide stabilization contributes to CRS4C-1 microbicidal activity against certain bacterial targets. Although the disulfide pairings of CRS4C dimers have not been determined definitively, the dimer lacks free thiol groups, and evidence suggests that the two monomer strands are oriented as parallel strands that are stabilized by three or more intermolecular disulfide bonds.7 Because analyses of several α-defensins have shown that the canonical α-defensin disulfide array is not required for bactericidal peptide activity (44), we tested whether the disulfide bonding is a functional determinant in structurally distinct CRS4C-1(54–72). Accordingly, CRS4C-1 was modified by reduction and acetylation to eliminate all disulfide bonds, and its bactericidal peptide activity was compared with native CRS4C-1, with Crp-4 and pro-Crp-4, which served as respective positive and negative control peptides. In assays against Vibrio cholerae and Staphylococcus aureus (Fig. 6, A and D), both peptides were bactericidal, although the activity against S. aureus was lower. However, disulfide bond disruption eliminated the in vitro bactericidal activity against Salmonella enterica serovar typhimurium CS022, S. typhimurium 14028S, and Listeria monocytogenes (Fig. 6, C, E, and F) and attenuated bactericidal activity against E. coli ML35 (Fig. 6B). These findings are representative of assays performed two to three times on different days. In summary, alkylation of CRS4C-1 resulted in peptide bactericidal activity that differed with the microbial cell target, being fully active against V. cholerae (Fig. 6A) but abrogated against L. monocytogenes (Fig. 6C). Thus, although the outcome of disulfide mutagenesis is attenuation of activity against certain bacterial species, no generally applicable effect can be drawn from these results. Because in both Crp-4 and RMAD-4, Ala for Cys substitutions induce sensitivity to in vitro proteolysis by their convertases (19, 44), we considered testing for comparable effects on CRS4C. Although native CRS4C-1 is not proteolyzed by MMP-7 (Fig. 4A), this peptide is highly sensitive to trypsin and is degraded to such an extent that peptide fragments cannot be detected (data not shown). Thus, unlike α-defensins, which are inherently resistant to proteolysis (19, 44, 56), disulfide bonds in native CRS4C-1 do not confer comparable stability.
FIGURE 6.

Bactericidal peptide activities of native and alkylated CRS4C. Exponentially growing V. cholerae (A), E. coli (B), L. monocytogenes (C), S. aureus (D), S. typhimurium 14028s (E), and S. typhimurium CS022 (F) were exposed to the peptide concentrations shown at 37 °C in 50 ml of 10 mm PIPES buffer supplemented with 1% trypticase soy broth for 1 h. Following peptide exposure, the bacteria were plated on trypticase soy broth and agar and incubated overnight at 37 °C. Surviving bacteria were counted as colony-forming units (CFU)/ml at each peptide concentration, and each panel is representative of three independent experiments. Values at or below 1 × 103 colony-forming units/ml signify that no colonies were detected. ●, Crp-4; ○, pro-Crp-4; ▿, native CRS4C-1(54–72); ▾, alkylated CRS4C-1(54–72).
CRS4C(54–72) Mediates Membrane-disruptive Effects
Most α-defensins mediate bactericidal effects via membrane-disruptive mechanisms (8–11, 39, 57, 58). However, the strikingly different primary and quaternary structures of CRS4C and α-defensins suggested that CRS4C might kill bacteria by a different, perhaps unique, mechanism. To determine the mechanism of CRS4C-1 bactericidal action, we measured the ability of CRS4C-1 to permeabilize live E. coli ML35 cells by monitoring intracellular conversion of ONPG to ONP as a function of peptide concentration and exposure time (see “Experimental Procedures” and Fig. 7A). As reported previously (11, 23, 59), Crp-4 induced robust and rapid ONPG conversion in E. coli at 1.5 and 3 μm peptide concentrations, whereas CRS4C-1 induced no ONP production at those concentrations (Fig. 7A, upper panel, and data not shown). CRS4C-1-mediated cell permeabilization was evident at a minimum peptide concentration of 6 μm, under which conditions neither precursor had detectable cell permeabilizing activity (Fig. 7A, lower panel). Also, the kinetics of CRS4C-induced ONPG conversion were markedly slower than for Crp-4, and longer peptide exposure times were needed for CRS4C-1 to reach maximal ONP production (Fig. 7A, lower panel). Thus, although CRS4C-1 induces E. coli cell killing via a membrane disruption mechanism, the distinct CRS4C-1 dose response and kinetics of ONP production suggest that its disruptive mechanisms differ from those of Crp-4 and α-defensins.
