

Abstract
We have developed a strategy for the identification of peptides able to functionally replace a zinc finger domain in a transcription factor. This strategy could have important ramifications for basic research on gene regulation and for the development of therapeutic agents. In this study in yeast, we expressed chimeric proteins that included a random peptide combinatorial library in association with two zinc finger domains and a transactivating domain. The library was screened for chimeric proteins capable of activating transcription from a target sequence in the upstream regulatory regions of selectable or reporter genes. In a screen of approximately 1.5 × 107 transformants we identified 30 chimeric proteins that exhibited transcriptional activation, some of which were able to discriminate between wild-type and mutant DNA targets. Chimeric library proteins expressed as glutathione S-transferase fusions bound to double-stranded oligonucleotides containing the target sequence, suggesting that the chimeras bind directly to DNA. Surprisingly, none of the peptides identified resembled a zinc finger or other well-known transcription factor DNA binding domain.
Keywords: protein-DNA interaction, gene expression
Transcription factors are key regulators of gene expression. These proteins are usually modular in structure with distinct DNA-binding and trans-activating domains (1). In nature, several highly conserved protein structural motifs are used to mediate specific DNA-protein recognition; these motifs include zinc fingers (Zifs), leucine zippers, helix–turn–helix structures, homeodomains, and β-sheet structures (1–3). It would be of considerable interest to extend the repertory of DNA-binding protein motifs by identifying novel peptides capable of sequence-specific recognition of DNA. These entities could enhance our understanding of the physical basis of protein-DNA interactions. As well, they might find a variety of uses in experimental biology and in therapeutics, in terms of either positive or negative regulation of gene expression (4).
Combinatorial library strategies offer powerful tools for the identification of novel molecular structures (5–9). One such strategy, phage display, already has been applied to the task of finding protein structures with novel DNA-binding capabilities (10–15). Thus, several research groups have randomized critical regions of zinc finger DNA-binding domains, expressed the regions as fusions with bacteriophage coat proteins, and have screened the resulting phage populations for clones having the ability to bind to specific double-stranded oligonucleotides (12–15). These studies demonstrated that it is possible to use phage combinatorial libraries to identify protein domains with novel and specific DNA binding abilities. It should be noted that this approach, while randomizing certain critical amino acid residues, maintained the essential structure of the zinc finger. Other groups (10, 11) have used fully random peptide libraries expressed as fusions with phage coat proteins; these efforts resulted in the identification of DNA-binding peptides having only modest sequence specificity.
Recently, another selection system, the yeast one-way hybrid, has been developed for screening cDNA libraries for genes encoding proteins that bind to specific DNA sequences (16–20). We decided to explore the possibility of incorporating a combinatorial approach into the yeast one-hybrid system, thus creating a strategy for identification of novel, sequence-specific DNA-binding peptides. This approach has allowed us to directly select for peptides that support transcriptional activation in vivo. As demonstrated below, the yeast library approach has been successful in identifying several peptides that can functionally substitute for a zinc finger domain. Interestingly, the peptides selected thus far have novel sequences not resembling zinc fingers, nor any known transcription factor DNA-binding domains.
MATERIALS AND METHODS
Materials.
Yeast strain yWAM2 (MATαΔgal4Δgal80 URA3::GAL1-lacZlys2801amberhis3-Δ200trp1-Δ63leu2ade2– 101ochreCYH2) was provided by P. Hieter (Johns Hopkins, Baltimore) (21). Yeast strain yM4271 (MATa,ura-52, his3–200,ade2–101,lys2–801,leu2–3,112,trp1–903,tyr1–501) was purchased from CLONTECH. Yeast shuttle vectors pRS313 and pRS314 were purchased from the American Type Culture Collection. The yeast shuttle vectors pPC82 and pRS315HIS were provided by R. Reed (Johns Hopkins, Baltimore) (22). An Sp1 cDNA plasmid was provided by R. Tjian (University of California, Berkeley) (23). The pLG670Z vector was provided by R. West (University of Colorado) (24). Escherichia coli strain KC8 (pyrF::Tn5 hsdR leuB600 trpC9830 lacd74 strA galK hisB436) was provided by K. Struhl (Harvard) (25). E. coli BL21 competent cells were purchased from Novagen. The glutathione S-transferase (GST) fusion protein vector (pGEX4T-1) was purchased from Pharmacia. Oligonucleotides were synthesized by Oligos Etc. (Wilsonville, OR). All restriction enzymes and calf alkaline phosphatase were purchased from Boehringer Mannheim. Sequenase was purchased from United States Biochemicals.
Construction of Expression Vectors for Yeast Combinatorial Peptide Libraries.
