

Abstract
Human cytomegalovirus (HCMV) gpUL18 is a HLA class I (HLA-I) homologue with high affinity for the inhibitory receptor LIR-1/ILT2. The previously described 67 kDa form of gpUL18 is shown here to be sensitive to endoglycosidase-H (EndoH). A novel form of gpUL18 with a molecular mass of ~160 kDa and resistance to EndoH was identified in cells infected with HCMV strain AD169 or the low passage HCMV isolates Merlin and Toledo. The 67 kDa EndoH-sensitive gpUL18 glycoform was detected earlier in a productive infection (from 24 h post-infection) than the slower-migrating EndoH-resistant glycoform (from 72 h post-infection). Deletion of the US2–US11 region from the HCMV genome was associated with a substantial up-regulation of endogenous HLA-I in infected cells, but had no obvious effect on the gpUL18 expression pattern. Vaccinia virus and adenovirus vectors were used to further analyse gpUL18 expression. Depending on the delivery vector system, differences in the electrophoretic motility of the EndoH-resistant >105 kDa form of gpUL18, but not the EndoH-sensitive 67 kDa form, were observed; post-translational modification of the higher molecular mass glycoform appears to be influenced by active virus infection and vector delivery. The EndoH-sensitive 67 kDa gpUL18 had a rapid turnover, while the maturation to the EndoH-resistant >105 kDa form was relatively slow and inefficient. However, synthesis of the EndoH-resistant >105 kDa form was enhanced with elevated levels of β2-microglobulin. When expressed by using an adenovirus vector, both the EndoH-sensitive 67 kDa and the EndoH-resistant >105 kDa gpUL18 forms could be detected on the cell surface.
INTRODUCTION
Human cytomegalovirus (HCMV) is a β-herpesvirus with the largest genome (~236 kb) of any characterized human virus. Although its genetic content is not fully characterized, a substantial proportion has been shown to, or is predicted to, encode functions capable of modulating the human immune system. HCMV encodes four genes (US2, US3, US6 and US11) that down-regulate cell surface HLA class I (HLA-I) expression (Tortorella et al., 2000), three genes that modulate natural killer (NK) cell functions (UL16, UL40, UL141) (Cosman et al., 2001; Tomasec et al., 2000, 2005), an interleukin 10 (IL10) homologue (UL111A) (Kotenko et al., 2000), chemokine homologues (UL146, UL147) (Penfold et al., 1999), potential (UL33, UL78, US27) (Chee et al., 1990) or recognized (US28) chemokine receptors (Neote et al., 1993), inhibitors of interferon (TRS1, IRS1, UL83) (Child et al., 2004), inhibitors of apoptosis (UL36, UL37, IE1, IE2) (Zhu et al., 1995) and a tumour necrosis factor receptor homologue (UL144) (Benedict et al., 1999). HCMV is ubiquitous worldwide, with the vast majority of primary infections being subclinical and followed by lifelong asymptomatic carriage of the virus. Thus, even though HCMV encodes an impressive array of immunomodulatory functions, infections are normally efficiently controlled by immune surveillance. Clinical disease is found primarily in individuals whose immune systems have not fully developed (congenital transmission resulting in cytomegalic inclusion disease) or are severely compromised, most notably transplant recipients and AIDS patients. Although the different arms of the immune system act in concert to control infection, individuals with genetic defects affecting NK cell function are particularly susceptible to HCMV disease (Arase et al., 2002; Biron et al., 1989; Gazit et al., 2004). In vivo pathogenesis studies with murine CMV indicate that not only is the NK cell response crucial to controlling disease but also the virus-encoded genes that modulate NK cell functions are important virulence determinants (Abenes et al., 2004; Arase et al., 2002; Farrell et al., 1997).
UL18 was identified as a HLA-I homologue during the analysis of the strain AD169 sequence (Beck & Barrell, 1988) and, like HLA-I, was found to form a trimeric complex with β2-microglobulin (β2m) and endogenous peptides (Browne et al., 1990; Fahnestock et al., 1995). The leukocyte immunoglobulin-like receptor-1 (LIR-1, also referred to as ILT2) was initially identified by its interaction with gpUL18 (Cosman et al., 1997), and was then found to be an inhibitory receptor that recognized an exceptionally broad range of classical (HLA-A, HLA-B, HLA-C) and non-classical (HLA-E, HLA-F, HLA-G) HLA-I molecules (Chapman et al., 1999; Lepin et al., 2000; Shiroishi et al., 2003). LIR-1 has >1000-fold higher affinity for gpUL18 than for HLA-I molecules, thus even low levels of gpUL18 could be expected to compete efficiently for binding (Chapman et al., 1999). The prediction that gpUL18 would suppress NK cell recognition has been both supported (Kim et al., 2004; Reyburn et al., 1997) and questioned (Leong et al., 1998; Odeberg et al., 2002) by experimental data in published studies. LIR-1 expression is restricted to myeloid cells, B cells, and subpopulations of T and NK cells (Young et al., 2001). Interestingly, the proportion of both NK and T cells expressing LIR-1 increases in frequency in lung-transplant recipients with HCMV disease (Berg et al., 2003). On CD8+ T lymphocytes, LIR-1 is detected preferentially on highly differentiated activated and memory T cells (Young et al., 2001). Saverino et al. (2004) have reported that UL18 expression stimulated non-HLA-restricted killing by CD8+ cytotoxic T lymphocytes, via an interaction with LIR-1, although the mechanism responsible for enhanced recognition and its role in HCMV infection have yet to be elucidated.
UL18 is non-essential for HCMV replication in vitro (Browne et al., 1992), and is transcribed with late-phase kinetics in human fibroblasts infected with HCMV (Park et al., 2002). Analysis of gpUL18 function has been hampered by the apparent inability to detect gpUL18 in HCMV-infected cells, and a failure to generate stable cell lines expressing the UL18 open reading frame (ORF). When continuous cell lines stably transfected with US2, US3, US6 or US11 were infected with vaccinia virus UL18 vector, gpUL18 was expressed efficiently and was detected as a 67 kDa glycoprotein (Park et al., 2002). In this study, we have further characterized gpUL18 in the context of productive HCMV infection, and also when expressed using vaccinia virus (v-UL18) and replication-deficient adenovirus (RAdUL18) vectors. The previously reported 67 kDa form of gpUL18 is shown here to be sensitive to digestion with endoglycosidase-H (EndoH), whereas a novel, EndoH-resistant form of gpUL18 migrating with an apparent molecular mass in excess of 105 kDa is described.