FIGURE 7.
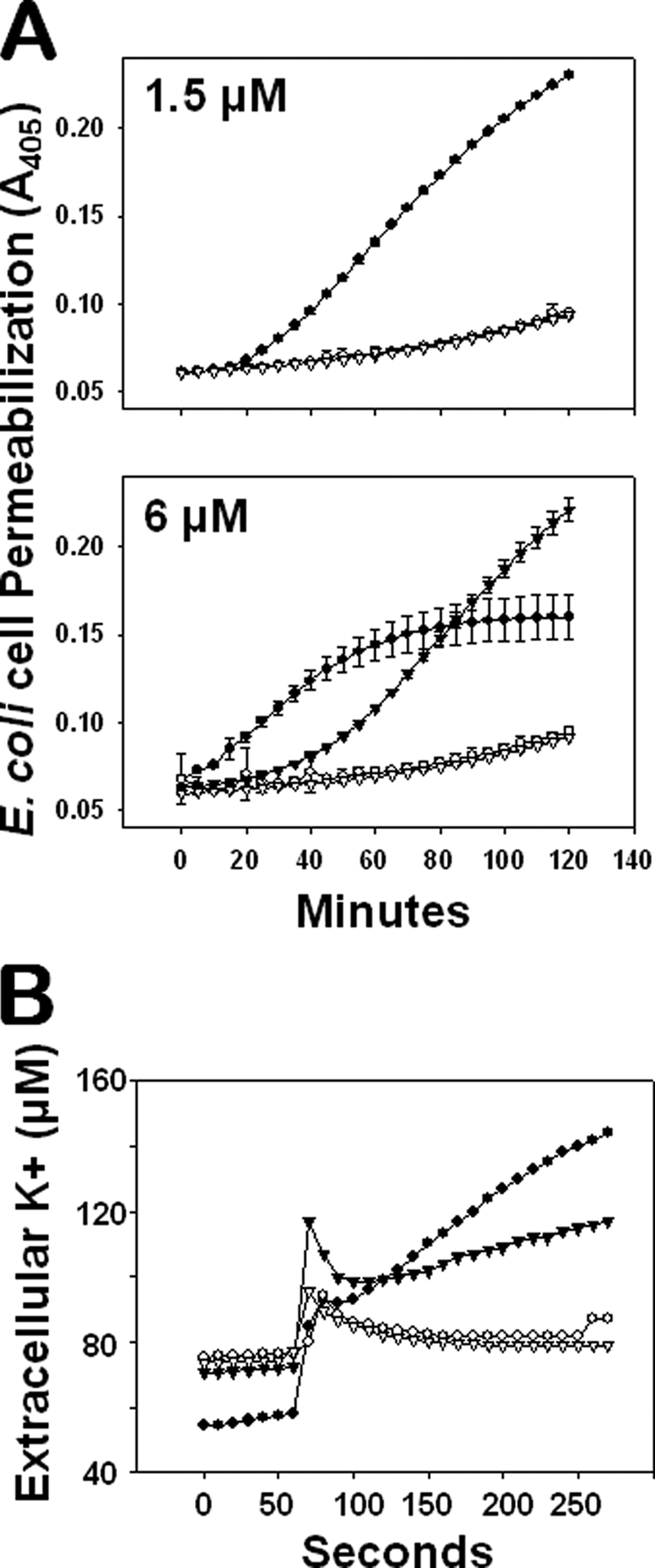
CRS4C-mediated bactericidal peptide activity is mediated via a membrane disruptive mechanism. A, E. coli ML35 cells were exposed to 1.5 μm (upper panel) and 6.0 μm (lower panel) pro-Crp-4 (○), Crp-4 (●), pro-CRS4C-1 (▿), and CRS4C-1 (▾) in the presence of 2.5 mm ONPG for 2 h at 37 °C. Membrane permeabilization was measured spectrophotometrically at A405 nm. Mature CRS4C-1 and Crp-4, but not their precursors, induce target cell membrane leakage. B, membrane efflux of K+ from live E. coli cells was measured in response to peptide exposure as an index of membrane disruption and cell death. Exponential-growing E. coli ML35 cells were incubated for 1 min in 10 mm PIPES (pH 7.4) supplemented with 0.1% (v/v) trypticase soy broth and subsequently exposed to 7 μm pro-Crp-4 (○), Crp-4 (●), pro-CRS4C-1 (▿), and CRS4C-1 (▾) in the same buffer for 4 min at 37 °C. Extracellular levels of potassium ions were measured using an ion-selective electrode sensitive for K+ (see “Experimental Procedures”). Mature CRS4C-1 and Crp-4, but not their precursors, induce K+ efflux of target cells.
The release or efflux of potassium ions from bacterial cells indicates that cell membrane physiology is disrupted and that the death of the organism has occurred (47, 48, 60). Accordingly, we also assayed efflux of bacterial cell K+ as an index of membrane-permeabilizing mechanisms of the CRS4C and Crp-4 peptides. Extracellular K+ levels were measured over time using an ion-selective electrode in incubation mixtures containing bacterial cells mixed with CRS4C-1 or Crp-4 as well as their respective precursors (see “Experimental Procedures”). As predicted, neither precursor induced K+ efflux from E. coli cells (Fig. 7B), but extracellular K+ levels increased substantially in the presence of either of the mature