The general strategy used was to construct a yeast shuttle vector capable of expressing the yeast GAL4 transactivating domain fused to any one of several DNA binding domains. The potential DNA binding domains included: (i) the complete three zinc finger DNA binding domain from the Sp1 transcription factor as a positive control; (ii) two zinc fingers from Sp1 as a negative control; (iii) two zinc fingers from Sp1 fused to a fully randomized 30-aa sequence (the peptide library). A spacer region from Sp1 also was placed between the transactivating and DNA-binding domains. The choice of 30 amino acids as the length of the library peptides was influenced by the approximate size of authentic Zif domains (1). To facilitate the construction of the various vectors used in this project, a parent vector coding for the GAL4 transactivating domain, the Sp1 spacer region, and containing a poly linker was constructed; this vector is termed pXC1 and is depicted in Fig. 1A. The negative control vector, termed pXCF2, was constructed by in-frame ligation of a PCR product of Sp1 containing two zinc fingers (Zifs 1 and 2) into pXC1 (Fig. 1B). A positive control vector, termed pXCF3, was constructed by in-frame ligation of the three zinc fingers (Zifs 1, 2, and 3) of Sp1 into the pXC1 vector (Fig. 1C). All constructs were confirmed by sequencing of both strands.
Figure 1.
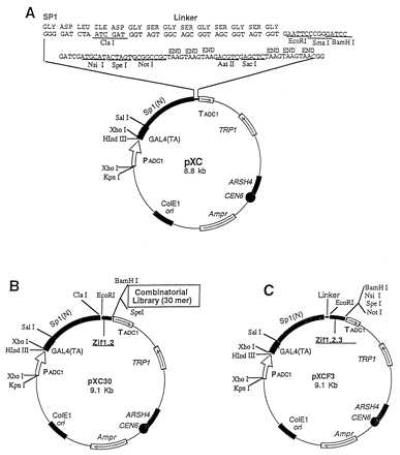
Construction of a yeast combinatorial library. The structure of the yeast combinatorial peptide library used here consists of three key parts: a yeast GAL4 activation domain, a partial DNA binding domain (two zinc fingers) from Sp1, and a peptide library. A spacer comprised of a portion of the N-terminal region (185 amino acids) of Sp1 and a flexible linker of 10 amino acids were inserted between the GAL4 domain and the partial Sp1 DNA binding domain. (A) The pXC1 vector includes a GAL4 activation domain, a spacer sequence, a flexible linker of 10 amino acids, a multiple cloning site, and selective marker genes. (B) The library pXC30. The control vector pXCF2 was constructed by inserting the first two zinc fingers of Sp1 into pXC1 at EcoRI and BamH sites. To construct the library, a 100-bp degenerate double-stranded oligonucleotide encoding 30 random amino acids was inserted into pXCF2, giving rise to the yeast library pXC30. (C) The positive control vector pXCF3. Three zinc fingers of Sp1 were inserted into the pXC1 vector at EcoRI and BamHI sites.
Use of Degenerate Oligonucleotides to Form the pXC30 Library.
The yeast combinatorial library, termed pXC30, was constructed by placing a degenerate oligonucleotide able to code for 30 amino acids between the BamHI and SpeI sites located at the 3′ end of the two zinc fingers in pXCF2. A 100-bp single-stranded oligonucleotide containing terminal BamHI and XbaI sites and fully degenerate at the remaining (NNK)30 positions was synthesized, converted to double-stranded form and purified by gel electrophoresis (26). The restricted oligonucleotides were ligated into the pXCF2 vector, and the ligated DNA was used to transform E. coli DH10B competent cells (25). The transformants (about 1.3 × 107) were harvested, and the pXC30 DNA library was purified from the transformants by using Qiagen columns.
Establishing Yeast Strains Containing Sp1 Target Sites.
The target DNA sequences contain either two (W2) or four (W4) copies of a wild-type (WT) Sp1 binding site (27). The control sequences (Mu2 and Mu4) are made by mutating the third zinc finger binding site from GAG to AAA. The approximate binding positions of the three zinc fingers (Z1, Z2, and Z3) are indicated over the last Sp1 site of the W4 oligonucleotide. The sequences are shown below with the Sp1 sites bolded:
Double-stranded oligonucleotides were inserted upstream of the minimal promoters of two reporter vectors, pRS315HIS and pLG670Z, at BamHI and XhoI sites, respectively. The vector pRS315HIS contains a GAL1 minimum promoter driving expression of a HIS3 gene as well as a LEU2 selectable marker gene driven by a constitutive promoter. The vector pLG670Z contains a CYC1 minimum promoter driving a lacZ gene as well as a URA3 selectable marker gene. The newly constructed vectors were named pRS315W4, pRS315W2, pRS315M4, pRS315M2, pLG670W4, pLG670W2, pLG670M4, and pLG670M2. Yeast strains yW315W4, yW315W2, yW315M4, and yW315M2 were established by transformation (25) of the yeast yWAM2 strain with pRS315W4, pRS315W2, pRS315M4, and pRS315M2, respectively. Yeast strains, yM670W4, yM670W2, yM670M4, and yM670M2 were established by transformation of yeast yM4271 with pLG670W4, pLG670W2, pLG670M4, and pLG670M2 vectors, respectively.