METHODS
Cells and viruses
Human fetal foreskin fibroblasts (HFFFs) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % fetal calf serum (FCS) and antibiotics (Invitrogen). Transporter associated with antigen processing (TAP)-deficient (NP) fibroblasts were derived from a skin biopsy of a patient with Bare Lymphocyte Syndrome, a genetic disease associated with severe down-regulation of surface MHC-I expression. NP cells, kindly provided by V. Cerundolo (University of Oxford, Oxford, UK), have a defect in TAP-1 expression (Tomasec et al., 2000). Unless otherwise indicated, cells were infected with HCMV (m.o.i. of 10) 96 h prior to analysis. HCMV infections were performed using strains AD169, Toledo, Merlin, the AD169 UL18 insertional mutant (△UL18) (Browne et al., 1992) or the AD169 US2–US11 deletion mutant (RV798) (Jones et al., 1995). The vaccinia virus vector encoding UL18 (v-UL18) has been described previously (Browne et al., 1990). The UL18 ORF was amplified by PCR and inserted into a replication-deficient human adenovirus-5 vector (RAdUL18) under the control of the HCMV major IE promoter, as described by McGrory et al. (1988) and Wilkinson & Akrigg (1992). Unless otherwise indicated, HFFFs were infected with RAdUL18 or the same vector without an insert (RAdCtrl) at an m.o.i. of 100 for 72 h prior to analysis.
Immunofluorescence and flow cytometry
Cells on glass cover-slips were washed in PBS and fixed with 2 % paraformaldehyde for 15 min at room temperature. The cells were washed in PBS and then, to ensure sustained permeabilization of cells, all subsequent treatments were done in PBS containing 3 % BSA, 0·1 % saponin. Next, the cells were incubated with the gpUL18 antibody M71 (2 μgml−1) for 1 h at room temperature, washed and stained with fluorescein isothiocyanate (FITC)-conjugated anti-mouse immunoglobulin G (IgG) for 1 h at 37 °C. The cells, still on the coverslips, were washed and mounted on slides with 2 % DABCO, 10 % (v/v) glycerol, PBS (Sigma). Fluorescence was visualized using a Leica DM IRBE microscope with a Hamamatsu ORCA-ER camera and Improvision Openlab software.
One-colour flow cytometry was used to detect surface gpUL18 or HLA-I expression. Cells were washed twice with cold PBS, 2 % FCS and incubated for 30 min at 4 °C with monoclonal antibodies (mAbs) specific for gpUL18 (1 : 50 dilution; 10C7/CRL-2430) (Fahnestock et al., 1995) or HLA-I (1 : 100 dilution; W6/32). Normal mouse IgG was used as a negative control. The cells were washed twice with cold PBS, 2 % FCS and then labelled with FITC-conjugated anti-mouse Fab fragments (Sigma) on ice for 30 min. Cells were washed twice with cold PBS, and fixed with 2 % paraformaldehyde. A total of 10 000 events were collected by the FACSCalibur cytometer and analysed with CellQuest Pro software (BD Biosciences).
Immunoblotting
To prepare extracts, cells were first washed with PBS, before being resuspended in NP-40 lysis buffer (1 % NP-40, 150 mM NaCl, 5 mM EDTA, 50 mM Tris pH 8) supplemented with P8340 protease inhibitor cocktail (Sigma). Cells were lysed during an incubation for 30 min, on ice, and clarified by centrifugation for 30 min at 13 000 r.p.m. in a Heraeus Biofuge Fresco at 4 °C. For glycosidase treatments, samples were adjusted to 0·05 % SDS, 0·1% β-mercaptoethanol, and protein denatured by heating at 100 °C for 10 min, before being digested with either 1000 U Endo H in 5 mM sodium citrate (pH 5·5) or 500 U peptide: N-glycosidase F (PNGase F) in 5 mM sodium phosphate (pH 7) for 12 h according to the manufacturer’s instructions (NEB). Enzyme was omitted from mock-digested samples. All samples were boiled after the addition of SDS-PAGE loading buffer, and proteins separated by SDS-PAGE using a Mini Protean II gel apparatus (Bio-Rad). Following electrophoresis, protein was transferred to nitrocellulose membrane (Amersham Hybond-C), by wet transfer at 100 V for 1 h using a Bio-Rad mini trans-blot system. The membrane was washed with Tris-buffered saline (TBS), blocked for 16 h at 4 °C in 5 % BSA, TBST, and for a further 8 h at 4 °C in Superblock (Pierce), before being incubated with mAbs specific for gpUL18 (M71; 2·5 μgml−1), HLA-I (HC10; 1 : 200 dilution) or HLA-E (1 : 200 dilution; Serotec) for 16 h in 10 % (v/v) Superblock, TBST. The membrane was washed in TBST, 5 % dried milk, then again in TBST, before being incubated with goat anti-mouse horseradish peroxidase (HRP) antibody (Bio-Rad) (1 : 1000 dilution) in 5 % dried milk, TBST for 1 h at room temperature. Supersignal substrate (Pierce) was used to detect the HRP signal and the membrane exposed to Kodak BioMax MR film.
Immunoprecipitation
HFFFs were infected with v-UL18 or v-Ctrl (m.o.i. of 10) for 6 h, then cultured in methionine-free medium for 30 min. Cells were labelled with 50 μCi (1·85 MBq) [35S]methionine ml−1 (ICN) for 4 h prior to immunoprecipitation, or pulse-labelled with [35S]methionine for 15 min, then chased for the times indicated. When harvested, cells were resuspended in NP-40 lysis buffer (1 % NP-40, 1 % protease inhibitor (Sigma), 150 mM NaCl, 50 mM Tris, pH 8) for 30 min at 4 °C, precleared by adding 100 μl protein G agarose (Pierce) for 45 min at 4 °C and centrifuged at 13 000 r.p.m. in a Heraeus Biofuge Fresco for 30 min at 4 °C. Supernatants were then incubated with 10 μg M71 mAb overnight at 4 °C, then with 50 μl protein G agarose for 45 min at 4 °C. The protein G agarose was then washed, and the complexed proteins released by resuspending the beads in SDS denaturing buffer (NEB) and heating to 100 °C. Where indicated, precipitated proteins were digested with EndoH or PNGaseF, and then subjected to SDS-PAGE and fluorography.
