

Abstract
Many germ line antibodies have asparagine residues at specific sites to achieve specific antigen recognition. To study the role of asparagine residues in the stabilization of antigen-antibody complexes, we examined the interaction between hen egg white lysozyme (HEL) and the corresponding HyHEL-10 variable domain fragment (Fv). We introduced Ala and Asp substitutions into the Fv side chains of l-Asn-31, l-Asn-32, and l-Asn-92, which interact directly with residues in HEL via hydrogen bonding in the wild-type Fv-HEL complex, and we investigated the interactions between these mutant antibodies and HEL. Isothermal titration calorimetric analysis showed that all the mutations decreased the negative enthalpy change and decreased the association constants of the interaction. Structural analyses showed that the effects of the mutations on the structure of the complex could be compensated for by conformational changes and/or by gains in other interactions. Consequently, the contribution of two hydrogen bonds was minor, and their abolition by mutation resulted in only a slight decrease in the affinity of the antibody for its antigen. By comparison, the other two hydrogen bonds buried at the interfacial area had large enthalpic advantage, despite entropic loss that was perhaps due to stiffening of the interface by the bonds, and were crucial to the strength of the interaction. Deletion of these strong hydrogen bonds could not be compensated for by other structural changes. Our results suggest that asparagine can provide the two functional groups for strong hydrogen bond formation, and their contribution to the antigen-antibody interaction can be attributed to their limited flexibility and accessibility at the complex interface.
Keywords: Antibodies, Antigen, Calorimetry, Crystal Structure, Protein-Protein Interactions
Introduction
The specific recognition of ligands by proteins is a fundamental biological phenomenon (1), and the interaction between antigen and antibody in the immune system is a typical example. Antibodies acquire their affinity and specificity for various target antigens by changing the amino acid residue composition of their six hyper-variable regions, known as complementarity-determining regions (CDRs)3 (2). Despite the small number of amino acid residues composing the CDRs, antibodies can precisely bind a large number of target antigens (3). Some of these residues, serine and asparagine, for example, are located at specific positions in the CDRs in the germ line protein (supplemental Fig. S1). Many structural studies have shown that these residues are essential for the affinity and specificity of the antigen-antibody interaction (4–9, 11).
Recent high resolution analyses of three-dimensional structures of protein-ligand complexes, including the antigen-antibody complexes, and analyses of the kinetic and thermodynamic parameters underlying these interactions (12–19) have indicated that protein-ligand interactions require a good geometric fit according to the lock-and-key (20) and induced fit models (21) as well as a high degree of complementarity of hydrophobic and polar parts of each binding site (17, 22, 23). There are various forces determining the affinity and specificity of these interfacial complementarities, including noncovalent bonds, such as hydrogen bonds, salt bridges, and van der Waals interactions (17, 22–24). Among those noncovalent bonds, hydrogen bonds play a unique and functionally important role in molecular associations because of its involvement in both thermodynamic and kinetic processes. First, hydrogen bonds are strong and directional enough to control and direct the structures within molecular assemblies. Second, from a mechanistic point of view, the energy of hydrogen bonds, which is between that of van der Waals interactions and covalent bonds, allows biomolecules to associate and dissociate quickly at room temperature. These features of hydrogen bonds form the basis by which specific recognition is achieved quickly with moderate affinity (25). Many studies show that hydrogen bonds play a significant role in a wide range of biomolecular interactions in terms of generating affinity and specific recognition (26–31). It is also reported that hydrogen bonds can make a favorable enthalpic contribution to protein-ligand interactions (32–34). Thus, the subject of hydrogen bonding is of major interest in biological research.
Here, we investigated the involvement of side chains of asparagine and serine in hydrogen bond formation. To elucidate the contribution of hydrogen bonds to the affinity and specificity of antigen-antibody interactions, mutagenesis in combination with both structural and thermodynamic analyses has been a productive and powerful strategy (35). X-ray crystallographic studies can provide information on the structural complementary of the mutant antibody (or antigen) and its interaction with its counterpart (36–39), and measurement of thermodynamic parameters can quantify the energetic effect of the mutated residues on the interaction (40, 41). Approaches based on multiple analyses are essential to further our understanding of the mechanisms underlying the roles of hydrogen bonds in antigen-antibody interactions.
We focused on the interaction between hen egg white lysozyme (HEL) and the variable domain fragment (Fv) of the anti-HEL monoclonal antibody HyHEL-10, which is one of the most studied proteinaceous antigen-antibody interactions in terms of structural and functional features (42–50). The bacterial expression system for the HyHEL-10 Fv fragment has been established (51–53), and the Fv-HEL interactions have been investigated by using the wild-type and/or mutant Fv fragments (42–44, 46, 47, 54), including the x-ray crystal structure of its complex with HEL (45, 47, 48, 55). In the wild-type Fv-HEL complex, Kondo et al. (45) observed 12 water molecules bridging the imperfect antigen-antibody interface as well as 20 direct hydrogen bonds between residues of the antibody and antigen at the interface. In a previous study, we examined the role of indirect hydrogen bonds via interfacial water molecules in the HyHEL-10 Fv-HEL interaction by thermodynamic analysis and x-ray structural analysis in combination with mutagenesis (48). We discovered that hydrogen bonds made a minor contribution by providing an enthalpic advantage to the interaction, despite the partial offset caused by entropy loss resulting from the hydrogen bonding stiffening the antigen-antibody complex (48). Here, we further examined the role of hydrogen bonds in stiffening the antigen-antibody complex by focusing on the three residues Asn-31, Asn-32, and Asn-92 in the light chain, which have side-chain amide groups that participate in the formation of direct hydrogen bonds with residues in HEL (Fig. 1). Mutational analyses achieved by truncating these amide groups in the antibody side chains should give further insight into the effect of direct hydrogen bonding on complex formation. We constructed six Fv mutants, LN31D, LN31A, LN32D, LN32A, LN92D, and LN92A, and performed thermodynamic analyses of the interaction between these HyHEL-10 Fv mutants and HEL by means of isothermal titration calorimetry (ITC) in combination with x-ray crystallographic analysis of the mutant Fv-HEL complexes. Based on our results, we elucidated the contribution of direct hydrogen bonds at the atomic level to the antigen-antibody interaction. We also discussed the role of interfacial asparagine residues in the antigen-antibody interaction with respect to their role in achieving specificity and affinity of antibodies for target antigens.
FIGURE 1.

Interaction between HyHEL-10 Fv and HEL. A, overall structure of the wild-type HyHEL-10 Fv-HEL complex. The C-α schematic diagrams of VL, VH, and HEL are shown in green, cyan, and pink, respectively. The residues investigated in this study are shown in orange. Interfacial water molecules bridging Fv and HEL are represented by red balls. B and C, local structure around the target sites investigated in this study. Interfacial Asn residues at sites 31, 32 (B), and 92 (C) in the VL participate in the antigen-antibody interaction by the formation of direct hydrogen bonds with the antigen. The contacting residues in VL and HEL are shown by green and pink sticks, respectively. Direct hydrogen bonds and indirect hydrogen bonds (via interfacial water molecules) between the antigen and antibody are indicated by red dotted lines. The figures were generated with WebLab Viewer (Molecular Simulations Inc., San Diego).
EXPERIMENTAL PROCEDURES
Materials
All enzymes for genetic engineering were obtained from Takara Shuzo (Kyoto, Japan), Toyobo (Osaka, Japan), and New England Biolabs (Beverly, MA). Isopropyl β-d-thiogalactopyranoside was obtained from Wako Fine Chemicals Inc. (Osaka, Japan). All other reagents were of biochemical research grade. The HEL antigen purchased from Seikagaku-Kogyo (Tokyo, Japan) was purified by ion exchange chromatography on SP-Sepharose FF (GE Healthcare), followed by gel filtration on Superdex 75 pg equilibrated with phosphate-buffered saline. Eluted antigen was lyophilized and dissolved in water at a concentration of 0.54 mm prior to use.
Site-directed Mutagenesis
The gene structure of the light chain variable region (VL) and heavy chain variable region (VH) coexpression vector of the HyHEL-10 Fv fragment is described in our previous paper (51). Site-directed mutagenesis was performed with phagemid pTZ18U (Bio-Rad) according to the method of Kunkel et al. (56). The DNA oligonucleotide primers for mutation of Asn to Ala, and Asn to Asp, at sites 31, 32, and 92 of VL were 5′-GTCGATCGGCGCCAACCTCCAC-3′, 5′-GTCGATCGGCGACAACCTCCAC-3′, 5′-GATCGGCAACGCCCTCCACTGG-3′, 5′-GATCGGCAACGACCTCCACTGG-3′, 5′-CAGCAGTCGGCCAGCTGGCCG-3′, and 5′- CAGCAGTCGGACAGCTGGCCG-3′, respectively (mutated sites are underlined). The correctness of the intended mutations was confirmed by DNA sequencing (ABI 310 Genetic Analyzer, Applied Biosystems, Tokyo, Japan).