peptides. As in the ONPG conversion assays (Fig. 7A), 7 μm Crp-4 evoked a more rapid K+ efflux from E. coli ML35 cells than equimolar CRS4C-1 (Fig. 7B). Although CRS4C-1 and Crp-4 both disrupt bacterial cell membranes, the reduced kinetics of CRS4C-1-mediated K+ efflux suggest that its effects on E. coli viability are less immediate than Crp-4-induced cell killing.
DISCUSSION
In the mouse small intestine, enteric α-defensins and CRS4C peptides are Paneth cell-specific products that are stored within secretory granules for release into the crypt lumen. CRS4C-coding Defcr-rs genes exhibit a pattern of expression that differs both regionally and in relative abundance to that of mouse Paneth cell α-defensins, with the highest CRS4C mRNA levels found in the ileum and barely detectable transcript and protein levels in the proximal small intestine. The CRS4C expression pattern resembles that of Crp-4 in that Crp-4 mRNA levels also are highest in Paneth cells of the distal small intestine (49). Additionally, the levels of CRS4C mRNA and peptide are markedly higher in Paneth cells of ileitis-prone SAMP1/YitFc mice compared with non-prone strains. Furthermore, the increased CRS4C expression in SAMP1/YitFc ileum occurs prior to any histologic evidence of ileitis. Accordingly, we investigated the mechanism of pro-CRS4C processing and its bactericidal mode of action. Inactive pro-CRS4C peptides are processed by MMP-7 in activating cleavage events that are identical to those involved in the processing of pro-α-defensins. The absence of processed enteric α-defensin and CRS4C peptides in MMP-7 null mice demonstrates the in vivo requirement for this enzyme in peptide processing. ONPG membrane leakage and potassium efflux assays demonstrated that the mechanism of action for CRS4C-1 occurs via membrane disruption, an activity that pro-CRS4C-1 lacks.