Biotinylation of cell surface proteins
Cells were washed in cold PBS then incubated according to the manufacturer’s instructions for 45 min at 4 °C with 0·5 mg sulfo-NHS-biotin ml−1 (Pierce). Cells were washed and lysed in NP-40 buffer, and the lysates cleared by centrifugation. Biotinylated proteins were captured with streptactin-sepharose (IBA), and washed with NP-40 and RIPA (150 mM NaCl, 50 mM Tris pH8, 0·5 % deoxycholate, 0·1 % SDS, 1 % NP-40) buffers. Bound proteins were eluted in SDS denaturing buffer and analysed by immunoblotting.
RESULTS
Identification of a highly glycosylated form of gpUL18
The first analysis of the UL18 ORF using the vaccinia virus recombinant v-UL18 demonstrated that the gene encoded a 67 kDa glycoprotein that formed a complex with β2m (Browne et al., 1990). In subsequent studies using v-UL18, an additional slower-migrating protein with an apparent molecular mass >105 kDa was detected by immunoprecipitation with a gpUL18-specific mAb (Fig. 1). This gpUL18 species was consistently observed as a diffuse band following denaturing SDS-PAGE, under reducing conditions. To analyse the rate of gpUL18 synthesis and maturation, v-UL18-infected cells were pulsed with [35S]methionine, and labelled gpUL18 was then followed over time. Maximum levels of the 67 kDa gpUL18 were detected immediately following the labelling, with levels declining over time, indicating that this species had a relatively short half-life. In contrast, the >105 kDa gpUL18 form was not apparent until 6 h after labelling, consistent with its probable synthesis from the 67 kDa precursor and having an exceptionally slow rate of maturation (Fig. 2a). Endogenous HLA-I molecules were immunoprecipitated in parallel, demonstrating that the stability and the kinetics of HLA-I maturation were normal, and thus under these conditions not affected by the vaccinia virus vector (Figs 2a, b, respectively). Significantly, the 67 kDa gpUL18 was susceptible to EndoH digestion, whereas the >105 kDa form was resistant, consistent with it being further post-translationally processed in the Golgi apparatus from the 67 kDa precursor. Levels of HLA-I heavy chain remained constant over the duration of the time course, while gpUL18 levels decreased markedly. The endogenous HLA-I was thus more stable than gpUL18, while both the rate and efficiency with which it was converted to an EndoH-resistant form exceeded that observed for gpUL18. Like HLA-I, gpUL18 forms a trimeric complex with β2m and peptide. Cell surface gpUL18 levels were enhanced by co-expressing β2m from a second vaccinia virus recombinant (Browne et al., 1990). By its association with newly synthesized gpUL18, β2m may have acted to protect it from degradation and promote its maturation. In line with this model, we observed that co-infection of v-UL18 and v-β2m recombinant viruses enhanced the relative levels of the EndoH-resistant >105 kDa form of gpUL18 as determined by both immunoblotting (Fig. 3a) and immunoprecipitation (Fig. 3b) assays.
Fig. 1.

Immunoblot showing expression of gpUL18 using a vaccinia virus vector (v-UL18). HFFFs were infected with v-Ctrl or v-UL18, or mock infected. At 4 h p.i. the cells were labelled with [35S]methionine for 6 h. Cell lysates were divided into three equal parts and immunoprecipitated with mouse IgG (mIg), the gpUL18 specific mAb M71 (M71) or without primary antibody (–). Both a fast- (single arrowhead) and slow-migrating (double arrowhead) form of gpUL18 were apparent.
Fig. 2.

Immunoblot showing a time course of the expression of gpUL18 using a vaccinia virus construct (v-UL18). (a) HFFFs were infected with v-Ctrl or v-UL18. At 4 h p.i. the cells were starved in methionine-free medium, pulse-labelled for 15 min with [35S]methionine and chased with cold methionine for the indicated times. Immunoprecipitations were performed using anti-UL18 mAb M71 or anti-HLA-I mAb W6/32. (b) One half of each sample was also treated with EndoH.
Fig. 3.

Expression of the >105 kDa gpUL18 form was enhanced by β2m. (a) Immunoblot of cell lysates prepared from HFFFs infected with v-UL18, or co-infected with v-UL18 and v-β2m as indicated. Cell lysates were divided into three equal parts and either left mock treated (–), or digested with EndoH (+E) or PNGaseF (+P). gpUL18 was detected using the mAb M71. (b) HFFFs were infected with v-UL18 or v-Ctrl alone, or co-infected with v-UL18 and v-β2m as indicated. Immunoprecipitation was performed using the mAb M71, one half of each sample was then treated with EndoH.
UL18 expression from an adenovirus vector
Using the v-UL18 vector, transgene expression took place against a background of a productive vaccinia-virus infection. To express gpUL18 efficiently yet free of lytic virus infection, the UL18 gene was inserted into a replication-deficient human adenovirus-5 vector, designated RAdUL18. Following infection of fibroblasts with RAdUL18, gpUL18 immunofluorescence exhibited a predominantly intracellular staining that was excluded from the nucleus (Fig. 4a, b). In immunoblotting experiments, again, not only the 67 kDa gpUL18 species could be detected, but also a slower migrating form with an apparent molecular mass of ~110 kDa (Fig. 4C) could be detected. The 67 kDa protein was again found to be EndoH sensitive, and the higher molecular mass form EndoH resistant (Fig. 4c).
Fig. 4.

UL18 expression using a recombinant adenoviral vector (RAdUL18). Immunofluorescence of human fibroblasts infected with RAdUL18 when labelled with (a) control murine polyclonal antibody or (b) the gpUL18-specific mAb M71. (c) Immunoblotting experiment using extracts of HFFFs infected with either RAdCtrl or RAdUL18. Extracts were divided into three equal parts and either left mock treated (–), or digested with EndoH (+E) or PNGaseF (+P). gpUL18 was detected using the mAb M71.