Preparation of HyHEL-10 Mutant Fv Fragments
We obtained wild-type and mutant Fv fragments by using the Escherichia coli BL21 (DE3) expression system. BL21 (DE3) cells harboring the appropriate expression plasmid were precultured in 3 ml of LB medium, which was then used to inoculate in 3 liters of 2× YT medium containing 100 mg/liter ampicillin. The culture was shaken overnight at 28 °C and centrifuged at 3000 × g for 20 min, and the bacteria pellet was resuspended in 3 liters of 2× YT medium containing 100 mg/liter ampicillin and isopropyl 1-thio-β-d-galactopyranoside at a final concentration of 1 mm. The culture was again shaken overnight at 28 °C. The culture was then centrifuged at 3000 × g for 20 min, and the collected supernatant was subjected to ammonium sulfate precipitation at 80% saturated ammonium sulfate, followed by centrifugation. The protein pellet was solubilized in 30–40 ml of phosphate-buffered saline buffer and then dialyzed against phosphate-buffered saline buffer. Fv fragments were purified by affinity chromatography. The protein solution was loaded onto an HEL-Sepharose column (51), and the column was washed with phosphate-buffered saline buffer and then wash buffer (50 mm Tris-HCl, pH 8.5, containing 0.5 m NaCl). Fv fragments were eluted with elution buffer (0.1 m Gly-HCl, pH 2.0, containing 0.2 m NaCl) and then buffered rapidly with 1 m Tris-HCl, pH 7.5. Fv-containing fractions were centrifuged, and minor impurities were removed by gel filtration with a Sephacryl S-200 column (GE Healthcare) pre-equilibrated with 50 mm Tris-HCl, pH 7.5, containing 0.2 m NaCl. The purity of isolated proteins was confirmed by SDS-PAGE in the buffer system described by Laemmli (57). The purified Fv fragments were concentrated using a Centriprep-10 column (Millipore, Billerica, MA).
Inhibition Assay of HEL Enzymatic Activity
The experimental procedure for the inhibition assay was essentially as described by Ueda et al. (51). Briefly, various concentrations of the Fv fragment were mixed with 1.5 μm HEL and incubated at 25 °C for 1 h in 30 μl of phosphate-buffered saline. Each mixture was then added to 970 μl of 50 mm NaH2PO4 buffer (pH 6.2, adjusted with NaOH) containing 340 μg of Micrococcus luteus cells. The initial rate of the decrease in A540 nm was monitored at 25 °C.
Isothermal Titration Calorimetry
Thermodynamic parameters of the interaction between HEL and wild-type or mutant HyHEL-10 Fv fragments were determined by ITC using a VP-ITC microcalorimeter (MicroCal, Inc., Northampton, MA). HEL at 5 μm in 50 mm phosphate buffer, pH 7.2, containing 0.2 m NaCl, was placed into the calorimeter cell and was titrated with a 50 μm solution of the Fv fragment in the same buffer at four different temperatures (25, 30, 35, or 40 °C) for the LN31A, LN31D, LN32D, and LN92D mutants, and at one temperature (30 °C) for the LN32A mutant. The solution containing the Fv fragments was injected 25 times in 10-μl aliquots over 20 s. Thermograms were analyzed with Origin 5 software (MicroCal, Inc.) after correcting for the buffer contribution. The enthalpy change (ΔH) and binding constant (Ka) for each antigen-antibody interaction were obtained directly from the experimental titration curve. The Gibbs free energy change (ΔG = −RT ln Ka) and the entropy change (ΔS = (−ΔG + ΔH)/T) for the association were calculated from the ΔH and Ka. The heat capacity change (ΔCp) was estimated from the temperature dependence of the enthalpy change.
Estimation of Protein Concentration
The concentration of HEL was estimated by using A1%280 = 26.5 (58). The concentrations of wild-type and mutant HyHEL-10 Fv fragments were estimated by using A1%280 = 20.6 (51).
Crystallization, Data Collection, and Structural Determination of the HyHEL-10 Mutant Fv-HEL Complexes
Fv fragment-HEL complexes for the three HyHEL-10 mutants LN31D, LN32D, and LN92D were crystallized under conditions similar to those used for the wild-type Fv-HEL complex (45). The best crystals were grown in 0.1 m Hepes buffer, pH 7.6–7.8, 9–11% w/v polyethylene glycol 6000, and 7–9% (w/v) 2-methyl-2,4-pentanediol. The resultant crystals were elongated bipyramid shapes. For the LN31A-HEL complex, a micro needle-like crystal was obtained in 0.1–0.2 m ammonium sulfate, 22.5–27.5% (w/v) polyethylene glycol 4000, and 0.1–0.2 m sodium acetate trihydrate, pH 4.6; however, it was too small to be used for obtaining data sets of x-ray diffraction images. Furthermore, for the LN32A-HEL complex, the sample of LN32A Fv fragment was too poor to crystallize. All crystallization conditions included glycerol at a final concentration of 15% as a cryoprotectant.
Data sets for all mutant Fv-HEL complexes were obtained at 100 K using the synchrotron x-ray source at beamline BL6A at the Photon Factory (Tsukuba, Japan). The diffraction images were processed by the interactive data processing package DPS/MOSFLM/CCP4. Integration was carried out using the MOSFLM software (59); scaling was carried out using SCALA software (60), and the final file of structural factors was obtained by using TRUNCATE (61) and MTZ2VARIOUS in the CCP4 program suite (62). The structures of the Fv-HEL complexes were determined by a molecular replacement method using the wild-type complex (Protein Data Bank code 2DQJ) as a model structure and refined by using the CNS program (63). The graphic program O (64) was used for making adjustments to the molecular model. Crystallographic and refinement data for each Fv mutant-HEL complex are summarized in the supplemental Table S1.
Calculations of the root mean square deviation (r.m.s.d.) for structural comparison were performed using the programs LSQKAB (65) and COMPAR in the CCP4 software suite. Interfacial areas were calculated with AREAIMOL in the CCP4 software suite. Determination of contacting atoms between Fv and HEL was performed with the CONTACT program in the CCP4 suite. Figures were drawn with the program WebLab Viewer Lite (Accelrys Inc., San Diego).
Atomic coordinates and structural factors for each mutant Fv-HEL complex were deposited in the Protein Data Bank. The Protein Data Bank accession codes are 3A67 for LN31D, 3A6B for LN32D, and 3A6C for LN92D.
RESULTS
Expression and Purification of Fv Fragments
To elucidate the role of interfacial asparagine residues in direct hydrogen bond formation in HyHEL-10 Fv-HEL complexes, we constructed, expressed, and purified six mutant Fv fragments named LN31A, LN31D, LN32A, LN32D, LN92A, and LN92D. With the exception of LN32A and LN92A, we obtained purities of greater than 95%, and the final yields were greater than 10 mg/liter of culture. LN92A could not be expressed using the established expression system. Furthermore, the yield of LN32A was low following purification, despite the level of expression, and thus only a limited number of experiments were performed for LN32A.
Inhibition of Enzymatic Activity of Hen Lysozyme by Mutant Fv Fragments
Tsumoto and co-workers (42, 51, 52) demonstrated that the HyHEL-10 Fv fragment inhibits the enzymatic activity of its antigen, HEL, in the presence of a slight molar excess of the Fv fragment. Thus, we investigated the inhibition of the enzymatic activity of HEL by the wild-type Fv fragment and the five of the mutant Fv fragments LN31A, LN31D, LN32A, LN32D, and LN92D (Fig. 2). The wild-type Fv fragment and LN31D showed a similar level of inhibition of HEL enzymatic activity, and for LN92D this level was only slightly lower than that of the wild type. By comparison, the level of inhibition of HEL enzymatic activity shown by LN32D was notably lower than that of the wild type. LN31A and LN32A displayed no inhibitory activities toward HEL. These results suggest that the mutations harbored by LN31A, LN32A, and LN32D play an important role in target antigen affinity.
FIGURE 2.