Although MMP-7 activates both mouse enteric α-defensin and CRS4C precursors at the same three residue positions, the efficiency of MMP-7-induced cleavage at the Ser58↓Leu59 and Ala58↓Leu59 sites in Crp-4 and CRS4C-1 sequences, respectively, (Fig. 4) appears to differ. For example, native mouse enteric α-defensins purified from intestinal tissue of wild-type mice are fully processed, with Leu59 at the N terminus (28). However, analyses of native CRS4C peptides detected CRS4C peptides with Leu54 or Leu59 at their N termini, and peptides with Leu54 N termini were more prevalent (16). Although the in vitro digest products are the same, perhaps in vivo, MMP-7-induced cleavage at the Ala58↓Leu59 residue positions in CRS4C precursors is less efficient than at the corresponding position in mouse Paneth cell pro-α-defensins. It is possible that the striking differences in primary, secondary, and tertiary structures between CRS4C and Crps influence access to this cleavage site and account for the difference in cleavage efficiency. MMP-7 cleavage sites are defined by a sequence of preferred residues. Pro-Leu-Glu↓Leu-Arg-Ala was shown to be an optimal hexapeptide substrate for MMP-7-mediated proteolysis (61). With Arg at the P2′ position, the Ser58↓Leu59 site in mouse pro-Crp proregions appears to match the preferred cleavage site sequence better than the corresponding site in pro-CRS4Cs. Reduced binding affinity and/or a slower rate of catalysis of CRS4C precursors due to a less optimal sequence may account for the less efficient MMP-7-mediated processing at this site.
Despite marked structural differences between the mature peptides, the proregions of both the CRS4C and Crp peptide families appear to have similar inhibitory roles. Pro-CRS4C and pro-Crps are activated to bactericidal forms by MMP-7-mediated cleavage of the proregion (Fig. 4), and assays of cell permeabilization and membrane-disruptive behavior (Fig. 7) show that CRS4C-1 is active whereas its precursor is not (Fig. 4). It seems remarkable that nearly identical prosegment sequences are able to perform the same function on peptides with almost no sequence or structural similarity. However, the high level of electropositive charge common to the two peptide families may represent a feature that allows the proregions to neutralize charge and inhibit interactions with electronegative bacterial cell envelopes.
The inhibition of CRS4C membrane-disruptive activity by the CRS4C proregion may provide a protective role in the Paneth cell during peptide biosynthesis. α-Defensins interact with model membranes and permeabilize and disrupt bacterial cell membranes with electronegative surfaces (10, 62). CRS4C also induces bacterial cell death and permeabilization by a similar general mechanism (Fig. 7). The inner leaflet of eukaryotic cells is enriched with phophatidylserine, and it has been proposed that mature α-defensins may perturb cellular inner membranes during biosynthesis in the absence of a charge-neutralizing proregion (24). Proregion charge neutralization, therefore, could attenuate certain of the cytotoxic properties of defensins, such as the ability of HNP-1 to permeabilize K562 leukemia cell membranes (20). The cytotoxicity of mature CRS4C has not been characterized, but it appears reasonable to speculate that pro-CRS4C proregions could mask potential cytotoxic properties.
The CRS4C-coding Defcr-rs genes exist uniquely in the mouse genome. Thus, the association of CRS4C overproduction with the development of ileitis in the SAMP1/YitFc mouse model of Crohn disease cannot apply directly to human disease per se. However, the extraordinary overexpression of these highly cationic, disulfide-bonded peptides, e.g. CRS4C, provides a system for investigating the role of such Paneth cell gene products in the induction or exacerbation of chronic intestinal inflammation of relevance to human disease. Possibly, unusually high levels of CRS4C peptide secretion into the SAMP1/YitFc mouse ileum could influence the composition of the microbiome, leading to ileitis in these mice. Certainly, the repertoire of Crp peptides secreted by Paneth cells has profound effects on the composition of resident small bowel flora (63).