In order to function effectively as a HLA-I mimic capable of interacting directly with its receptors on lymphoid cells, gpUL18 has to be present at the cell surface. To investigate this aspect, HFFFs were infected with RAdUL18, and non-permeabilized cells analysed by flow cytometry. The fluorescence observed with RAdUL18-infected cells using the gpUL18-specific mAb 10C7 was significantly higher than for cells infected with an equivalent vector without an insert (RAdCtrl) or when a control IgG was used (Fig. 5a). While gpUL18 clearly reached the surface of RAdUL18-infected cells, flow cytometry was unable to differentiate between the two forms of gpUL18 (Fig. 5b). We hypothesized that the ~110 kDa species corresponded to a mature EndoH-resistant form of the protein, which would be preferentially expressed on the cell surface. Cell surface proteins on RAdUL18-infected cells were labelled with biotin and purified with streptactin beads. HSP40, being an intracellular protein associated with the endoplasmic reticulum, was used as a negative control for this assay. HSP40 was not detected in the biotinylated fraction of cell surface proteins, although it was present in cell extracts (Fig. 5c). Both the 67 kDa and ~110 kDa forms of gpUL18 were clearly biotinylated, and thus both appear to be present on the surface of RAdUL18-infected cells (Fig. 5d).
Fig. 5.

gpUL18 expression on the cell surface. (a) HFFFs were infected with RAdUL18 or RAdCtrl, and at 72 h p.i. stained using the gpUL18 mAb 10C7. The flow cytometry histogram shows expression of gpUL18 on the surface of RAdUL18 infected cells. Mouse IgG was used as a negative control. (b) A proportion of cells was also analysed by immunoblotting, using the mAb M71, showing the expression of gpUL18. Cell surface proteins of RAdUL18- or RadCtrl-infected fibroblasts were biotinylated and purified with streptactin-sepharose; total cell extracts were prepared simultaneously. Extracts were analysed by immunoblotting using (c) anti-HSP40 antibody or (d) the gpUL18-specific mAb M71. (e) Histogram showing expression of gpUL18; NP human fibroblasts were analysed exactly as in (a). (f) Immunoblotting analysis showing expression of gpUL18; NP cells were analysed exactly as in (b).
During productive HCMV infection, US6 interferes with the TAP. As peptide binding is required for maturation of endogenous HLA-I molecules, US6 acts to down-regulate cell surface expression of HLA-I. To test whether TAP-dependent peptide translocation was required for efficient gpUL18 expression, NP fibroblasts were also infected with RAdUL18 and analysed in a FACS experiment. NP fibroblasts have a genetic defect in TAP-1 function. gpUL18 was expressed efficiently on the cell surface in the absence of TAP-1 (Fig. 5e), albeit at a lower level than that observed in HFFFs when analysed in parallel (Fig. 5a). Significantly, both glycoforms of gpUL18 were detected in NP cells (Fig. 5f). TAP function, therefore, does not appear to be essential for normal expression of gpUL18.
gpUL18 expression during an HCMV infection
Analysis of UL18 during a productive HCMV infection is problematical due to a combination of extremely low gpUL18 levels and confounding antibody interactions with virus-encoded Fc receptors. Nevertheless, human fibroblasts infected with HCMV-AD169 or the AD169△UL18 mutant (△UL18) for 96 h (m.o.i. of 10) were analysed by immunoblotting. gpUL18 levels were much lower in HCMV-infected cells than those observed with v-UL18- or RAdUL18-infected cells; however, both gpUL18 species, 67 kDa and ~160 kDa, were evident (Fig. 6a). To generate AD169△UL18, the Escherichia coli lacZ was inserted within the UL18-encoding gene (Browne et al., 1992). β-Galactosidase was expressed at such high levels from the HCMV β2·7 early promoter (Spaete & Mocarski, 1987) that it was detected by a non-specific antibody interaction (Fig. 6a).
Fig. 6.
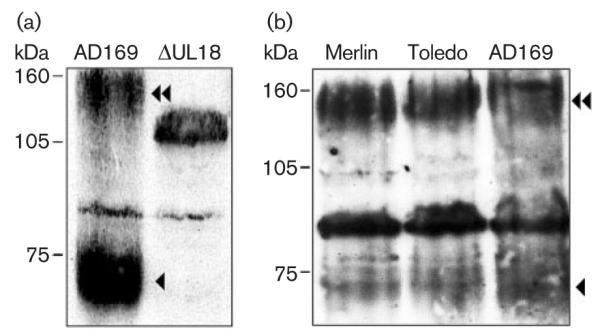
gpUL18 expression in HCMV-infected cells. (a) Immunoblot of HFFFs infected with HCMV AD169 or △UL18 at 96 h p.i., using mAb M71. (b) Immunoblot of HFFFs infected with HCMV AD169, Merlin or Toledo at 96 h p.i., using mAb M71.
It proved problematical to determine a precise molecular mass for the EndoH-resistant glycoform of gpUL18, as it was observed as a diffuse band on immunoblots, and its migration effectively depended on the virus vector used to infect cells. However, the mass of gpUL18 expressed during productive HCMV infection was by far the greatest migrating close to the 160 kDa molecular mass marker, while the mass of gpUL18 expressed from RAdUL18 vector was the smallest and close to the 105 kDa marker (see Supplementary Fig. S1 available in JGV online). Vaccinia virus and HCMV infections are both recognized to exert gross effects on the host cells metabolism, and both may potentially influence the cellular glycosylation machinery.
HCMV strain AD169 has undergone genetic changes during passage in vitro (Akter et al., 2003); however, the UL18 amino acid sequence exhibits a high level of conservation (95·1–100 % identity) between strains, with there being no sequence-predicted defect associated with strain AD169 to account for its low level of expression (see Supplementary Figs S2 and S3 available in JGV online). When assayed directly, levels of gpUL18 expression, gpUL18 migration on gels and the existence of dual forms of the protein were similar in cells infected with HCMV AD169, Toledo or Merlin (Fig. 6b). When expressed from either strain AD169 or Toledo, the 67 kDa gpUL18 was again susceptible to EndoH digestion, whereas the ~160 kDa form was not affected by EndoH but was susceptible to PNGaseF treatment (not shown). Thus, the highly glycosylated, EndoH-resistant gpUL18 glycoform was typical, and not restricted to the laboratory strain AD169. The 67 kDa form of gpUL18 was detected at 24 h post-infection (p.i.) and increased in abundance to reach a peak by 72 h p.i. The levels were then sustained through the late phase of infection. The ~160 kDa form of gpUL18 appeared at 48 h p.i. and also reached peak levels by 72 h p.i (Fig. 7a). gpUL18 was thus expressed most efficiently at a time when endogenous HLA-I had been effectively eliminated. At no point during this time-course assay were we able to detect gpUL18 on the cell surface of HCMV-infected cells (Fig. 7b, Supplementary Fig. S4 available in JGV online). A major effort was taken to optimize staining conditions to enable detection of gpUL18 on the surface of cells undergoing lytic HCMV infection, as this was considered central to the investigation of its function. A small increase in staining with the UL18-specific mAb relative to control IgG was detected; however, a similar effect was observed when using cells infected with the △UL18 mutant, and was thus consistent with an enhanced level of non-specific fluorescence associated with HCMV infection (Fig. 7b).