Screening for Peptides Able to Support Transcription at an Sp1 Site.
The pXC30 library was screened to identify plasmids coding for chimeric proteins that could interact with WT Sp1 binding sites driving a HIS3 gene. Transformed yW315W4 cells were plated onto medium lacking histadine, leucine, and tryptophan (SD-Trp-Leu-His) and containing 0.05 mM 3-amino-1,2,4 triazole (3-AT); strong positive colonies were picked for further studies.
β-Galactosidase Assay.
A luminometry-based assay was used for quantitative determination of β-galactosidase activity. Total yeast proteins (25) were measured by using the BCA protein assay reagent (Pierce). Quantitation was carried out by using a luminescent detection kit (CLONTECH), and the values were read by using a Monolight 2010 instrument (Analytical Luminescence Laboratory, San Diego).
GST Fusion Proteins and Electrophoretic Mobility-Shift Assay.
Yeast plasmids derived from the pXC30 library were isolated from positive yeast clones as described (29) and were used to transform bacterial KC8 cells by electroporation (25). Purified plasmids able to activate both HIS3 in yW315W4 and LacZ in yM670W4 were restricted with EcoRI and NotI. The DNA fragments were purified and subcloned into pGEX-4T-1 restricted between EcoRI and NotI, thus creating vectors capable of expressing chimeric proteins comprised of GST, Zifs 1 and 2, and the combinatorial peptide. Expression and purification of GST fusion proteins were carried out as described (30). The electrophoretic mobility shift assay using GST fusion proteins and 32P-end-labeled W4 oligonucleotide was performed by using a standard protocol (31). A monoclonal anti-GST antibody (Sigma) was used for super-shifting experiments.
Competition Assays.
GST fusion proteins (10 μg) were briefly preincubated with various concentrations of unlabeled W4 or Mu4 oligonucleotide in buffer at 4°C. Approximately 50 fmols of 32P-end-labeled W4 oligonucleotide was added, and the incubation continued for 1 hr at 25°C. Magnetic beads (Dynal, Great Neck, NY) coated with anti-mouse IgG and then with mouse monoclonal anti-GST antibody were added and incubated for 1 hr. The beads were separated from the incubation mixture by using a magnetic device and then washed with buffer. Bound 32P oligonucleotide was quantitated by scintillation counting.
Analysis of Sequences.
Plasmid DNAs from positive library clones were sequenced by using a Sequenase kit, and the corresponding peptide sequences were analyzed by using the GCG computer program.
RESULTS
Feasibility of the Yeast Combinatorial Library Approach.
The essence of the current approach is to use a combinatorial library to identify novel peptides that can fully or partially substitute for the third Zif of the Sp1 transcription factor. To pursue this approach we needed to establish its overall feasibility. Thus we needed to determine that we could construct chimeric transcription factors that could effectively activate selectable and reporter genes in yeast, and that these chimeric transcription factors could discriminate WT from mutant Sp1 target sites. Finally, we needed to know the selection stringency, that is, the “window” of transcriptional activation between our positive control, a chimeric protein with the complete three Zifs of Sp1, and our negative control, a protein with only two Sp1 Zifs, anticipating that the transcriptional activity of the chimeric library proteins likely would fall into this “window.”
To explore these issues, the pXCF3 or pXCF2 vectors were transfected into yeast strains containing WT or mutant Sp1 binding sites driving either the HIS3 or lacZ genes. As seen in Fig. 2A, pXCF3 efficiently activated HIS3 expression in a yeast strain containing WT target sites, but not in a strain having mutated sites. The pXCF2 vector was unable to activate sufficient HIS3 expression from either WT or mutant sites to permit cell growth. As seen in Fig. 2B, pXCF3 also could activate lacZ expression in strains containing WT but not mutant Sp1 sites, whereas pXCF2 failed to activate in either case. Thus the chimeric transcription factor having three Zifs can function effectively in activating HIS3 or LacZ in yeast and can discriminate WT from mutant targets. By contrast, the chimera having only two Zifs was ineffective in activating transcription.
Figure 2.