It is equally plausible that the potential consequences of CRS4C hyper abundance may not involve peptide-mediated membrane disruption or direct effects of the peptide on the luminal flora. Rather, overexpression of this peptide could induce inflammation by disrupting cellular homeostasis. For example, in pro-HNP-1, the covalently associated proregion greatly facilitates defensin folding by solubilizing and interacting with the C-terminal, mature form of the peptide (64). Disruption of Paneth cell homeostasis by deficient autophagy (65–67) or by severe disruption of the unfolded protein response and the attendant induced apoptosis (68) increases sensitivity to proinflammatory stimuli or causes fulminant ileitis, respectively. Several observations are consistent with the disruption of cellular homeostasis in this model. First, CRS4C peptide accumulation is already very high before ileitis is histologically detectable in SAMP1/YitFc mice. Second, SAMP1/YitFc mice are able to develop attenuated ileitis in the absence of intestinal microflora (69). Third, the membrane-active CRS4C peptide is highly unstable in solution and folds inefficiently in vitro. These facts lead us to speculate that high intracellular levels of CRS4C may affect Paneth cell ER homeostasis adversely, perhaps increasing sensitivity to additional proinflammatory stimuli and initiating the ileitis. Therefore, although CRS4C is unique to the mouse, the exceptionally high levels of CRS4C in SAMP1/YitFc mouse ileum may influence inflammation by disrupting homeostatic mechanisms common to all cells and thus have relevance to the human condition.
Supplementary Material
Acknowledgments
We thank Drs. Michael E. Selsted and Dat Tran for useful discussions and Xiaoqing Qu for excellent technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 DK044632 and R01 AI059346 and by the Human Frontiers Science Program (to A. J. O.) and by National Institutes of Health Grants R01 DK064751 and P01 DK57880 (to S. M. C.) and through the Molecular Biology and Morphology Cores of the University of Virginia National Institutes Diabetes and Digestive and Kidney Diseases/National Institutes of Health Digestive Diseases Research Core Center Grant P30 DK067629.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4.
M. T. Shanahan, unpublished observations.
- Crp
- cryptdin
- MMP-7
- matrix metalloproteinase-7
- CRS4C
- cysteine-rich sequence 4C
- pro-Crp
- pro-cryptdin
- AU-PAGE
- acid-urea PAGE
- HPLC
- high performance liquid chromatography
- MALDI-TOF
- matrix-assisted laser desorption ionization time-of-flight
- PIPES
- 1,4-piperazinediethanesulfonic acid
- ONPG
- 2-ortho-nitrophenyl β-d-galactopyranoside
- ONP
- o-nitrophenol.
REFERENCES
- 1.Lehrer R. I. (2007) Curr. Opin. Hematol. 14, 16–21 [DOI] [PubMed] [Google Scholar]
- 2.Selsted M. E., Ouellette A. J. (2005) Nat. Immunol. 6, 551–557 [DOI] [PubMed] [Google Scholar]
- 3.Brogden K. A., Ackermann M., McCray P. B., Jr., Tack B. F. (2003) Int. J. Antimicrob. Agents 22, 465–478 [DOI] [PubMed] [Google Scholar]
- 4.Schutte B. C., McCray P. B., Jr. (2002) Annu. Rev. Physiol. 64, 709–748 [DOI] [PubMed] [Google Scholar]