Fig. 7.
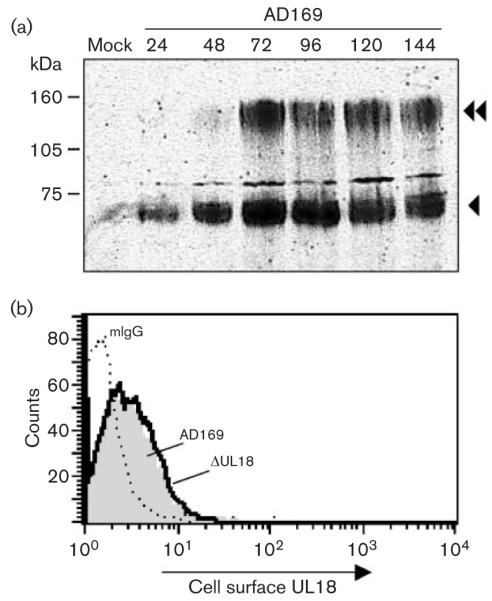
gpUL18 expression in HCMV-infected cells. (a) HFFFs were mock infected or infected with HCMV AD169, and extracts were prepared at the times (h p.i.) indicated. Immunoblot using mAb M71 showing the time course of gpUL18 expression during productive infection. (b) At no time after infection, was it possible to detect gpUL18 on the surface of HCMV-infected cells. The histogram shows the 96 h time point and illustrates the non-specific staining of mAb 10C7 to the infected cells. Grey shading, HCMV AD169.
Given the low levels of gpUL18 expression during lytic HCMV infection, it still remained possible that one or more of the HLA-I down-regulatory genes (US2, US3, US6 or US11) were suppressing, but not eliminating gpUL18 expression. To address this issue directly in the context of HCMV infection, HFFFs were infected with HCMV AD169, △UL18 or the AD169 mutant RV798 deleted in sequences encoding US2–US11, and then analysed 96 h p.i. by immunoblotting (Fig. 8). At this late phase of infection, similar levels of the two gpUL18 forms were consistently detected in cells infected with strain AD169 or the strain AD169 US2–US11 deletion mutant (Fig. 8a). This was at a time when infection with AD169 had efficiently down-regulated HLA-I expression, while the infection with HCMV RV798 markedly stimulated HLA-I expression both at the cell surface (Fig. 8b) and overall (Fig. 8c), relative to mock-infected controls. While US2–US11 genes efficiently down-regulated HLA-I, they did not appear to affect gpUL18 expression during the late phase of a productive infection in fibroblasts. HLA-E has been reported to be resistant to US2 and US11, when co-expressed in isolation (Llano et al., 2003). In agreement with these observations, total cellular levels of the non-classical HLA-E increased during HCMV strain AD169 infection, in contrast to classical HLA-I, but not during infection with HCMV RV798 (Fig. 8d).
Fig. 8.

The effect of HCMV US2, US3, US6 and US11 on gpUL18 and HLA-I expression. HFFFs were infected with HCMV AD169, △UL18 or RV798 (△US2–US11). (a) Immunoblot using mAb M71 demonstrating that US2–US11 expression during productive HCMV infection had no apparent effect upon gpUL18 expression. (b) Cell surface staining, using mAbs W632, and (c) immunoblot assays, using mAb mAbs W632, showing US2–US11 expression during productive HCMV infection had a significant effect upon cell surface HLA-I. (d) Immunoblot, using the mAb MEM-E/02, showing HLA-E synthesis was significantly increased following infection with HCMV AD169, but not the US2–US11 mutant RV798.
DISCUSSION
The study of UL18 function has been frustrated by issues both of expression and detection. UL18 constitutive expression is not well-tolerated in long-term stable cell lines (Davis et al., 1997; Kim et al., 2004; Reyburn et al., 1997) and, to our knowledge, there has previously been no definitive demonstration of gpUL18 expression during HCMV infection. The UL18 ORF encodes 13 consensus N-linked glycosylation sites, whereas classical HLA-I molecules typically have a single functional site. When treated with PNGaseF, the deglycosylated UL18 protein exhibited an apparent molecular mass of 35 kDa as estimated by SDS-PAGE, compared with 41·7 kDa predicted from the sequence. Glycosylation increased the molecular mass to 67 kDa for the EndoH-sensitive protein, which further increased to >105 kDa for the EndoH-resistant, PNGaseF-sensitive form, size being dependent on the expression system. A similar pattern of gpUL18 expression was observed in cells infected with a range of low and high passage HCMV strains, with the EndoH-resistant glycoform consistently estimated to have a molecular mass of ~160 kDa, a result consistent with the UL18 gene coding sequence being relatively conserved (Dolan et al., 2004; Valés-Gómez et al., 2005; Supplementary Figs S2 and S3 available in JGV online). The majority of the apparent mass of the fully glycosylated gpUL18, therefore, appears to be derived from carbohydrates added during post-translational modifications.
During productive HCMV infection, we were only able to demonstrate expression of gpUL18 by immunoblotting, at the detection limit of the assay. By flow cytometry, gpUL18 could not be detected on the cell surface. Higher levels of gpUL18 were obtained by the use of other viral vectors, which then resulted in detection of gpUL18 on the cell surface, suggesting the constraints of expression could at least be partially overcome by enhanced levels of transcription. The synthesis and processing of the fully glycosylated gpUL18 appeared to be exceptionally slow and inefficient when compared to endogenous HLA-I. A requirement for limiting chaperone functions to stabilize the UL18 polypeptide undergoing glycosylation and maturation within the ER may have contributed towards its inefficient expression. Elevating the levels of β2m has previously been shown to promote cell surface expression of β2m and gpUL18 (Browne et al., 1990), and we have shown here that the availability of β2m was a factor affecting the rate of synthesis of the >105 kDa form of gpUL18. In contrast to HLA-I, gpUL18 was expressed well in both TAP-1 positive or negative fibroblasts, suggesting that TAP-dependent peptide loading may not be essential for gpUL18 maturation, or sufficient levels of gpUL18-binding peptide can be scavenged independently of TAP. In this context, it is interesting to note that gpUL18 expression had previously been shown to be susceptible to down-regulation by HSV-1 ICP47 but not HCMV US6 (Park et al., 2002), both genes being inhibitors of TAP function. Also, peptide binding is thought not to be required for the interaction with LIR-1 (Chapman et al., 1999; Willcox et al., 2003).