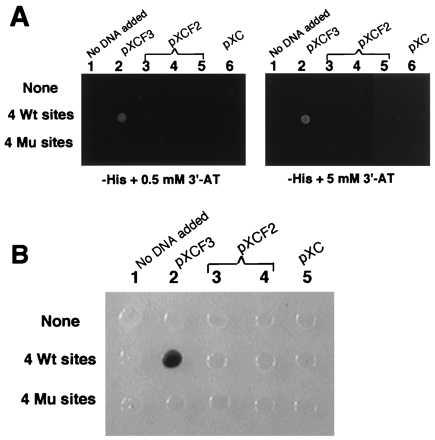
Transactivation by the positive and negative control vectors. (A) The yeast strains yWAM2 (None = no target sites), yW315W4 (four WT sites), and yW315M4 (four mutant sites) each containing the HIS3 selectable marker were transformed with the vectors pXC1, pXCF2, or pXCF3. The transformants were tested for their ability to grow on histadine-deficient plates containing either 0.5 mM or 5 mM 3-AT. Growth is indicated by a white spot. (B) The same vectors were used to transform yeast strains having four WT or four mutant sites driving the lacZ gene. The transformants were tested for color generation on 5-bromo-4-chloro-3-indolyl β-d-galactoside containing plates.
To evaluate the size of the “screening window,” we examined the ability of constructs expressing three Zifs, two Zifs, or two Zifs plus the peptide library to activate HIS3 and promote cell growth in the presence of increasing concentrations of 3-AT, an inhibitor of the histadine pathway (32). Background growth of yeast cells transformed with the negative control vector pXCF2 could be totally eliminated with low concentrations of 3-AT (0.05 mM). By contrast, yeast transformed with the three-Zif vector pXCF3 grew well at up to 5 mM 3-AT. Thus there is at least a 100-fold difference in sensitivity to 3-AT between the positive (pXCF3) and negative (pXCF2) controls. Yeast transfected with the pXC30 library displayed some isolated colonies but no generalized growth in the presence of 3-AT, suggesting that only a few of the peptides in the library would permit transcriptional activation of the target gene.
Screening the Yeast Combinatorial Peptide Library.
The pXC30 library was screened against a target comprised of four WT Sp1 binding sites upstream of HIS3. After transformation, yeast were spread on SD-Trp-Leu-His plates containing 0.05 mM 3-AT. About 1.5 × 107 transformants were examined, and 30 positive clones were isolated. The initial positive clones were tested for their ability to grow in the presence of increasing 3-AT concentrations. As shown in Fig. 3, a number of clones grew well even at high concentrations of 3-AT. DNA was isolated from these clones and used to transform E. coli KC8 cells. Plasmids were isolated from individual bacterial colonies and used to transform a second yeast strain (yM670W4) containing the lacZ gene driven by WT Sp1 binding sites. Only clonally isolated plasmids positive for both HIS3 and LacZ activation were used in further studies.
Figure 3.

Activation of HIS3 by candidate positive clones. The pXC30 library was transformed into yW315W4 cells, and transformants were screened for their ability to grow on histadine-deficient plates containing 0.05 mM 3-AT; 30 positive clones were picked. The positive clones were retested for their ability to grow on histadine-deficient plates containing increasing concentrations of 3-AT. Clones transformed with the positive (pXCF3) or negative (pXCF2) control vectors also were tested. (A) Diagram identifying the positions of individual clones. (B) Growth with 0.1 mM 3-AT. (C) Growth with 0.3 mM 3-AT. (D) Growth with 3 mM 3-AT.
Testing the Specificity of Candidate DNA Binding Proteins in Vivo.
Yeast strains yM670W4 and yM670M4, containing LacZ driven by WT (W4) or mutant (Mu4) target sequences, were used to test if peptides expressed by the candidate positive clones have specificity for WT Sp1 binding sites. The two strains were transformed with plasmid DNA from the candidate clones, pXCF3, or pXCF2. Transformed yeast were dropped onto synthetic dextrose plates without tryptophan, but containing 5-bromo-4-chloro-3-indolyl β-d-galactoside. As shown in Fig. 4A, the three-Zif vector (pXCF3) is very specific, strongly activating lacZ expression from the WT Sp1 site but not from a mutant site. By contrast, the two-Zif vector (pXCF2) produces only weak activation at both WT and mutant sites. Most of the candidate positive plasmids isolated from the pXC30 library showed a strong preference for the WT Sp1 binding site versus the mutant Sp1 binding site. However, one clone (C29) had very strong transcriptional activity at both the WT and mutant Sp1 binding sites.
Figure 4.
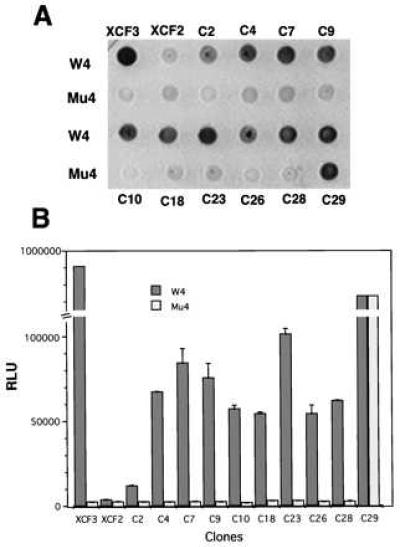
Ability of candidate positive clones to activate lacZ. Yeast strains yM670W4 and yM670M4, containing WT or mutant Sp1 binding sites driving lacZ, were used to test the specificity of positive clones from the pXC30 library. The two strains were transformed with selected library plasmid DNAs (Fig. 3), or with pXCF3 or pXCF2. (A) Transformants were tested for color formation on 5-bromo-4-chloro-3-indolyl β-d-galactoside plates and photographed. (B) Transformants were tested for β-galactosidase activity by using a quantitative luminescence assay. Total cell protein was normalized for each sample. y-axis, RLU = relative luminescence units; x-axis, identity of the transformants. Results are the means and standard errors of three determinations.