- 5.Tomasinsig L., Zanetti M. (2005) Curr. Protein. Pept. Sci. 6, 23–34 [DOI] [PubMed] [Google Scholar]
- 6.Selsted M. E. (2007) Curr. Pharm. Des. 13, 3061–3064 [DOI] [PubMed] [Google Scholar]
- 7.Selsted M. E., Harwig S. S. (1989) J. Biol. Chem. 264, 4003–4007 [PubMed] [Google Scholar]
- 8.White S. H., Wimley W. C., Selsted M. E. (1995) Curr. Opin. Struct. Biol. 5, 521–527 [DOI] [PubMed] [Google Scholar]
- 9.Wimley W. C., Selsted M. E., White S. H. (1994) Protein Sci. 3, 1362–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Satchell D. P., Sheynis T., Shirafuji Y., Kolusheva S., Ouellette A. J., Jelinek R. (2003) J. Biol. Chem. 278, 13838–13846 [DOI] [PubMed] [Google Scholar]
- 11.Hadjicharalambous C., Sheynis T., Jelinek R., Shanahan M. T., Ouellette A. J., Gizeli E. (2008) Biochemistry 47, 12626–12634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ganz T., Selsted M. E., Szklarek D., Harwig S. S., Daher K., Bainton D. F., Lehrer R. I. (1985) J. Clin. Invest. 76, 1427–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Porter E. M., Bevins C. L., Ghosh D., Ganz T. (2002) Cell. Mol. Life Sci. 59, 156–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Selsted M. E., Miller S. I., Henschen A. H., Ouellette A. J. (1992) J. Cell Biol. 118, 929–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Porter E. M., Liu L., Oren A., Anton P. A., Ganz T. (1997) Infect. Immun. 65, 2389–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Putsep K., Axelsson L. G., Boman A., Midtvedt T., Normark S., Boman H. G., Andersson M. (2000) J. Biol. Chem. 275, 40478–40482 [DOI] [PubMed] [Google Scholar]
- 17.Weeks C. S., Tanabe H., Cummings J. E., Crampton S. P., Sheynis T., Jelinek R., Vanderlick T. K., Cocco M. J., Ouellette A. J. (2006) J. Biol. Chem. 281, 28932–28942 [DOI] [PubMed] [Google Scholar]
- 18.Shirafuji Y., Tanabe H., Satchell D. P., Henschen-Edman A., Wilson C. L., Ouellette A. J. (2003) J. Biol. Chem. 278, 7910–7919 [DOI] [PubMed] [Google Scholar]
- 19.Kamdar K., Maemoto A., Qu X., Young S. K., Ouellette A. J. (2008) J. Biol. Chem. 283, 32361–32368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valore E. V., Martin E., Harwig S. S., Ganz T. (1996) J. Clin. Invest. 97, 1624–1629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ganz T., Liu L., Valore E. V., Oren A. (1993) Blood 82, 641–650 [PubMed] [Google Scholar]
- 22.Valore E. V., Ganz T. (1992) Blood 79, 1538–1544 [PubMed] [Google Scholar]
- 23.Figueredo S. M., Weeks C. S., Young S. K., Ouellette A. J. (2009) J. Biol. Chem. 284, 6826–6831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michaelson D., Rayner J., Couto M., Ganz T. (1992) J. Leukoc. Biol. 51, 634–639 [DOI] [PubMed] [Google Scholar]
- 25.Elphick D., Liddell S., Mahida Y. R. (2008) Am. J. Pathol. 172, 702–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghosh D., Porter E., Shen B., Lee S. K., Wilk D., Drazba J., Yadav S. P., Crabb J. W., Ganz T., Bevins C. L. (2002) Nat. Immunol. 3, 583–590 [DOI] [PubMed] [Google Scholar]
- 27.Wilson C. L., Ouellette A. J., Satchell D. P., Ayabe T., López-Boado Y. S., Stratman J. L., Hultgren S. J., Matrisian L. M., Parks W. C. (1999) Science 286, 113–117 [DOI] [PubMed] [Google Scholar]