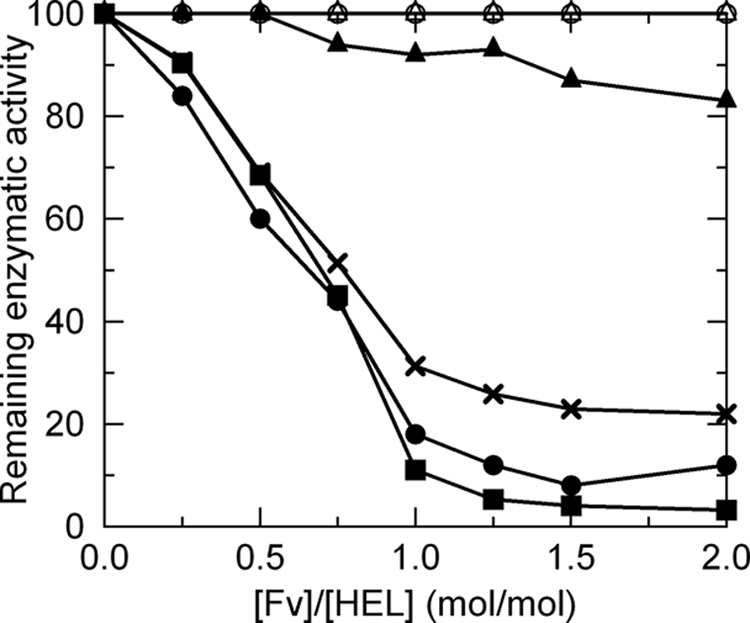
Inhibition of lysozyme enzymatic activity by HyHEL-10 Fv. Experimental conditions are provided in the text. Symbols used are as follows: solid squares, wild type; open circles, LN31A; solid circles, LN31D; open triangle, LN32A; solid triangles, LN32D; solid crosses, LN92D.
Thermodynamic Analyses
To investigate the interactions between the Fv fragment mutants and HEL from a thermodynamic viewpoint, we carried out an ITC study of the association between the mutant Fv fragments and lysozyme (42–44, 46–48, 55). The thermogram for each experiment was obtained by titrating the HEL solution with the Fv solution and then subtracting the base line obtained from titrating buffer with the Fv solution (Fig. 3). Thermodynamic parameters are summarized in Table 1. In the Asp substitution mutants LN31D (l-Asn-31) and LN92D (l-Asn-92), thermodynamic analysis revealed that a small loss in binding enthalpy (ΔΔH, 6.9 kJ mol−1 for LN31D, and ΔΔH, 5.1 kJ mol−1 for LN92D) and a smaller (close to zero) gain in binding entropy led to a minor loss in Gibbs energy of binding compared with the wild-type-HEL interaction, and it resulted in a small decrease in the binding affinity constants for LN31D (Ka, 17.8 × 107 m−1) and LN92D (Ka, 14.0 × 107 m−1). By comparison, for LN31A, LN32A, and LN32D there was a large decrease in negative binding enthalpy, and there was a decrease in binding entropy loss, which notably decreased the Gibbs energy of binding. These results indicated that the interaction between HEL and each Ala or Asp substitution in the Fv fragments at l-Asn-31, l-Asn-32, and l-Asn-92 led to unfavorable enthalpy changes and favorable entropy changes and that the enthalpy-entropy compensation reduced the loss in the Gibbs energy change, to some degree. For LN31A, LN32A, and LN32D, the large decrease in enthalpy change failed to maintain affinity for HEL, resulting in marked 600-, 500-, and 100-fold decreases in their binding affinity constant, respectively, compared with that of the wild type. The changes in heat capacity for LN31A, LN31D, LN32D, and LN92D, estimated from the values of enthalpy change of temperature dependence, were −2.35, −1.70, −1.01, and −1.68 kJ mol−1 K−1, respectively (supplemental Fig. S2).
FIGURE 3.

Thermodynamic analyses of interactions between HyHEL-10 Fv mutants and HEL by isothermal titration calorimetry. Thermogram and titration curves for LN31A-HEL (A), LN32D-HEL (B), and LN92D-HEL (C) are shown. The base line obtained by titrating each mutant Fv solution (50 μm) with buffer was subtracted from the thermogram obtained by titrating the corresponding Fv solution with the HEL solution (5 μm).
TABLE 1.
Thermodynamic parameters of mutant Fv-HEL interactions at 30 °C and pH 7.2 in phosphate buffer
Experimental protocols are described in the text. Data represent the average of at least three independent measurements. Errors for all values were within 5% for several experiments. The abbreviations used are as follows: n, stoichiometry; Ka, binding constant; ND, not determined; ΔG, ΔH, ΔS, and ΔCp, changes in Gibbs energy, binding enthalpy, entropy, and heat capacity, respectively.
| Mutant | n | Ka | ΔG | ΔΔG | ΔH | ΔΔH | TΔS | TΔΔS | ΔS | ΔΔS | ΔCpa | ΔΔCpa |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ×107m−1 | kJ mol−1 | kJ mol−1 | kJ mol−1 | kJ mol−1K−1 | kJ mol−1K−1 | |||||||
| Wild type | 1.05 | 82.1 | −51.7 | 0 | −99.7 | 0 | −48.0 | 0 | −0.158 | 0 | −1.53 | 0 |
| LN31A | 0.96 | 0.13 | −35.4 | 16.3 | −48.9 | 50.8 | −13.5 | 34.5 | −0.045 | 0.113 | −2.35 | −0.82 |
| LN31D | 0.94 | 17.8 | −47.9 | 3.8 | −92.8 | 6.9 | −44.9 | 3.1 | −0.148 | 0.010 | −1.70 | −0.17 |
| LN32A | 0.84 | 0.17 | −36.1 | 15.6 | −74.0 | 25.7 | −37.9 | 10.1 | −0.125 | 0.033 | NDb | NDb |
| LN32D | 0.97 | 0.93 | −40.3 | 11.4 | −47.2 | 52.5 | −6.9 | 41.1 | −0.023 | 0.135 | −1.01 | 0.52 |
| LN92D | 0.85 | 14.0 | −47.2 | 4.5 | −94.6 | 5.1 | −47.4 | 0.6 | −0.156 | 0.002 | −1.68 | −0.15 |
a The changes in heat capacity were calculated by performing measurements at four different temperatures (25, 30, 35, and 40 °C) except for LN32A.
b The measurement for the association between the LN32A and HEL was performed only at one temperature (30 °C), because the expressed and purified amount of LN32A mutant protein was too small for further analyses.
Crystal Structure of Mutant Fv-HEL Complexes
The crystal structures of mutant HyHEL-10 Fv-HEL complexes were solved at resolutions sufficient for determining local structural differences (1.8 Å) (supplemental Table S1). Most of the interfacial water molecules, which mediate the Fv-HEL interaction, were conserved among the mutant Fv-HEL and wild-type complexes (supplemental Table S2). Additional interfacial water molecules appear in the LN32D-HEL and LN92D-HEL complexes.
Crystal structures of mutant HyHEL-10 Fv-HEL complexes were superimposed onto the wild-type Fv-HEL complex by means of the LSQKAB (65) and COMPER programs in the CCP4 software suite (62). The resultant r.m.s.d. between C-α atoms of each mutant-HEL complex and that of the wild-type Fv-HEL complex are shown in Table 2. The structure of the LN31D-HEL complex is similar to that of the wild-type Fv-HEL complex, apart from the region containing 17–19 residues in HEL adjacent to its epitope, which is out of alignment in the other mutant Fv-HEL complexes (48). The crystal structure of the LN32D-HEL and LN92D-HEL complexes is relatively distinct from that of the wild-type Fv-HEL complex. In LN32D-HEL and LN92D-HEL, the r.m.s.d. value for each polypeptide obtained by superposing the corresponding polypeptide and the r.m.s.d. value for the Fv fragment obtained by superposing Fv chains are very low. These observations indicate that the Asp substitution at l-Asn-32 and l-Asn-92 did not introduce drastic structural changes to the respective domains. However, the r.m.s.d. values for VL and/or VH, in the case of HEL fitting, and the r.m.s.d. values for HEL, in the case of Fv fitting, especially for the l-Asn-32 Fv-HEL complex, are moderately large. This finding shows that the orientation of VL and/or VH (in other words Fv) with HEL in the LN32D-HEL and LN92D-HEL complexes is different from that in the wild-type Fv-HEL complex and that the difference in relative orientation of VL, VH, and HEL in the LN32D-HEL complex is greater than that in the LN92D-HEL complex (Table 2).