To evaluate binding strength and specificity in a more quantitative fashion, a luminescence-linked β-galactosidase assay was used. As seen in Fig. 4B, there was a 200- to 300-fold difference in activity of β-galactosidase in yeast strains having WT or mutant Sp1 binding sites upstream of LacZ when transformed with pXCF3. By contrast, transformation with pXCF2 resulted in very modest activation in either reporter yeast strain. Compared with pXCF3, the transcriptional activities of most of the library candidates were weaker, but they were distinctly stronger than that of pXCF2. Most of the candidate clones from the XC30 library expressed proteins that clearly preferred WT Sp1 binding site rather than mutant sites, with 5- to 50-fold differences in β-galactosidase activities in yeast strains having WT or mutant binding sites.
In vitro Evaluation of DNA Binding by Chimeric Library Proteins.
The candidate clones were used to construct fusion proteins including the GST domain, two Zifs, and a library peptide. Positive and negative controls were chimeras having, respectively, three or two Zifs fused to GST. The fusion proteins were tested (Fig. 5A) for their ability to bind to oligonucleotides containing WT Sp1 sites by using an electrophoretic mobility shift assay; the samples also were tested for super-shifting behavior by using an antibody to GST. Consistent with the in vivo data, the GST-3 Zif fusion protein showed strong binding to an oligonucleotide containing WT sites (Fig. 5A, lane 3), with the identity of the binding protein confirmed by super-shifting (Fig. 5A, lane 4). The fusion protein prepared from the candidate nonspecific clone (C29) also showed strong binding to the oligonucleotide probe (Fig. 5A, lanes 5 and 6). Fusion proteins from the candidate specific clones from the library also showed binding that was clearly stronger than that of the two-Zif fusion protein (Fig. 5A, compare lanes 7, 9, 11, and 13 to lane 2), although weaker than that of the three-Zif protein. In most cases the library peptide fusion proteins apparently bound to the oligonucleotide as monomers, whereas in some cases larger aggregates were observed. Binding to the oligonucleotide probe is clearly because of the various GST fusion proteins, because the anti-GST antibody super-shifts the patterns (Fig. 5A, lanes 8, 10, 12, and 14). Thus, these results suggest that several of the chimeric library proteins are able to directly bind to DNA.
Figure 5.
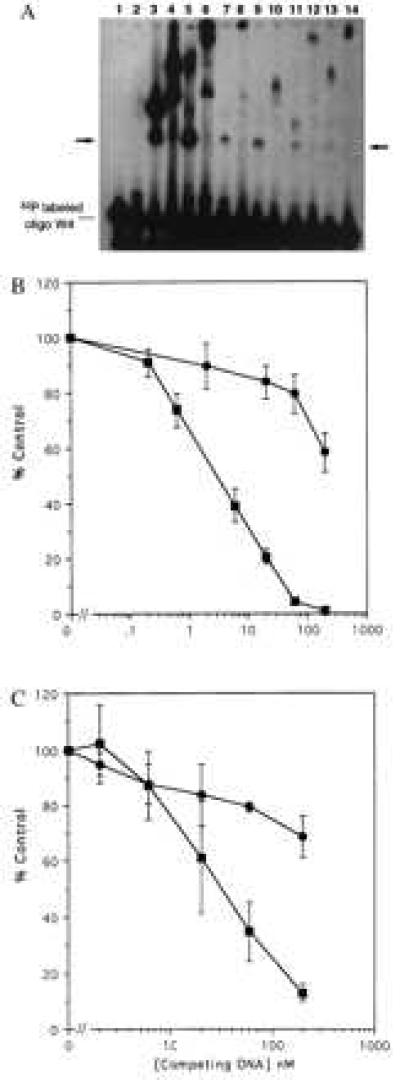
In vitro DNA binding by GST fusion proteins containing library peptides. Candidate DNA binding peptides identified by screening the pXC30 library were fused to GST, expressed, and purified by using standard techniques. (A) Equal amounts (10 μg) of the GST chimeric proteins were incubated with a double-stranded 32P-labeled oligonucleotide (50 fmols) having four WT Sp1 sites. Formation of protein-DNA complexes was monitored by electrophoretic mobility-shift assay. In some cases anti-GST was used to “super-shift” the complexes. The unmarked lane is the free 32P oligonucleotide. The arrow marks the expected migration position of oligonucleotide binding to a fusion protein monomer. Lane 1, unmodified GST protein. Lane 2, two-Zif GST fusion protein. Lane 3, three-Zif fusion protein. Lane 4, the same as lane 3 plus anti-GST antibody. Lane 5, GST fusion with the nonspecific C29 library peptide. Lane 6, the same as lane 5 plus anti-GST. Lanes 7, 9, 11, and 13 are GST fusions with library peptides from clones C4, C7, C23, and C28, respectively. Lanes 8, 10, l2, and 14 are the same as the odd numbered lines plus anti-GST. (B) A radioligand binding competition assay was used to evaluate specificity of DNA binding. Unlabeled double-stranded oligonucleotides having four WT Sp1 sites (▪) or mutated sites (•) were used to competitively displace a 32P oligonucleotide having four WT sites. (Upper) Data for the GST chimera having three Sp1 Zifs. (Lower) Data for the chimera having two Sp1 Zifs linked to the C7 library peptide. The abcissa shows the concentration of unlabeled competitor in nanomolar units. The ordinate shows percent of control radioligand binding (100% = no competitor). The results represent means and standard errors of three determinations.