- 28.Ayabe T., Satchell D. P., Pesendorfer P., Tanabe H., Wilson C. L., Hagen S. J., Ouellette A. J. (2002) J. Biol. Chem. 277, 5219–5228 [DOI] [PubMed] [Google Scholar]
- 29.Patil A., Hughes A. L., Zhang G. (2004) Physiol. Genomics 20, 1–11 [DOI] [PubMed] [Google Scholar]
- 30.Ouellette A. J., Selsted M. E. (1996) FASEB J. 10, 1280–1289 [DOI] [PubMed] [Google Scholar]
- 31.Huttner K. M., Selsted M. E., Ouellette A. J. (1994) Genomics 19, 448–453 [DOI] [PubMed] [Google Scholar]
- 32.Huttner K. M., Ouellette A. J. (1994) Genomics 24, 99–109 [DOI] [PubMed] [Google Scholar]
- 33.Hornef M. W., Pütsep K., Karlsson J., Refai E., Andersson M. (2004) Nat. Immunol. 5, 836–843 [DOI] [PubMed] [Google Scholar]
- 34.Ouellette A. J., Lauldi J. C. (1990) J. Biol. Chem. 265, 9831–9837 [PubMed] [Google Scholar]
- 35.Rivera-Nieves J., Bamias G., Vidrich A., Marini M., Pizarro T. T., McDuffie M. J., Moskaluk C. A., Cohn S. M., Cominelli F. (2003) Gastroenterology 124, 972–982 [DOI] [PubMed] [Google Scholar]
- 36.Ouellette A. J., Hsieh M. M., Nosek M. T., Cano-Gauci D. F., Huttner K. M., Buick R. N., Selsted M. E. (1994) Infect. Immun. 62, 5040–5047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanabe H., Yuan J., Zaragoza M. M., Dandekar S., Henschen-Edman A., Selsted M. E., Ouellette A. J. (2004) Infect. Immun. 72, 1470–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harwig S. S., Chen N. P., Park A. S., Lehrer R. I. (1993) Anal. Biochem. 208, 382–386 [DOI] [PubMed] [Google Scholar]
- 39.Satchell D. P., Sheynis T., Kolusheva S., Cummings J., Vanderlick T. K., Jelinek R., Selsted M. E., Ouellette A. J. (2003) Peptides 24, 1795–1805 [DOI] [PubMed] [Google Scholar]
- 40.Vidrich A., Buzan J. M., Barnes S., Reuter B. K., Skaar K., Ilo C., Cominelli F., Pizarro T., Cohn S. M. (2005) Am. J. Pathol. 166, 1055–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ouellette A. J., Satchell D. P., Hsieh M. M., Hagen S. J., Selsted M. E. (2000) J. Biol. Chem. 275, 33969–33973 [DOI] [PubMed] [Google Scholar]
- 42.Hagen S. J. (1990) J. Electron Microsc. Tech. 16, 37–44 [DOI] [PubMed] [Google Scholar]
- 43.Rice W. G., Ganz T., Kinkade J. M., Jr., Selsted M. E., Lehrer R. I., Parmley R. T. (1987) Blood 70, 757–765 [PubMed] [Google Scholar]
- 44.Maemoto A., Qu X., Rosengren K. J., Tanabe H., Henschen-Edman A., Craik D. J., Ouellette A. J. (2004) J. Biol. Chem. 279, 44188–44196 [DOI] [PubMed] [Google Scholar]
- 45.Tran D., Tran P. A., Tang Y. Q., Yuan J., Cole T., Selsted M. E. (2002) J. Biol. Chem. 277, 3079–3084 [DOI] [PubMed] [Google Scholar]
- 46.Lehrer R. I., Barton A., Ganz T. (1988) J. Immunol. Methods 108, 153–158 [DOI] [PubMed] [Google Scholar]
- 47.Tincu J. A., Menzel L. P., Azimov R., Sands J., Hong T., Waring A. J., Taylor S. W., Lehrer R. I. (2003) J. Biol. Chem. 278, 13546–13553 [DOI] [PubMed] [Google Scholar]
- 48.Orlov D. S., Nguyen T., Lehrer R. I. (2002) J. Microbiol. Methods 49, 325–328 [DOI] [PubMed] [Google Scholar]
- 49.Ouellette A. J., Darmoul D., Tran D., Huttner K. M., Yuan J., Selsted M. E. (1999) Infect. Immun. 67, 6643–6651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karlsson J., Pütsep K., Chu H., Kays R. J., Bevins C. L., Andersson M. (2008) BMC Immunol. 9, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ge Y. B., Yang D. H., Ohmori J., Tsuyama S., Kim B. S., Kim J. B., Murata F. (1997) Arch. Histol. Cytol. 60, 133–142 [DOI] [PubMed] [Google Scholar]