TABLE 2.
r.m.s.d. in the C-α atoms of each chain (Å)
r.m.s.d. were obtained by superposing the C-α atom coordinates of each polypeptide chain (VL, VH, or HEL), the Fv portion (VL and VH), or all chains on the corresponding chain of the wild-type complex. r.m.s.d. were calculated with LSQKAB and COMPAR in the CCP4 suite. WT indicates wild type.
| Complex | VL fit | VH fit | HEL fit | Fv fit | All fit |
|---|---|---|---|---|---|
| LN31D vs. WT | |||||
| VL | 0.08 | 0.13 | 0.14 | 0.09 | 0.11 |
| VH | 0.15 | 0.08 | 0.10 | 0.09 | 0.09 |
| HEL | 0.25 | 0.13 | 0.08 | 0.17 | 0.09 |
| LN32D vs. WT | |||||
| VL | 0.12 | 0.20 | 0.99 | 0.16 | 0.20 |
| VH | 0.33 | 0.14 | 1.16 | 0.16 | 0.30 |
| HEL | 0.59 | 0.87 | 0.16 | 0.74 | 0.39 |
| LN92D vs. WT | |||||
| VL | 0.10 | 0.13 | 0.67 | 0.11 | 0.13 |
| VH | 0.18 | 0.12 | 0.80 | 0.13 | 0.21 |
| HEL | 0.41 | 0.56 | 0.15 | 0.49 | 0.28 |
The interfacial areas within the crystal structures of mutant HyHEL-10 Fv-HEL complexes were calculated by means of the program AREAMOL in the CCP4 software suite and are listed in supplemental Table S3. In LN32D-HEL and LN92D-HEL, the total interfacial area is reduced mostly because of a decrease in the VL-HEL interfacial area, whereas in LN31D-HEL the domains (VL, VH, and HEL) and total interfacial area are similar to those in the wild-type Fv-HEL complex.
The direct contacts between mutant HyHEL-10 Fv and HEL in the mutant complexes were calculated by means of the program CONTACT in the CCP4 software suite (supplemental Table S4). In LN31D-HEL, the noncovalent bonds were well conserved with the wild-type Fv-HEL complex. By comparison, in the LN32D and LN92D-HEL complexes, multiple noncovalent bonds, which were largely distinct from those in the wild-type Fv-HEL complex, were observed. Some hydrogen bonds and van der Waals interactions between CDR-L3 and HEL were abolished. Four hydrogen bonds between CDR-H1 and CDR-H2 and HEL, multiple van der Waals contacts, and one salt bridge between CDR-H3 and CDR-L3 and HEL were newly introduced and increased the total number of interactions in the LN32D-HEL and LN92D-HEL complexes. These contact changes (loss and/or gain) (supplemental Table S5) and the subsequent local structural changes at the mutated sites (Fig. 4) are similar in the LN32D-HEL and LN92D-HEL complexes.
FIGURE 4.

Comparison of local structures at the mutation site between mutant Fv-HEL and wild-type Fv-HEL complexes. A, LN31D-HEL; B, LN32D-HEL; C, LN92D-HEL. C-α atoms of all polypeptide chains of each mutant complex are superimposed on those of the wild-type complex. Wild-type complex is shown in gray. Residues of VL, VH, and HEL in the mutant Fv-HEL complexes are shown in green, cyan, and pink, respectively. The positions marked W correspond to the water molecules (parentheses indicate wild-type water molecules) shown as red balls. Hydrogen bonds in the mutant Fv-HEL complexes and wild-type complex are depicted as red dotted lines and gray dotted lines, respectively. Salt bridges in the mutant complexes are depicted as blue broken lines. The hydrogen bonding (observed in the wild-type Fv-HEL complex) that is abolished in each mutation is represented as a gray thick dashed line.
Our results have led to the following conclusions: 1) the overall structure of LN31D-HEL, including its interfacial region that is the size of interfacial area, formations of atomic contacts, and interfacial water molecules, is almost identical to that of the wild-type Fv-HEL complex; 2) in LN32D-HEL and LN92D-HEL, the relative orientation of Fv (VL and VH) and HEL was notably altered by the mutations, resulting in the structure of the interfacial regions being different from that in the wild-type Fv-HEL complex; 3) the structural differences in LN32D-HEL and LN92D-HEL are similar, except for the hydrogen bonds generated by the amide group of the mutated side chains in LN32D and LN92D.
Structure of Mutant Fv-HEL Complexes
LN31D-HEL
The overall structure of LN31D-HEL, including the interfacial water molecules and the local structure around the site of mutation, is similar to that of the wild-type Fv-HEL complex (Table 2, supplemental Table S2-S5, and Fig. 4A). The structures of antigen-antibody interfacial sites, other than the site of mutation, are also similar to that of the wild-type complex. Thus, the removal of the hydrogen bond between N-δ2 in l-Asn-31 and O in His-15 of HEL, resulting from the substitution of l-Asn-31 with Asp, has little impact on the antigen-antibody interaction.
LN32D-HEL
The backbone structure of LN32D-HEL was in principle identical to the corresponding structure in the wild-type Fv-HEL complex. However, the relative orientation of VL, VH, and HEL was altered in the mutant complex. In particular, the difference in the orientation of Fv and HEL between the wild-type and LN32D complexes is great (Table 2), as described above. In addition, the local structures within the LN32D complex, especially at the antigen-antibody interface, are notably different from those in the wild-type complex (Table 2, supplemental Table S2-S5, and Fig. 4B). Notably, the atoms of O-δ2 in l-Asp-32 and of O in HEL-Gly-16, which were originally close enough to create a hydrogen bond in the wild-type complex, are widely separated from each other in the mutant complex because of indirect hydrogen bonds from a newly introduced interfacial molecular water (W34) located between them. Furthermore, within the same proximity as the mutated site, conformational changes were observed around multiple residues in LN32D and around the counterpart residues in HEL, including l-Asn-31 and His-15, because of newly formed hydrogen bonds between O-δ1 in l-Asn-31 and O in HEL-His-15. Therefore, around the mutated site of l-Asp-32 in LN32D-HEL, in which the target hydrogen bond was removed by the Asn to Asp substitution, the distance between l-Asp-32 and HEL-Gly-16 was increased thus resulting in many large structural changes, including the newly introduced interfacial water molecule, movement of interfacial water molecules, and the associated reorganization of interactions.
LN92D-HEL
In LN92D-HEL, the relative orientations of VL, VH, and HEL, in particular Fv and HEL, as well as the local structure around the site of mutation, are notably different from those in the wild-type complex and similar to those in LN32D-HEL (Table 2 and supplemental Tables S2–S5). The local conformational changes in LN92D-HEL around the site of mutation, such as the movement of side chains and water molecules, show a greater difference from those of the wild-type complex than do those in LN32D-HEL (Fig. 4C). However, differences in the r.m.s.d. and interfacial areas between the wild-type complex and LN92D-HEL were smaller than those between the wild-type complex and LN32D-HEL. The atoms of O-δ2 in l-Asp-92 and of O in HEL-Asn-19, which were originally close enough to create a hydrogen bond in the wild-type complex, were widely separated from each other in LN92D-HEL. A similar situation was observed for O-δ2 in l-Asp-32 and for O in HEL-Gly-16 in LN32D-HEL. The removal of direct hydrogen bonds resulted in conformational changes in the surrounding region and resulted in a newly introduced interfacial water molecule (W57), movement of multiple water molecules, and the reconstruction of interfacial hydrogen bonding networks accompanying these changes.
Comparison between LN32D-HEL and LN92D-HEL
The local conformational changes around l-Asn-92 in LN32D-HEL (Fig. 5A) are similar to those in LN92D-HEL, especially in the characteristic movement of the side chain of Asn-19 in HEL that interacts with l-Asn-92 (Fig. 4C). The local conformational changes around l-Asn-31 and l-Asn-32 in LN92D-HEL (Fig. 5B) are similar to those in LN32D-HEL in terms of their effects on the surrounding region, including a newly introduced interfacial water molecule at the coordinate W95, which corresponds to W34 in LN32D-HEL, movement of multiple water molecules, and the reconstruction of interfacial hydrogen bonding networks accompanying these changes. Thus, the only difference between the LN92D-HEL and LN32D-HEL complexes is the substantial conformational change resulting from the maintenance of the hydrogen bond between N-δ2 of l-Asn-32 and O of HEL-Gly-16 in the LN92D-HEL complex that is not present in the LN32D-HEL complex.
FIGURE 5.

Comparison of local structures at sites other than the mutation site between mutant Fv-HEL and wild-type Fv-HEL complexes. Local structures around l-Asn-92 in the LN32D-HEL complex (A) and around l-Asn-32 in the LN92D-HEL complex (B) are shown. Hydrogen bonds conserved in mutant and wild-type Fv-HEL complexes are omitted to facilitate visualization. Refer to Fig. 4 for details.