A radioligand competition assay was performed to establish the specificity of DNA binding by the GST chimeras (Fig. 5B). Various concentrations of unlabeled double-stranded oligonucleotides having either WT Sp1 sites or mutated sites were used to compete the binding of a 32P-labeled WT Sp1 oligonucleotide to a three-Zif GST (Fig. 5B, Upper), or to a chimera having two Zifs and the C7 library peptide (5B, Lower). Approximately 3 nM WT oligonucleotide inhibited radioligand binding by 50%, whereas more than 200 nM mutant oligonucleotide was required, indicating that the three-Zif GST chimera displays considerable sequence specificity of binding. Similarly, approximately 40 nM WT oligonucleotide inhibited binding by 50%, whereas more than 200 nM mutant oligonucleotide was required; this result indicates that the chimera containing two Zifs and the library peptide also displays sequence specificity of binding. That the concentration of WT competitor required for inhibition of radioligand binding was higher for the two-Zif plus library peptide chimera than for the three-Zif chimera indicates that the three-Zif protein has a higher affinity for the target DNA (33), consistent with the observed relative effects on transcriptional activation (Fig. 4).
Sequences of the Selected Peptides.
Plasmids from the candidate positive clones were sequenced; the corresponding peptide sequences are shown in Fig. 6. Thus far no significant homology has been found among the selected peptides. Search of the protein data banks has not yielded known proteins with a high homology score. In particular, the candidate DNA binding peptides identified thus far seem to have no obvious resemblance to Zifs or other known transcription factor DNA binding domains.
Figure 6.

Sequences of the peptides identified by screening the pXC30 library.
DISCUSSION
Molecules able to selectively manipulate gene expression would be powerful tools for experimental biology and might lead to the development of novel therapeutic agents for a variety of diseases. We suggest that the use of yeast combinatorial libraries to identify peptides capable of interacting with DNA in a sequence-specific manner may become an important approach for generating novel molecules for the manipulation of gene expression. Sequence-specific peptides targeted to promoters could interfere with transcriptional activation by competing for transcription factor binding sites; alternatively, they could be coupled to transcriptional repressor domains (34–36) and interfere in that manner. By judicious choice of the target sequence it should be possible to selectively inhibit the expression of a single gene. Similarly, such peptides could be used to target transactivating domains to specific promoters thus potentially activating genes that otherwise would be silent. The key issue for the evolution of this approach, however, is the identification of peptides capable of specific interactions with DNA.
In a previous study, we were unable to find sequence-specific DNA binding moieties in a random peptide phage display library (10). This result led us to explore a yeast-based system as an alternative approach for using random libraries to identify molecules capable of specifically interacting with DNA. Our approach contrasts with previous studies (12–15) that used libraries comprised of modified Zif domains displayed as bacteriophage coat fusion proteins. The yeast combinatorial peptide library system offers one clear advantage over phage display in that the chimeric proteins identified in the yeast system are able to interact with DNA within the chromatin of living cells. By contrast, phage display selects candidates that can bind in vitro, but these candidates must be subsequently evaluated for in vivo activity. However, there are also limitations to the yeast approach; thus the complexity of yeast libraries is inherently much lower than can be attained in phage library construction (5, 8, 37).