To act as a ligand for LIR-1 on NK or alternative target cells, gpUL18 must be present on the cell surface, and such was observed in experiments using either v-UL18 (Browne et al., 1990; Park et al., 2002) or RAdUL18-infected fibroblasts. We anticipated that the 67 kDa gpUL18, being a precursor of the >105 kDa EndoH-resistant form, would be preferentially expressed intracellularly, and the >105 kDa form would be preferentially targeted to the cell surface. However, Park et al. (2002) had previously demonstrated that the 67 kDa form was present on the surface of cells infected with v-UL18. Additionally, labelling of cell surface proteins with biotin confirmed that both forms of gpUL18 were equally well targeted to the surface of RAdUL18-infected cells. As both forms of gpUL18 could be expressed at the cell surface, they could both be involved in intercellular signalling. The extensive and differential glycosylation of gpUL18 could also result in gains of function by promoting lectin interactions. For example, high mannose forms of HIV gp120 generated by growth in PBMC, but not macrophages, promote virion binding to DC-SIGN on dendritic cells and DC-SIGNR on endothelial cells (Lin et al., 2003). Neither the 67 kDa nor the >105 kDa form of gpUL18 was found to be secreted or in purified HCMV virions (not shown). Furthermore, gpUL18 was not identified in a systematic proteomic analysis of HCMV virions (Varnum et al., 2004), consistent with it being a non-structural protein. Despite the results with viral vectors, surface expression of gpUL18 has yet to be demonstrated definitively in the context of HCMV infection. While some binding of UL18-specific mAb to the surface of HCMV-infected cells was detected, comparable binding was obtained using the △UL18 mutant in parallel. While gpUL18 may be expressed on the surface of HCMV-infected cells, its low level, weak immunogenicity and the co-expression of HCMV-encoded antibody-binding proteins (Keller et al., 1976), all hamper detection.
When using a vaccinia virus vector to express UL18, it was not down-regulated in continuous cell lines stably transfected with US2, US3, US6 or US11 genes (Park et al., 2002). However, when both UL18 and US11 were co-expressed using adenovirus vectors, UL18 expression was clearly suppressed by US11 (not shown), the higher levels of US11 possibly pushing the equilibrium towards suppression of gpUL18. It was, therefore, important to investigate the effects of US2, US3, US6 and US11 on UL18 expression more cautiously, during a productive HCMV infection. While deletion of US2–US11 in HCMV RV798 resulted in a typical up-regulation of endogenous classical HLA-I, relative to uninfected cells, it had no obvious effect on gpUL18 during the late phase of infection. Whilst US2–US11 may yet prove capable of modulating gpUL18 expression, this gene cluster clearly preferentially targets endogenous classical HLA-I in fibroblasts. Great care should clearly be taken in using HCMV US2–US11 deletion mutants in NK cell functional assays because of significant changes in both cellular classical HLA-I and non-classical HLA-E levels, uncharacteristic of conventional HCMV infections.
The migration of the EndoH-resistant gpUL18 form varied when expressed using different vector systems. The extreme level of gpUL18 glycosylation potentially makes it a sensitive indicator of cellular processes involved in modifying N-linked oligosaccharide side chains. While there is no indication that adenovirus, HCMV or vaccinia virus encode glycosyltransferases (Markine-Goriaynoff et al., 2004), HCMV infection has been shown to up-regulate transcription of multiple glycosyltransferases and the expression of the selectin ligand component of the Lewis and sialyl-Lewis antigen in endothelial cells (Cebulla et al., 2000). Furthermore, glycosylation of PVR is modified in HCMV-infected fibroblasts (Tomasec et al., 2005). We speculate that the host cell’s glycosylation systems may be modified during lytic infection with vaccinia virus and HCMV.
If gpUL18 can get to the surface it should be able to bind LIR-1 in trans and impart a suppressive signal to an NK or CD8+ effector cell. Analysing the role of UL18 in either stimulating or suppressing NK or T cell recognition is not straightforward. HCMV encodes multiple genes that have the potential to impact on the cellular immune system. NK cells are a heterogeneous population expressing a complex and variable array of activating and inhibitory receptors. By using a replication-deficient adenovirus vector to express UL18 efficiently on the cell surface of human fibroblasts it should now be possible to identify and characterize NK cell clones that are either inhibited or activated by gpUL18, and test them back against HCMV-infected cells.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
The authors are most grateful to the Amgen Corporation, Seattle, for generously providing the mAb M71, Tom Jones (Wyeth Research, Pearl River, New York) for providing the HCMV mutant RV978 and Lynne Neale (Dept Medical Microbiology, Cardiff) for low passage HCMV clinical isolates. Technical support was kindly provided by S. Llewellyn-Lacey (MRC Co-operative Tissue Culture Facility). This work was funded by a Division of Hospital Specialities PhD Studentship and the Wellcome Trust.
Footnotes
Four figures showing the mobility of highly glycosylated forms of UL18, UL18 sequence alignments and a time course of gpUL18 surface exprssion in HCMV-infected cells are available as supplementary material in JGV Online.