DISCUSSION
Here, we constructed six mutant HyHEL-10 Fv fragments named LN31A, LN31D, LN32A, LN32D, LN92A, and LN92D to elucidate the energetic contributions of direct (not via interfacial water molecules) hydrogen bonds, which are formed by amino acid residues in antibody-antigen interactions. We investigated the interactions between mutant Fv fragments and the HEL antigen by structural and thermodynamic analyses of the resultant complexes. The mutations do not lead to drastic structural changes of the Fv fragments, with the exception of LN92A, and do not alter their stability. Thus, the structural and thermodynamic changes we observed in the mutant antigen-antibody complexes do not originate from changes in the structure of the mutant Fv fragments in the antigen-free state. However, a slight and minor change in the structure of the antigen-free Fv fragment might have a strong impact on the interaction between the Fv fragment and the antigen. To address this, it is necessary to investigate the structural changes in the Fv fragment in the antigen-free state; structural analyses of antigen-free Fv will be reported in the near future.4 In the following sections, we discuss and correlate our thermodynamic and structural findings.
Thermodynamic Analysis of Mutant Fv-HEL Interactions
The values for the enthalpic (−ΔH) and entropic (−TΔS) contributions to the interaction between the mutants and HEL increased in the order LN92D, LN31D, LN32A, LN31A, and LN32D. The values of the binding constant (Ka) for the LN31D-HEL and LN92D-HEL interactions were slightly lower than that for the wild-type Fv-HEL interaction, whereas the Ka for the LN31A-HEL, LN32A-HEL, and LN32D-HEL interactions were markedly lower than that for the wild-type Fv-HEL interaction, resulting in a smaller change in the Gibbs energy (−ΔG). These results indicate that removal of the direct hydrogen bonds formed through interfacial Asn residues on the light chain of the antibody in the HyHEL-10 Fv-HEL complex is enthalpically unfavorable and entropically favorable. Thus, the interfacial hydrogen bonds appeared to make an enthalpic contribution to the HyHEL-10 Fv-HEL interaction similar to the finding in a previous study for indirect hydrogen bonds via interfacial water molecules (48). For the LN31A-HEL, LN32A-HEL, and LN32D-HEL interactions, the advantage in binding entropy relative to that in the wild type cannot compensate for the loss in binding enthalpy and so causes a decrease in their affinities compared with the wild-type interaction.
We estimated the change in heat capacity (ΔCp) from the temperature dependence of the enthalpy changes for interactions between LN31A, LN31D, LN32D, and LN92D and the antigen. The ΔCp values for the interactions between LN31D and LN92D and HEL (−1.70 and −1.68 kJ mol−1, respectively) were similar to that for the interaction between the wild type and HEL (−1.53 kJ mol−1), whereas those for the LN31A-HEL and LN32D-HEL interactions were lower (−2.35 kJ mol−1) and greater (−1.01 kJ mol−1), respectively, than the wild-type Fv-HEL interaction. It has been reported that the large negative heat capacity change is related to the decrease in the nonpolar accessible surface area (ΔASAapolar) for water molecules, i.e. the hydrophobic effect of the molecule association (66–69). The ΔCp values for the LN31A-HEL and LN32D-HEL interactions suggest that the conformational changes and/or hydration structure changes introduced by the interaction of the mutant antibodies with HEL could be different from those introduced in the wild-type Fv-HEL interaction (68, 70, 71).
l-Asn-31, Contribution of the Polar but Noncharged Side Chain Is Favorable for the Interaction
The overall structure, the size of interfacial area, formations of atomic contacts, position of interfacial water molecules, and the local structure around the mutation site of the LN31D-HEL complex are similar to that of the wild-type complex (Table 2, supplemental Tables S2–S5, and Fig. 4A). These results suggested that the effects of the loss of hydrogen bonds caused by the substitution of l-Asn-31 with Asp on the interaction between the mutant Fv and HEL are directly reflected in the changes to the thermodynamic parameters (ΔG, ΔH, ΔS, and ΔCp) (Table 1). Thermodynamically, the substitution of l-Asn-31 with Asp leads to a decrease in the negative change in binding enthalpy (ΔΔH; 6.9 kJ mol−1) accompanied by a small decrease in the binding constant (Ka; 1.78 × 108 mol−1). These thermodynamic changes might originate from the removal of a hydrogen bond between the N-δ2 atom of l-Asn-31 and the O atom of HEL-His-15 and from the conversion of a hydrogen bond between the O-δ1 atom of l-Asn-31 and the N-ζ atom of HEL-Lys-96 to a salt bridge. The hydrogen bond present in the wild-type Fv-HEL complex, but absent from the LN31D-HEL complex, is expected to be weak because the distance between the two atoms involved in this bond are close to the limit for hydrogen bonding to occur. Therefore, the contribution of this hydrogen bond to the strength of the antibody-antigen interaction is most likely to be relatively small compared with other bonds. On the contrary, the hydrogen bond that was converted to a salt bridge in the mutant Fv was found by free energy simulations to be highly polarized (72). Thus, the effect of deleting this hydrogen bond is expected to be greater than the former. Furthermore, the changes in thermodynamic parameters of the antibody-antigen interaction are most likely caused by the substitution of a hydrogen bond with a salt bridge. The free energy simulations described above (72), however, suggest that the substitution of a hydrogen bond with a salt bridge between LN31D and HEL-Lys-96 or between LN31E and HEL-Lys-96 is expected to stabilize the interaction between HyHEL-10 and HEL. This indicates a discrepancy between the predicted result (calculated data) and our present experimental data, which shows that the substitution destabilizes the interaction (ΔΔG, 3.8 kJ mol−1). However, our data are supported by a previous study showing that the substitutions of l-Asn-31 with Asp and with Glu using the single-chain variable fragment of the HyHEL-10 mutant antibodies, LN31D scFv and LN31E scFv, destabilize the interaction between the mutant antibodies and HEL (ΔΔG = 1.4 ± 0.3 kcal mol−1 (5.9 ± 1.3 kJ mol−1) and ΔΔG = 5.7 ± 0.1 kcal mol−1 (23.8 ± 0.4 kJ mol−1), respectively) (49). These thermodynamic parameters might indicate that the charged antigen epitope residues with a positive charge, corresponding to Lys-96 of HEL in this case, are more stable by coupling with the donor and/or acceptor of the hydrogen bond in the neutral state, corresponding to the side chain of l-Asn-31 in this case, than by coupling with its counterpart charge, corresponding to the side chain of l-Asp-31 with a negative charge in this case (49). Furthermore, analysis of charged amino acid side chains (Arg, Lys, Glu, and Asp) buried at the intermolecular interfaces indicates that oriented dipoles are usually preferred over countercharges in stabilizing these buried residues (69).
Thermodynamics analysis of the interaction between LN31A and HEL suggests that the Ala substitution at l-Asn-31 leads to a drastic decrease in binding enthalpy (ΔΔH; 50.8 kJ mol−1), which cannot be compensated for by the large decrease in entropic loss (TΔΔS; 34.5 kJ mol−1), thus resulting in a decrease in Gibbs energy (ΔΔG; 16.3 kJ mol−1). It has been suggested that a large negative change in enthalpy mostly originates from the formation of hydrogen bond and/or van der Waals interactions during binding (32, 33, 43, 55). Thus, it is conceivable that in the LN31A-HEL interaction the substitution of Asn with Ala might introduce an interfacial structure different from that in the wild-type Fv-HEL complex as well as many changes in the interfacial noncovalent bonds between the antibody and antigen, including loss of van der Waals interactions and hydrogen bonds with large energetic contributions to binding in the wild-type Fv-HEL complex. The binding constant of the LN31A-HEL interaction is much lower than that of the wild-type interaction by about 3 orders of magnitude, despite the large decrease in entropic loss that results partly from a reduction in the conformational flexibility of the antibody upon complexation (Table 1) (73, 74). Unexpectedly, the binding constant is lower for the LN31A-HEL interaction than for the HY33A-HEL interaction, in which the mutation site (H-Tyr-33) is considered as a hot spot of the paratope in the HyHEL-10 Fv-HEL interaction (55). These findings suggest that the hydrogen bond between the O-δ1 atom of l-Asn-31 and the N-ζ atom of HEL-Lys-96 and/or the deleted van der Waals contacts between atoms on the side chain of l-Asn-31 (O-δ1, N-δ2, and C-γ atoms) and HEL might confer an entropy disadvantage, but it confers an enthalpy advantage and thus plays a crucial role in the affinity between the wild-type antibody and the HEL target. Thus, we conclude that l-Asn-31 is one of the energetic hot spots (75) in the HyHEL-10 Fv-HEL interaction.