In this study, a random peptide library associated with the GAL4 transactivating domain and with two Zifs from the Sp1 transcription factor was expressed. In essence the peptide library replaces the third Zif of the intact Sp1 DNA binding domain. Chimeras having only two Zifs were ineffective in DNA binding and transactivation, whereas chimeras with three Zifs, or with two Zifs in association with certain library peptides, were quite effective. This finding indicates that the presence of the library peptide confers the ability to interact with DNA in a sequence-specific manner. Somewhat to our surprise, none of the peptides selected in our screen had any clear-cut similarity to authentic Zifs or other well-known DNA binding domains. Thus they may represent novel solutions to the DNA recognition problem. The interactions of the sequence-specific library chimeric proteins with the DNA target sequence were clearly weaker than that of the native Sp1 transcription factor. The chimeric protein containing library peptide C29 activated almost as strongly as native Sp1, but it failed to discriminate between WT and mutated target sequences. Although in this study we did not find a peptide that was fully equivalent to an authentic Zif, some of the sequences identified were quite specific and effective, with up to 50-fold greater transcriptional activation from the WT than the mutant target sequence. It seems possible, through the use of more complex libraries, more stringent selection, or by use of peptide libraries with conformational constraints, that one may be able to use the yeast library approach to select novel DNA binding peptides whose affinity and specificity approach that of authentic transcription factor DNA binding domains.
The peptide sequences selected thus far reveal few obvious clues concerning the nature of their interactions with DNA. Some of the peptides contain a number of positive charges, especially the strong, but nonspecific, peptide from clone 29. However, among the specific peptides, there are ones with both net negative (C9) and net positive charges. It remains possible that the peptides capable of specific DNA binding share a similar structural motif, although they differ greatly in amino acid sequence. Thus, in future studies, we will need to use physical techniques to develop a more detailed picture of the three-dimensional structures of these molecules. Another interesting issue concerns the size of a peptide needed to recognize a given number of base pairs in DNA. In one instance (C7) we found a peptide of only 20 residues able to provide specific recognition of a 3-bp motif. This result seems similar to recent work using a peptide-like molecule (a polyamide), in that a relatively small molecule was able to provide sequence-specific recognition of a six-base target (38). Expressing combinatorial peptide libraries in vivo to identify molecules capable of specific intermolecular interactions is a powerful and easily generalizable strategy (28, 39). It seems likely that the use of combinatorial libraries in yeast will provide an interesting approach for the identification and characterization of novel peptide motifs capable of interacting with a variety of DNA targets.

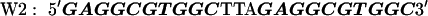
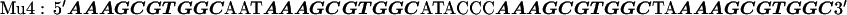
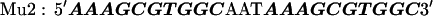
Acknowledgments
We thank Randall R. Reed, Philip A. Hieter, Robert West, Roger Brent, and Robert Tjian for generously providing plasmids and yeast strains. We also thank Brian Kay for encouragement and astute advice. This work was supported by National Institutes of Health Grant CA47044 and by a grant from the North Carolina Biotechnology Center to R.L.J.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: GST, glutathione S-transferase; Zif, zinc finger; WT, wild type; 3-AT, 3-amino-1,2,4 triazole.
References
- 1.Pabo C O, Sauer R T. Annu Rev Biochem. 1992;61:1053–1095. doi: 10.1146/annurev.bi.61.070192.005201. [DOI] [PubMed] [Google Scholar]
- 2.Struhl K. Trends Biochem Sci. 1989;14:137–140. doi: 10.1016/0968-0004(89)90145-X. [DOI] [PubMed] [Google Scholar]
- 3.Ptashne M. Nature (London) 1988;335:683–689. doi: 10.1038/335683a0. [DOI] [PubMed] [Google Scholar]
- 4.Sager R. Curr Opin Cell Biol. 1992;4:155–160. doi: 10.1016/0955-0674(92)90026-9. [DOI] [PubMed] [Google Scholar]
- 5.Devlin J J, Panganiban L C, Devlin P E. Science. 1990;249:404–406. doi: 10.1126/science.2143033. [DOI] [PubMed] [Google Scholar]