- 52.Mukherjee S., Vaishnava S., Hooper L. V. (2008) Cell. Mol. Life Sci. 65, 3019–3027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Selsted M. E. (1993) Genet. Eng. 15, 131–147 [DOI] [PubMed] [Google Scholar]
- 54.Rajabi M., de Leeuw E., Pazgier M., Li J., Lubkowski J., Lu W. (2008) J. Biol. Chem. 283, 21509–21518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang M. S., Pang J. S., Selsted M. E. (1997) Anal. Biochem. 253, 225–230 [DOI] [PubMed] [Google Scholar]
- 56.Mastroianni J. R., Ouellette A. J. (2009) J. Biol. Chem. 284, 27848–27856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hristova K., Selsted M. E., White S. H. (1996) Biochemistry 35, 11888–11894 [DOI] [PubMed] [Google Scholar]
- 58.Miszta A., Machán R., Benda A., Ouellette A. J., Hermens W. T., Hof M. (2008) J. Pept. Sci. 14, 503–509 [DOI] [PubMed] [Google Scholar]
- 59.Baroncelli S., Negri D. R., Rovetto C., Belli R., Ciccozzi M., Catone S., Michelini Z., Borghi M., Leone P., Fagrouch Z., Heeney J., Cara A. (2007) AIDS Res. Hum. Retroviruses 23, 287–296 [DOI] [PubMed] [Google Scholar]
- 60.Lambert P. A., Hammond S. M. (1973) Biochem. Biophys. Res. Commun. 54, 796–799 [DOI] [PubMed] [Google Scholar]
- 61.Smith M. M., Shi L., Navre M. (1995) J. Biol. Chem. 270, 6440–6449 [DOI] [PubMed] [Google Scholar]
- 62.Clarke L. L., Gawenis L. R., Bradford E. M., Judd L. M., Boyle K. T., Simpson J. E., Shull G. E., Tanabe H., Ouellette A. J., Franklin C. L., Walker N. M. (2004) Am. J. Physiol. Gastrointest. Liver. Physiol. 286, G1050–G1058 [DOI] [PubMed] [Google Scholar]
- 63.Salzman N. H., Hung K., Haribhai D., Chu H., Karlsson-Sjöberg J., Amir E., Teggatz P., Barman M., Hayward M., Eastwood D., Stoel M., Zhou Y., Sodergren E., Weinstock G. M., Bevins C. L., Williams C. B., Bos N. A. (2010) Nat. Immunol. 11, 76–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu Z., Li X., Ericksen B., de Leeuw E., Zou G., Zeng P., Xie C., Li C., Lubkowski J., Lu W. Y., Lu W. (2007) J. Mol. Biol. 368, 537–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cadwell K., Liu J. Y., Brown S. L., Miyoshi H., Loh J., Lennerz J. K., Kishi C., Kc W., Carrero J. A., Hunt S., Stone C. D., Brunt E. M., Xavier R. J., Sleckman B. P., Li E., Mizushima N., Stappenbeck T. S., Virgin H. W., 4th (2008) Nature 456, 259–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cadwell K., Patel K. K., Komatsu M., Virgin H. W., 4th, Stappenbeck T. S. (2009) Autophagy 5, 250–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stappenbeck T. S. (2009) Gastroenterology 137, 30–33 [DOI] [PubMed] [Google Scholar]
- 68.Kaser A., Lee A. H., Franke A., Glickman J. N., Zeissig S., Tilg H., Nieuwenhuis E. E., Higgins D. E., Schreiber S., Glimcher L. H., Blumberg R. S. (2008) Cell 134, 743–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barnes S. L., Vidrich A., Wang M. L., Wu G. D., Cominelli F., Rivera-Nieves J., Bamias G., Cohn S. M. (2007) J. Immunol. 179, 7012–7020 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.