REFERENCES
- Abenes G, Chan K, Lee M, Haghjoo E, Zhu J, Zhou T, Zhan X, Liu F. Murine cytomegalovirus with a transposon insertional mutation at open reading frame m155 is deficient in growth and virulence in mice. J Virol. 2004;78:6891–6899. doi: 10.1128/JVI.78.13.6891-6899.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akter P, Cunningham C, McSharry BP. Two novel spliced genes in human cytomegalovirus. J Gen Virol. 2003;84:1117–1122. doi: 10.1099/vir.0.18952-0. 8 other authors. [DOI] [PubMed] [Google Scholar]
- Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science. 2002;296:1323–1326. doi: 10.1126/science.1070884. [DOI] [PubMed] [Google Scholar]
- Beck S, Barrell BG. Human cytomegalovirus encodes a glycoprotein homologous to MHC class-I antigens. Nature. 1988;331:269–272. doi: 10.1038/331269a0. [DOI] [PubMed] [Google Scholar]
- Benedict CA, Butrovich KD, Lurain NS, Corbeil J, Rooney I, Schneider P, Tschopp J, Ware CF. Cutting edge: a novel viral TNF receptor superfamily member in virulent strains of human cytomegalovirus. J Immunol. 1999;162:6967–6970. [PubMed] [Google Scholar]
- Berg L, Riise GC, Cosman D, Bergström T, Olofsson S, Kärre K, Carbone E. LIR-1 expression on lymphocytes, and cytomegalovirus disease in lung-transplant recipients. Lancet. 2003;361:1099–1101. doi: 10.1016/S0140-6736(03)12855-3. [DOI] [PubMed] [Google Scholar]
- Biron CA, Byron KS, Sullivan JL. Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med. 1989;320:1731–1735. doi: 10.1056/NEJM198906293202605. [DOI] [PubMed] [Google Scholar]
- Browne H, Smith G, Beck S, Minson T. A complex between the MHC class I homologue encoded by human cytomegalovirus and β2 microglobulin. Nature. 1990;347:770–772. doi: 10.1038/347770a0. [DOI] [PubMed] [Google Scholar]
- Browne H, Churcher M, Minson T. Construction and characterization of a human cytomegalovirus mutant with the UL18 (class I homolog) gene deleted. J Virol. 1992;66:6784–6787. doi: 10.1128/jvi.66.11.6784-6787.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebulla CM, Miller DM, Knight DA, Briggs BR, McGaughy V, Sedmak DD. Cytomegalovirus induces sialyl Lewisx and Lewisx on human endothelial cells. Transplantation. 2000;69:1202–1209. doi: 10.1097/00007890-200003270-00027. [DOI] [PubMed] [Google Scholar]
- Chapman TL, Heikeman AP, Bjorkman PJ. The inhibitory receptor LIR-1 uses a common binding interaction to recognize class I MHC molecules and the viral homolog UL18. Immunity. 1999;11:603–613. doi: 10.1016/s1074-7613(00)80135-1. [DOI] [PubMed] [Google Scholar]
- Chee MS, Bankier AT, Beck S. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr Top Microbiol Immunol. 1990;154:125–169. doi: 10.1007/978-3-642-74980-3_6. 7 other authors. [DOI] [PubMed] [Google Scholar]
- Child SJ, Hakki M, De Niro KL, Geballe AP. Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J Virol. 2004;78:197–205. doi: 10.1128/JVI.78.1.197-205.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosman D, Fanger N, Borges L, Kubin M, Chin W, Peterson L, Hsu ML. A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity. 1997;7:273–282. doi: 10.1016/s1074-7613(00)80529-4. [DOI] [PubMed] [Google Scholar]
- Cosman D, Mullberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, Kubin M, Chalupny NJ. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity. 2001;14:123–133. doi: 10.1016/s1074-7613(01)00095-4. [DOI] [PubMed] [Google Scholar]
- Davis DM, Reyburn HT, Pazmany L, Chiu I, Mandelboim O, Strominger JL. Impaired spontaneous endocytosis of HLA-G. Eur J Immunol. 1997;27:2714–2719. doi: 10.1002/eji.1830271035. [DOI] [PubMed] [Google Scholar]
- Dolan A, Cunningham C, Hector RD. Genetic content of wild type human cytomegalovirus. J Gen Virol. 2004;85:1301–1312. doi: 10.1099/vir.0.79888-0. 12 other authors. [DOI] [PubMed] [Google Scholar]
- Fahnestock ML, Johnson JL, Feldman RM, Neveu JM, Lane WS, Bjorkman PJ. The MHC class I homolog encoded by human cytomegalovirus binds endogenous peptides. Immunity. 1995;3:583–590. doi: 10.1016/1074-7613(95)90129-9. [DOI] [PubMed] [Google Scholar]
- Farrell HE, Vally H, Lynch DM, Fleming P, Shellam GR, Scalzo AA, Davis-Poynter NJ. Inhibition of natural killer cells by a cytomegalovirus MHC class I homologue in vivo. Nature. 1997;386:510–514. doi: 10.1038/386510a0. [DOI] [PubMed] [Google Scholar]
- Gazit R, Garty BZ, Monselise Y. Expression of KIR2DL1 on the entire NK cell population: a possible novel immunodeficiency syndrome. Blood. 2004;103:1965–1966. doi: 10.1182/blood-2003-11-3796. 9 other authors. [DOI] [PubMed] [Google Scholar]
- Jones TR, Hanson LK, Sun L, Slater JS, Stenberg RM, Campbell AE. Multiple independent loci within the human cytomegalovirus unique short region down-regulate expression of major histocompatibility complex class I heavy chains. J Virol. 1995;69:4830–4841. doi: 10.1128/jvi.69.8.4830-4841.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller R, Peitchel R, Goldman JN, Goldman M. An IgG-Fc receptor induced in cytomegalovirus-infected human fibroblasts. J Immunol. 1976;116:772–777. [PubMed] [Google Scholar]
- Kim JS, Choi SE, Yun IH. Human cytomegalovirus UL18 alleviated human NK-mediated swine endothelial cell lysis. Biochem Biophys Res Commun. 2004;315:144–150. doi: 10.1016/j.bbrc.2004.01.027. 8 other authors. [DOI] [PubMed] [Google Scholar]
- Kotenko SV, Saccani S, Izotova LS, Mirochnitchenko OV, Pestka S. Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10) Proc Natl Acad Sci U S A. 2000;97:1695–1700. doi: 10.1073/pnas.97.4.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong CC, Chapman TL, Bjorkman PJ, Formankova D, Mocarski ES, Phillips JH, Lanier LL. Modulation of natural killer cell cytotoxicity in human cytomegalovirus infection: the role of endogenous class I major histocompatibility complex and a viral class I homolog. J Exp Med. 1998;187:1681–1687. doi: 10.1084/jem.187.10.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepin EJ, Bastin JM, Allan DS. Functional characterization of HLA-F and binding of HLA-F tetramers to ILT2 and ILT4 receptors. Eur J Immunol. 2000;30:3552–3561. doi: 10.1002/1521-4141(200012)30:12<3552::AID-IMMU3552>3.0.CO;2-L. 8 other authors. [DOI] [PubMed] [Google Scholar]