- 6.Scott J K, Smith G P. Science. 1990;249:386–390. doi: 10.1126/science.1696028. [DOI] [PubMed] [Google Scholar]
- 7.Kenan D, Tsai D, Keene J. Trends Biochem Sci. 1994;19:57–64. doi: 10.1016/0968-0004(94)90033-7. [DOI] [PubMed] [Google Scholar]
- 8.Kay B K, Adey N B, He Y S, Manfredi J P, Mataragnon A H, Fowlkes D M. Gene. 1993;128:59–65. doi: 10.1016/0378-1119(93)90153-t. [DOI] [PubMed] [Google Scholar]
- 9.Gold L, Polisky B, Uhlenbeck O, Yarus M. Annu Rev Biochem. 1995;64:763–797. doi: 10.1146/annurev.bi.64.070195.003555. [DOI] [PubMed] [Google Scholar]
- 10.Cheng X, Kay B K, Juliano R L. Gene. 1996;171:1–8. doi: 10.1016/0378-1119(95)00889-6. [DOI] [PubMed] [Google Scholar]
- 11.Wang B, Dickinson L A, Koivuneh E, Ruoslahti E, Kohwi-Shigematsu T. J Biol Chem. 1995;270:23239–23242. doi: 10.1074/jbc.270.40.23239. [DOI] [PubMed] [Google Scholar]
- 12.Rebar E J, Pabo C O. Science. 1994;263:671–673. doi: 10.1126/science.8303274. [DOI] [PubMed] [Google Scholar]
- 13.Choo Y, Klug A. Proc Natl Acad Sci USA. 1994;91:11163–11167. doi: 10.1073/pnas.91.23.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choo Y, Klug A. Proc Natl Acad Sci USA. 1994;91:11168–11172. doi: 10.1073/pnas.91.23.11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greisman H A, Pabo C O. Science. 1997;275:657–660. doi: 10.1126/science.275.5300.657. [DOI] [PubMed] [Google Scholar]
- 16.Sieweke M H, Tekotte H, Frampton J, Graf T. Cell. 1996;85:49–60. doi: 10.1016/s0092-8674(00)81081-8. [DOI] [PubMed] [Google Scholar]
- 17.Strubin M, Newell J W, Matthias P. Cell. 1995;80:205–214. doi: 10.1016/0092-8674(95)90500-6. [DOI] [PubMed] [Google Scholar]
- 18.Gstaiger M, Knoepfel L, Georgiev O, Schaffner W, Hovens C M. Nature (London) 1995;373:360–362. doi: 10.1038/373360a0. [DOI] [PubMed] [Google Scholar]
- 19.Lehming N, Thanos D, Brickman J M, Ma J, Maniatis T, Ptashne M. Nature (London) 1994;371:175–179. doi: 10.1038/371175a0. [DOI] [PubMed] [Google Scholar]
- 20.Li J J, Herskowitz I. Science. 1993;262:1870–1873. doi: 10.1126/science.8266075. [DOI] [PubMed] [Google Scholar]
- 21.Sikorski R S, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang M M, Reed R R. Nature (London) 1993;364:121–126. doi: 10.1038/364121a0. [DOI] [PubMed] [Google Scholar]
- 23.Kadonaga J T, Carner K R, Masiarz F R, Tjian R. Cell. 1987;51:1079–1090. doi: 10.1016/0092-8674(87)90594-0. [DOI] [PubMed] [Google Scholar]
- 24.West R W, Jr, Yocum R R, Ptashne M. Mol Cell Biol. 1984;4:2467–2478. doi: 10.1128/mcb.4.11.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ausubel F M, Brent R, Kingston R E, Moore D, Smith J A, Seidman J G, Struhl K, editors. Current Protocols in Molecular Biology. New York: Wiley; 1995. [Google Scholar]
- 26.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 27.Jones K A, Kadonaga J T, Luciw P W, Tjian R. Science. 1986;232:755–759. doi: 10.1126/science.3008338. [DOI] [PubMed] [Google Scholar]
- 28.Harada K, Martin S S, Frankel A D. Nature (London) 1996;380:175–179. doi: 10.1038/380175a0. [DOI] [PubMed] [Google Scholar]
- 29.Kaiser P, Auer B. BioTechniques. 1993;14:552. [PubMed] [Google Scholar]
- 30.Smith D B, Johnson K S. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 31.Carey J. Methods Enzymol. 1991;208:103–117. doi: 10.1016/0076-6879(91)08010-f. [DOI] [PubMed] [Google Scholar]
- 32.Bartel P, Chien C T, Sternglanz R, Fields S. BioTechniques. 1993;14:920–924. [PubMed] [Google Scholar]
- 33.Limbird L. Cell Surface Receptors: A Short Course on Theory and Methods. Norwell, MA: Kluwer; 1996. [Google Scholar]
- 34.Fisher A L, Ohsako S, Caudy M. Mol Cell Biol. 1996;16:2670–2677. doi: 10.1128/mcb.16.6.2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Friedman J R, Fredricks W J, Jensen D E, Speicher D W, Huang X P, Neilson E G, Rauscher F J. Genes Dev. 1996;10:2067–2078. doi: 10.1101/gad.10.16.2067. [DOI] [PubMed] [Google Scholar]
- 36.Hsieh J J, Hayward S D. Science. 1995;268:560–563. doi: 10.1126/science.7725102. [DOI] [PubMed] [Google Scholar]
- 37.Ruden D M, Ma J, Wood K, Ptashne M. Nature (London) 1991;350:426–430. doi: 10.1038/350250a0. [DOI] [PubMed] [Google Scholar]
- 38.Trauger K W, Baird E E, Dervan P B. Nature (London) 1996;382:559–561. doi: 10.1038/382559a0. [DOI] [PubMed] [Google Scholar]
- 39.Colas P, Cohen B, Jessen T, Grishina I, MaCoy J, Brent R. Nature (London) 1996;380:548–550. doi: 10.1038/380548a0. [DOI] [PubMed] [Google Scholar]