l-Asn-32, Critical Contribution of N-δ2 in Stabilizing the Complex through Hydrogen Bond Formation
The crystal structure of the LN32D-HEL complex has many changes in structural features, when compared with the wild-type Fv-HEL complex, including a large difference in the orientation of HEL to VL and/or VH (Table 2), a decrease in the antigen-antibody interfacial area (supplemental Table S3), conformational changes in local structure at, near, and far from the mutation site, movement of interfacial water molecules accompanied by rearrangements of the hydrogen bonding network, as well as many changes (loss and/or gain) in the antigen-antibody interaction resulting from these structural changes (Fig. 4B, Fig. 5A, and supplemental Table S2 and S4). These structural differences between the LN32D-HEL and wild-type Fv-HEL complexes are similar to the differences between the LN92D-HEL and wild-type Fv-HEL complexes apart from the loss of the hydrogen bond between the N-δ2 atom of l-Asn-32 and the O atom of HEL-Gly-16 that resulted from the substitution of Asn with Asp in LN32D. This hydrogen bond is unexpectedly conserved in the LN92D-HEL complex, whereas all the other changes described above are similar to those in the LN32D-HEL complex. By comparison, the loss of the hydrogen bond between the N-δ2 atom of l-Asn-92 and the O atom of HEL-Asn-19 observed in the LN92D-HEL complex because of the substitution of Asn with Asp in LN92D is not conserved in the LN32D-HEL complex, despite the lack of mutation at l-Asn-92 in LN32D (Fig. 5A and supplemental Table S4). Taken together, these observations suggest that the formation of the hydrogen bond between the N-δ2 atom of l-Asn-32 and the O atom of HEL-Gly-16 induces the formation of other interactions, including the hydrogen bond between the N-δ2 atom of l-Asn-92 and the O atom of HEL-Asn-19. The number of noncovalent bonds between antibody and antigen was higher for the LN32D-HEL interaction than for the wild-type Fv-HEL interaction with higher affinity (supplemental Table S5). The LN32D-HEL interaction had a lower binding constant than the wild-type Fv-HEL interaction by 2 orders of magnitude (Table 1). Therefore, even if large conformational changes are introduced by the other interfacial residues on CDRs and form other interactions, these newly formed interactions cannot compensate for the energetic loss of the hydrogen bond between the N-δ2 atom of l-Asn-32 and the O atom of HEL-Gly-16.
At the interface of HyHEL-10 Fv-HEL, about 20 direct hydrogen bonds have been observed between the antigen and the antibody (supplemental Table S4) (45, 55). For example, H-Ser-52, H-Ser-54, and H-Ser-56 also contact the antigen by forming hydrogen bonds via their side-chain hydroxyl group. Thermodynamic analysis of the interaction between the Fv mutants HS52A, HS54A, and HS56A and HEL show that the small decrease in the favorable enthalpy change and in the unfavorable entropy change results in a slight increase in the value of the binding constant (supplemental Table S6).5 This result suggests that the hydrogen bonds, which have been deleted in the mutant antibody-antigen interactions, have an energetically unfavorable entropy effect in the antigen-antibody interaction, rather than supplemental minor contribution to the interaction. Taken together, these results suggest that the energetic contribution of hydrogen bonds to the antigen-antibody interaction varies enormously and ranges from strong to weak in terms of affinity. Our results suggest that the more buried the hydrogen bond is at the interface, the greater its energetic contribution. l-Asn-32 in the wild-type Fv-HEL complex is completely buried at the interface and has an accessible surface area of 0 Å2. By comparison, H-Ser-54 and H-Ser-56 are relatively exposed with accessible surface areas of 60 Å2 and 25 Å2, respectively. Several other reports support our notion that the more buried the hydrogen bond is at the interface, the stronger the bond. Strong hydrogen bonds buried at the interface are observed in the interaction between thermolysin and its inhibitor (28) and between hemagglutinin of a mutant influenza virus and its monoclonal antibody (76). In the interaction between the D1.3 antibody, the anti-HEL antibody which is the same as HyHEL-10 and the anti-D1.3 antibody E5.2, the binding energy of the strongest hydrogen bond among those buried at the interface is 4.3 kcal mol−1, whereas the binding energies of exposed hydrogen bonds are only 1.3–1.7 kcal mol−1 (77). The hydrogen bond between the N-δ2 atom of l-Asn-32 and the O atom of HEL-Gly-16 might make the interface rigid with its high energy and result in an unfavorable entropy effect in the interaction. However, this type of hydrogen bond might be considered to have a greater favorable enthalpy effect than an unfavorable entropy effect and thus make an important contribution to the acquirement of affinity for the antigen. We can conclude that this type of hydrogen bond is one of the most critical in the interaction and thus cannot be compensated for by other conformational changes or by formatting other noncovalent bonds.
In the interaction between LN32A and HEL, the substitution of Asn with Ala at l-32 leads to a large decrease in binding enthalpy (ΔΔH; 25.7 kJ mol−1), exceeding the decrease in entropic loss (TΔΔS; 10.1 kJ mol−1), resulting in a great loss in Gibbs energy (ΔΔG; 15.6 kJ mol−1). Thermodynamic parameters suggest that this mutation introduces changes to the interfacial structure and noncovalent bonds found in wild-type HEL, including loss of van der Waals interactions and hydrogen bonds with large energetic contributions to the Fv-HEL interaction, which are also seen in LN31A-HEL and LN32D-HEL interactions. In each case, the Asn to Ala substitution decreased the affinity of the antibody for the antigen, suggesting that the Asn residue at l-32 contributes to generating affinity and specificity of the HyHEL-10 Fv-HEL interaction as one of the “hot spot” residues in the interaction.
l-Asn-92, a Small Enthalpic Gain Results in Minor Contribution to Affinity
As already described, the structural differences between the LN92D-HEL and the wild-type Fv-HEL complex (Table 2, Fig. 4C, Fig. 5B, and supplemental Tables S2–S5) are similar to the differences between LN32D-HEL and the wild-type Fv-HEL complex. However, the thermodynamics parameters for LN92D-HEL are markedly different from those for LN32D-HEL. The substitution of Asn with Asp at the l-92 leads to a small decrease in binding enthalpy and no change in entropic loss, resulting in only a decrease in binding constant compared with the wild-type Fv-HEL interaction, whereas the substitution of Asn with Asp at l-32 leads to a drastic decrease in the binding enthalpy as well as a decrease in the binding constant. These results suggest that the difference in thermodynamic parameters between LN92D-HEL and LN32D-HEL interactions can be attributed to the presence and absence, respectively, of the hydrogen bond between the N-δ2 atom of l-Asn-32 and the O atom of HEL-Gly-16 and that the LN92D mutant maintains affinity for HEL because this hydrogen bond is maintained. It can be concluded that the hydrogen bond between the N-δ2 atom of l-Asn-92 and the O atom of HEL-Asn-19 abolished by the substitution of Asn with Asp at l-92, like that at l-31 for LN31D, has only an incremental enthalpic contribution to the interaction, the loss of which can be compensated by changes in interfacial structure and/or formations of other noncovalent bonds.
Insight into the Role of Asn Residues in Antigen-Antibody Interactions
Our findings can be summarized as follows. 1) Isothermal titration calorimetric analysis shows that all mutations involving Asn residues lead to a decrease in the negative enthalpy change as well as a decrease in the association constants of the interaction. 2) The contribution of two hydrogen bonds is small, the deletion of which leads to only a slight decrease in affinity of the antibody for the antigen. By comparison, two hydrogen bonds show enthalpic gain, despite entropic loss perhaps due to stiffening of the interface by the bonds, and play a major role in the antigen-antibody interaction and thus in the affinity of the antibody for the antigen. 3) Structural analyses revealed that the effects of mutations involving Asn residues on the structure of the antigen-antibody complexes were compensated for by conformational changes and/or by gains of other interactions. 4) Hydrogen bonding buried at the interfacial area, such as those of l-Asn-32, have an enthalpic advantage over those exposed at the interfacial area and thus contribute significantly to the affinity of the antibody for the antigen; the deletion of these hydrogen bonds cannot be compensated for by structural changes. These results suggest that the contribution of Asn residues to the antigen-antibody interaction depends on the structure of the local interfacial area, which is independent of their potential for hydrogen bond formation. Formation of a salt bridge between the side chains of Asp (and/or Glu) and Arg residues (and/or Lys) at the complex interface often contributes to the affinity and specificity of an interaction (78–80). Asp is used at a higher than expected frequency in CDRs (5, 11, 80). It has been reported that in the interaction between HyHEL-5 and HEL, the loss of a salt bridge at the interface causes a remarkable decrease in the binding constant (79). By comparison, some Asp residues make only a minor contribution to the affinity of the protein interaction through salt bridges that originate from enthalpy-entropy compensation arising from the introduction of interfacial water molecules (44). Lines of evidence from previous reports suggest that the contribution of charged residues to the strength of protein interactions also depends on interfacial structure.