- Lin G, Simmons G, Pohlmann S. Differential N-linked glycosylation of human immunodeficiency virus and Ebola virus envelope glycoproteins modulates interactions with DC-SIGN and DC-SIGNR. J Virol. 2003;77:1337–1346. doi: 10.1128/JVI.77.2.1337-1346.2003. 8 other authors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano M, Guma M, Ortega M, Angulo A, Lopez-Botet M. Differential effects of US2, US6 and US11 human cytomegalovirus proteins on HLA class Ia and HLA-E expression: impact on target susceptibility to NK cell subsets. Eur J Immunol. 2003;33:2744–2754. doi: 10.1002/eji.200324182. [DOI] [PubMed] [Google Scholar]
- Markine-Goriaynoff N, Gillet L, Van Etten JL, Korres H, Verma N, Vanderplasschen A. Glycosyltransferases encoded by viruses. J Gen Virol. 2004;85:2741–2754. doi: 10.1099/vir.0.80320-0. [DOI] [PubMed] [Google Scholar]
- McGrory WJ, Bautista DS, Graham FL. A simple technique for the rescue of early region I mutations into infectious human adenovirus type 5. Virology. 1988;163:614–617. doi: 10.1016/0042-6822(88)90302-9. [DOI] [PubMed] [Google Scholar]
- Neote K, DiGregorio D, Mak JY, Horuk R, Schall TJ. Molecular cloning, functional expression, and signaling characteristics of a C-C chemokine receptor. Cell. 1993;72:415–425. doi: 10.1016/0092-8674(93)90118-a. [DOI] [PubMed] [Google Scholar]
- Odeberg J, Cerboni C, Browne H, Karre K, Moller E, Carbone E, Soderberg-Naucler C. Human cytomegalovirus (HCMV)-infected endothelial cells and macrophages are less susceptible to natural killer lysis independent of the downregulation of classical HLA class I molecules or expression of the HCMV class I homologue, UL18. Scand J Immunol. 2002;55:149–161. doi: 10.1046/j.1365-3083.2002.01025.x. [DOI] [PubMed] [Google Scholar]
- Park B, Oh H, Lee S, Song Y, Shin J, Sung YC, Hwang S-Y, Ahn K. The MHC class I homolog of human cytomegalovirus is resistant to down-regulation mediated by the unique short region protein (US)2, US3, US6, and US11 gene products. J Immunol. 2002;168:3464–3469. doi: 10.4049/jimmunol.168.7.3464. [DOI] [PubMed] [Google Scholar]
- Penfold MET, Dairaghi DJ, Duke GM, Saederup N, Mocarski ES, Kemble GW, Schall TJ. Cytomegalovirus encodes a potent α chemokine. Proc Natl Acad Sci U S A. 1999;96:9839–9844. doi: 10.1073/pnas.96.17.9839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyburn HT, Mandelboim O, Valés-Gómez M, Davis DM, Pazmany L, Strominger JL. The class I MHC homologue of human cytomegalovirus inhibits attack by natural killer cells. Nature. 1997;386:514–517. doi: 10.1038/386514a0. [DOI] [PubMed] [Google Scholar]
- Saverino D, Ghiotto F, Merlo A. Specific recognition of the viral protein UL18 by CD85j/LIR-1/ILT2 on CD8+ T cells mediates the non-MHC-restricted lysis of human cytomegalovirus-infected cells. J Immunol. 2004;172:5629–5637. doi: 10.4049/jimmunol.172.9.5629. 12 other authors. [DOI] [PubMed] [Google Scholar]
- Shiroishi M, Tsumoto K, Amano K. Human inhibitory receptors Ig-like transcript 2 (ILT2) and ILT4 compete with CD8 for MHC class I binding and bind preferentially to HLA-G. Proc Natl Acad Sci U S A. 2003;100:8856–8861. doi: 10.1073/pnas.1431057100. 11 other authors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaete RR, Mocarski ES. Insertion and deletion mutagenesis of the human cytomegalovirus genome. Proc Natl Acad Sci U S A. 1987;84:7213–7217. doi: 10.1073/pnas.84.20.7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasec P, Braud VM, Rickards C. Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science. 2000;287:1031. doi: 10.1126/science.287.5455.1031. 7 other authors. [DOI] [PubMed] [Google Scholar]
- Tomasec P, Wang ECY, Davison AJ. Downregulation of natural killer cell-activating ligand CD155 by human cytomegalovirus UL141. Nat Immunol. 2005;6:181–188. doi: 10.1038/ni1156. 10 other authors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortorella D, Gewurz B, Schust D, Furman M, Ploegh H. Down-regulation of MHC class I antigen presentation by HCMV; lessons for tumor immunology. Immunol Invest. 2000;29:97–100. doi: 10.3109/08820130009062289. [DOI] [PubMed] [Google Scholar]
- Valés-Gómez M, Shiroishi M, Maenaka K, Reyburn HT. Genetic variability of the major histocompatibility complex class I homologue encoded by human cytomegalovirus leads to differential binding to the inhibitory receptor ILT2. J Virol. 2005;79:2251–2260. doi: 10.1128/JVI.79.4.2251-2260.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnum SM, Streblow DN, Monroe ME. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J Virol. 2004;78:10960–10966. doi: 10.1128/JVI.78.20.10960-10966.2004. 11 other authors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson GW, Akrigg A. Constitutive and enhanced expression from the CMV major IE promoter in a defective adenovirus vector. Nucleic Acids Res. 1992;20:2233–2239. doi: 10.1093/nar/20.9.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willcox BE, Thomas LM, Bjorkman PJ. Crystal structure of HLA-A2 bound to LIR-1, a host and viral major histocompatibility complex receptor. Nat Immunol. 2003;4:913–919. doi: 10.1038/ni961. [DOI] [PubMed] [Google Scholar]
- Young NT, Uhrberg M, Phillips JH, Lanier LL, Parham P. Differential expression of leukocyte receptor complex-encoded Ig-like receptors correlates with the transition from effector to memory CTL. J Immunol. 2001;166:3933–3941. doi: 10.4049/jimmunol.166.6.3933. [DOI] [PubMed] [Google Scholar]
- Zhu H, Shen Y, Shenk T. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J Virol. 1995;69:7960–7970. doi: 10.1128/jvi.69.12.7960-7970.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.