Based on our structural and thermodynamics analysis on mutations involving Asn residues described above, we will discuss why Asn residues are a preferred amino acid for stabilizing antigen-antibody complexes. First, the side chain of Asn makes less van der Waals contacts with the target antigen and has less of a tendency to form a hydrophobic environment than that of Trp and Tyr, which is the most common residue in CDRs (55). By comparison, Asn has the ability to make more contacts and more types of contacts than does Gly, Ala, or Ser. Second, the steric hindrance attributed to Asn because of its moderate size compared with other residues is low enough to reduce the loss in conformational entropy in the antigen-antibody interaction. Support for this notion comes from the report that Asn is used in antigen-antibody interactions at a higher frequency than Gln, which has similar properties to Asn but generates more steric hindrance because of its larger size (10, 80). Third, and most importantly, the side chain of Asn has a neutral and polar amide group consisting of the donor and acceptor of hydrogen bonding atoms O-δ1 and N-δ2 under neutral pH conditions. This functional group confers the ability for Asn to accommodate both positive and negative charges of the antigen and to form many hydrogen bond interactions with its target at the interface of the antibody. Thus, Asn can provide this functional group consisting of two atoms of O-δ1 and N-δ2 for strong hydrogen bond formation, and their contribution to the antigen-antibody interaction can be attributed to their limited flexibility and accessibility at the complex interface. Tsumoto et al. (43) and Shiroishi et al. (55) have shown that Tyr residues, the most common in CDRs, are energetic hot spots at some sites but can also provide only a minor contribution to the antigen-antibody interaction, and these properties of Tyr depend on the local structure. For specific antigen recognition, such properties might be critical for the preparation of binding sites, and therefore, Asn might be an appropriate residue for antigen recognition through hydrogen bond formation by using this functional group. Finally, it should be noted that Asn residues rarely appear at conserved positions in frameworks of VL, despite their high frequent appearance in CDRs (supplemental Fig. S1). This fact also might support that Asn is an amino acid specifically used for binding to an antigen and for stabilizing the antigen-antibody complexes.
Supplementary Material
Acknowledgments
We thank N. Sakabe, S. Wakatsuki, M. Suzuki, and N. Igarashi at the Photon Factory for their kind help with data collection.
This work was supported by grants-in-aid for general research (to K. T. and I. K.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1–S6 and Figs. S1 and S2.
The atomic coordinates and structure factors (codes 3A67, 3A6B, and 3A6C) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
T. Nakanishi and I. Kumagai, manuscript in preparation.
A. Yokota, unpublished data.
- CDR
- complementarity-determining region
- HEL
- hen egg white lysozyme
- VH
- variable region of immunoglobulin heavy chain
- VL
- variable region of immunoglobulin light chain
- Fv
- fragment of immunoglobulin variable regions
- ITC
- isothermal titration calorimetry
- r.m.s.d.
- root mean square deviation. The abbreviation used for the mutants is, for example, LN31A, which is the mutant of HyHEL-10 Fv in which Ala is substituted for Asn-31 in the VL chain.
REFERENCES
- 1.Böhm H. J., Schneider G. (eds) (2003) Protein-Ligand Interactions: From Molecular Recognition to Drug Design (Methods and Principles in Medicinal Chemistry) Vol. 19, 1st Ed., pp. 21–51, Wiley-VCH, Weinheim, Germany [Google Scholar]
- 2.Kabat E. A., Wu T. T., Bilofsky H. (1976) Proc. Natl. Acad. Sci. U.S.A. 73, 4471–4473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kabat E. A., Wu T. T, Bilofsky H. (1977) J. Biol. Chem. 252, 6609–6616 [PubMed] [Google Scholar]
- 4.Padlan E. A. (1990) Proteins Struct. Funct. Genet. 7, 112–124 [DOI] [PubMed] [Google Scholar]
- 5.Mian I. S., Bradwell A. R., Olson A. J. (1991) J. Mol. Biol. 217, 133–151 [DOI] [PubMed] [Google Scholar]
- 6.Sheriff S., Silverton E. W., Padlan E. A., Cohen G. H., Smith-Gill S. J., Finzel B. C., Davies D. R. (1987) Proc. Natl. Acad. Sci. U.S.A. 84, 8075–8079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chothia C., Lesk A. M. (1987) J. Mol. Biol. 196, 901–917 [DOI] [PubMed] [Google Scholar]
- 8.Padlan E. A., Silverton E. W., Sheriff S., Cohen G. H., Smith-Gill S. J., Davies D. R. (1989) Proc. Natl. Acad. Sci. U.S.A. 86, 5938–5942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilkinson R. A., Piscitelli C., Teintze M., Cavacini L. A, Posner M. R., Lawrence C. M. (2005) J. Virol. 79, 13060–13069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee K. H., Xie D., Freire E., Amzel L. M. (1994) Proteins 20, 68–84 [DOI] [PubMed] [Google Scholar]
- 11.Jackson R. M. (1999) Protein Sci. 8, 603–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clements C. S., Dunstone M. A., Macdonald W. A., McCluskey J., Rossjohn J. (2006) Curr. Opin. Struct. Biol. 16, 787–795 [DOI] [PubMed] [Google Scholar]
- 13.Wang Y., Shen B. J., Sebald W. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 1657–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brautigam C. A., Steitz T. A. (1998) Curr. Opin. Struct. Biol. 8, 54–63 [DOI] [PubMed] [Google Scholar]
- 15.Chothia C., Lesk A. M., Tramontano A., Levitt M., Smith-Gill S. J., Air G., Sheriff S., Padlan E. A., Davies D., Tulip W. R., Colman P. M., Spinelli S., Alzari P. M., Poljak R. J. (1989) Nature 342, 877–883 [DOI] [PubMed] [Google Scholar]
- 16.Davies D. R., Padlan E. A., Sheriff S. (1990) Annu. Rev. Biochem. 59, 439–473 [DOI] [PubMed] [Google Scholar]
- 17.Janin J., Chothia C. (1990) J. Biol. Chem. 265, 16027–16030 [PubMed] [Google Scholar]
- 18.Smith-Gill S. J. (1991) Curr. Opin. Biotechnol. 2, 568–575 [DOI] [PubMed] [Google Scholar]
- 19.Mariuzza R. A., Poljak R. J. (1993) Curr. Opin. Immunol. 5, 50–55 [DOI] [PubMed] [Google Scholar]
- 20.Fischer E. (1894) Ber. Dtsch. Chem. Ges. 27, 2984–2993 [Google Scholar]
- 21.Koshland D. E. (1958) Proc. Natl. Acad. Sci. U.S.A. 44, 98–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lo Conte L., Chothia C., Janin J. (1999) J. Mol. Biol. 285, 2177–2198 [DOI] [PubMed] [Google Scholar]
- 23.Jones S., Thornton J. M. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 13–208552589 [Google Scholar]
- 24.Chothia C., Janin J. (1975) Nature 256, 705–708 [DOI] [PubMed] [Google Scholar]
- 25.Desiraju G. R., Steiner T. (2001) The Weak Hydrogen Bond in Structural Chemistry and Biology, pp. 343–440, Oxford University Press, Oxford [Google Scholar]
- 26.Fersht A. R., Shi J. P., Knill-Jones J., Lowe D. M., Wilkinson A. J., Blow D. M., Brick P., Carter P., Waye M. M., Winter G. (1985) Nature 314, 235–238 [DOI] [PubMed] [Google Scholar]
- 27.Bartlett P. A., Marlowe C. K. (1987) Science 235, 569–571 [DOI] [PubMed] [Google Scholar]
- 28.Tronrud D. E., Holden H. M., Matthews B. W. (1987) Science 235, 571–574 [DOI] [PubMed] [Google Scholar]
- 29.Mandel-Gutfreund Y., Schueler O., Margalit H. (1995) J. Mol. Biol. 253, 370–382 [DOI] [PubMed] [Google Scholar]
- 30.Fields B. A., Goldbaum F. A., Dall'Acqua W., Malchiodi E. L., Cauerhff A., Schwarz F. P., Ysern X., Poljak R. J., Mariuzza R. A. (1996) Biochemistry 35, 15494–15503 [DOI] [PubMed] [Google Scholar]
- 31.James L. C., Tawfik D. S. (2003) Protein Sci. 12, 2183–2193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Torigoe H., Nakayama T., Imazato M., Shimada I., Arata Y., Sarai A. (1995) J. Biol. Chem. 270, 22218–22222 [DOI] [PubMed] [Google Scholar]
- 33.Connelly P. R., Aldape R. A., Bruzzese F. J., Chambers S. P., Fitzgibbon M. J., Fleming M. A., Itoh S., Livingston D. J., Navia M. A., Thomson J. A., Wilson K. P. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 1964–1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhat T. N., Bentley G. A., Boulot G., Greene M. I., Tello D., Dall'Acqua W., Souchon H., Schwarz F. P., Mariuzza R. A., Poljak R. J. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 1089–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Webster D. M., Henry A. H., Rees A. R. (1994) Curr. Opin. Struct. Biol. 4, 123–129 [Google Scholar]
- 36.Davies D. R., Cohen G. H. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 7–128552677 [Google Scholar]
- 37.Colman P. M. (1988) Adv. Immunol. 43, 99–132 [DOI] [PubMed] [Google Scholar]
- 38.Braden B. C., Poljak R. J. (1995) FASEB J. 9, 9–16 [DOI] [PubMed] [Google Scholar]
- 39.Padlan E. A. (1996) Adv. Protein Chem. 49, 57–133 [DOI] [PubMed] [Google Scholar]
- 40.Brummell D. A., Sharma V. P., Anand N. N., Bilous D., Dubuc G., Michniewicz J., MacKenzie C. R., Sadowska J., Sigurskjold B. W., Sinnott B., Young N. M., Bundle D. R., Narang S. A. (1993) Biochemistry 32, 1180–1187 [DOI] [PubMed] [Google Scholar]
- 41.Sundberg E. J., Urrutia M., Braden B. C., Isern J., Tsuchiya D., Fields B. A., Malchiodi E. L., Tormo J., Schwarz F. P., Mariuzza R. A. (2000) Biochemistry 39, 15375–15387 [DOI] [PubMed] [Google Scholar]
- 42.Tsumoto K., Ueda Y., Maenaka K., Watanabe K., Ogasahara K., Yutani K., Kumagai I. (1994) J. Biol. Chem. 269, 28777–28782 [PubMed] [Google Scholar]
- 43.Tsumoto K., Ogasahara K., Ueda Y., Watanabe K., Yutani K., Kumagai I. (1995) J. Biol. Chem. 270, 18551–18557 [DOI] [PubMed] [Google Scholar]
- 44.Tsumoto K., Ogasahara K., Ueda Y., Watanabe K., Yutani K., Kumagai I. (1996) J. Biol. Chem. 271, 32612–32616 [DOI] [PubMed] [Google Scholar]
- 45.Kondo H., Shiroishi M., Matsushima M., Tsumoto K., Kumagai I. (1999) J. Biol. Chem. 274, 27623–27631 [DOI] [PubMed] [Google Scholar]
- 46.Nishimiya Y., Tsumoto K., Shiroishi M., Yutani K., Kumagai I. (2000) J. Biol. Chem. 275, 12813–12820 [DOI] [PubMed] [Google Scholar]
- 47.Shiroishi M., Yokota A., Tsumoto K., Kondo H., Nishimiya Y., Horii K., Matsushima M., Ogasahara K., Yutani K., Kumagai I. (2001) J. Biol. Chem. 276, 23042–23050 [DOI] [PubMed] [Google Scholar]
- 48.Yokota A., Tsumoto K., Shiroishi M., Kondo H., Kumagai I. (2003) J. Biol. Chem. 278, 5410–5418 [DOI] [PubMed] [Google Scholar]
- 49.Pons J., Rajpal A., Kirsch J. F. (1999) Protein Sci. 8, 958–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rajpal A., Taylor M. G., Kirsch J. F. (1998) Protein Sci. 7, 1868–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ueda Y., Tsumoto K., Watanabe K., Kumagai I. (1993) Gene 129, 129–134 [DOI] [PubMed] [Google Scholar]
- 52.Tsumoto K., Nakaoki Y., Ueda Y., Ogasahara K., Yutani K., Watanabe K., Kumagai I. (1994) Biochem. Biophys. Res. Commun. 201, 546–551 [DOI] [PubMed] [Google Scholar]
- 53.Merk H., Stiege W., Tsumoto K., Kumagai I., Erdmann V. A. (1999) J. Biochem. 125, 328–333 [DOI] [PubMed] [Google Scholar]
- 54.Ueda H., Tsumoto K., Kubota K., Suzuki E., Nagamune T., Nishimura H., Schueler P. A., Winter G., Kumagai I., Mohoney W. C. (1996) Nat. Biotechnol. 14, 1714–1718 [DOI] [PubMed] [Google Scholar]
- 55.Shiroishi M., Tsumoto K., Tanaka Y., Yokota A., Nakanishi T., Kondo H., Kumagai I. (2007) J. Biol. Chem. 282, 6783–6791 [DOI] [PubMed] [Google Scholar]
- 56.Kunkel T. A., Roberts J. D., Zakour R. A. (1987) Methods Enzymol. 154, 367–382 [DOI] [PubMed] [Google Scholar]
- 57.Laemmli U. K. (1970) Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 58.Imoto T., Johnson T. M., North A. C., Phillips D. C., Rupley J. A. (1972) in The Enzymes (Boyer P. D. ed) Vol. 7, 3rd Ed., pp. 665–868, Academic Press, New York [Google Scholar]
- 59.Leslie A. G. (1992) Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography, No. 26, Warrington, UK [Google Scholar]
- 60.Evans P. R. (1993) Proceedings of CCP4 Study Weekend on Data Collection and Processing, pp. 114–122, Warrington, UK [Google Scholar]
- 61.French S., Wilson K. (1978) Acta Crystallogr. Sect. A 34, 517–525 [Google Scholar]
- 62.Collaborative Computational Project, No 4 (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–76315299374 [Google Scholar]
- 63.Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 64.Jones T. A., Zou J. Y., Cowan S. W., Kjeldgaard M. (1991) Acta. Crystallogr. A 47, 110–119 [DOI] [PubMed] [Google Scholar]
- 65.Kabsch W. (1976) Acta Crystallogr. Sect. A 32, 922–923 [Google Scholar]
- 66.Kauzmann W. (1959) Adv. Protein Chem. 14, 1–63 [DOI] [PubMed] [Google Scholar]
- 67.Livingstone J. R., Spolar R. S., Record M. T., Jr. (1991) Biochemistry 30, 4237–4244 [DOI] [PubMed] [Google Scholar]
- 68.Spolar R. S., Record M. T., Jr. (1994) Science 263, 777–784 [DOI] [PubMed] [Google Scholar]
- 69.Chothia C. (1974) Nature 248, 338–339 [DOI] [PubMed] [Google Scholar]
- 70.Ladbury J. E., Wright J. G., Sturtevant J. M., Sigler P. B. (1994) J. Mol. Biol. 238, 669–681 [DOI] [PubMed] [Google Scholar]
- 71.Myszka D. G., Sweet R. W., Hensley P., Brigham-Burke M., Kwong P. D., Hendrickson W. A., Wyatt R., Sodroski J., Doyle M. L. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 9026–9031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pomès R., Willson R. C., McCammon J. A. (1995) Protein Eng. 8, 663–675 [DOI] [PubMed] [Google Scholar]
- 73.Willcox B. E., Gao G. F., Wyer J. R., Ladbury J. E., Bell J. I., Jakobsen B. K., van der Merwe P. A. (1999) Immunity 10, 357–365 [DOI] [PubMed] [Google Scholar]
- 74.Murphy K. P., Freire E., Paterson Y. (1995) Proteins 21, 83–90 [DOI] [PubMed] [Google Scholar]
- 75.Clackson T., Wells J. A. (1995) Science 267, 383–386 [DOI] [PubMed] [Google Scholar]
- 76.Fleury D., Wharton S. A., Skehel J. J., Knossow M., Bizebard T. (1998) Nat. Struct. Biol. 5, 119–123 [DOI] [PubMed] [Google Scholar]
- 77.Goldman E. R., Dall'Acqua W., Braden B. C., Mariuzza R. A. (1997) Biochemistry 36, 49–56 [DOI] [PubMed] [Google Scholar]
- 78.Aitio O., Hellman M., Kesti T., Kleino I., Samuilova O., Pääkkönen K., Tossavainen H., Saksela K., Permi P. (2008) J. Mol. Biol. 382, 167–178 [DOI] [PubMed] [Google Scholar]
- 79.Wibbenmeyer J. A., Schuck P., Smith-Gill S. J., Willson R. C. (1999) J. Biol. Chem. 274, 26838–26842 [DOI] [PubMed] [Google Scholar]
- 80.Bogan A. A., Thorn K. S. (1998) J. Mol. Biol. 280, 1–9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.