

Abstract
Many of the ichthyoses are associated with inherited disorders of lipid metabolism. These disorders have provided unique models to dissect physiologic processes in normal epidermis and the pathophysiology of more common scaling conditions. In most of these disorders, a permeability barrier abnormality “drives” pathophysiology through stimulation of epidermal hyperplasia. Among primary abnormalities of nonpolar lipid metabolism, triglyceride accumulation in neutral lipid storage disease as a result of a lipase mutation provokes a barrier abnormality via lamellar/nonlamellar phase separation within the extracellular matrix of the stratum corneum (SC). Similar mechanisms account for the barrier abnormalities (and subsequent ichthyosis) in inherited disorders of polar lipid metabolism. For example, in recessive X-linked ichthyosis (RXLI), cholesterol sulfate (CSO4) accumulation also produces a permeability barrier defect through lamellar/nonlamellar phase separation. However, in RXLI, the desquamation abnormality is in part attributable to the plurifunctional roles of CSO4 as a regulator of both epidermal differentiation and corneodesmosome degradation. Phase separation also occurs in type II Gaucher disease (GD; from accumulation of glucosylceramides as a result of to β-glucocerebrosidase deficiency). Finally, failure to assemble both lipids and desquamatory enzymes into nascent epidermal lamellar bodies (LBs) accounts for both the permeability barrier and desquamation abnormalities in Harlequin ichthyosis (HI). The barrier abnormality provokes the clinical phenotype in these disorders not only by stimulating epidermal proliferation, but also by inducing inflammation.
Supplementary key words: ATP binding cassette transporter 12, arachidonate lipoxygenase, barrier function, epidermal lipids, harlequin ichthyosis, neutral lipid storage disease, recessive X-linked ichthyosis, stratum corneum, transepidermal water loss
One thing is certain. The sequencing of the genome will soon look like the easiest thing that biologists ever did. …what the genes actually do—constitutes the real code of living systems. To crack that code will take centuries, but getting there will be more than half the fun.
—Melvin Konner, “Weaving Life’s Pattern,” Nature 418: 279, 2002
Many of the ichthyoses are associated with inherited disorders of lipid metabolism. These disorders have provided unique models to dissect physiologic processes in normal epidermis and the pathophysiology of more common scaling conditions. In most of these disorders, a permeability barrier abnormality “drives” pathophysiology through the stimulation of epidermal hyperplasia. Among primary abnormalities of nonpolar lipid metabolism, triglyceride accumulation in neutral lipid storage disease as a result of a lipase mutation provokes a barrier abnormality via lamellar/nonlamellar phase separation within the extracellular matrix of the stratum corneum (SC). Similar mechanisms account for the barrier abnormalities (and subsequent ichthyosis) in inherited disorders of polar lipid metabolism. For example, in recessive X-linked ichthyosis (RXLI), cholesterol sulfate (CSO4) accumulation also produces a permeability barrier defect through lamellar/nonlamellar phase separation. However, in RXLI, the desquamation abnormality is in part attributable to the plurifunctional roles of CSO4 as a regulator of both epidermal differentiation and corneodesmosome degradation (2). Phase separation also occurs in type II Gaucher disease (GD; from the accumulation of glucosylceramides as a result of β-glucocerebrosidase deficiency) (3). Finally, failure to assemble both lipids and desquamatory enzymes into nascent epidermal lamellar bodies (LBs) accounts for both the permeability barrier and desquamation abnormalities in harlequin ichthyosis (HI). The barrier abnormality provokes the clinical phenotype in these disorders not only by stimulating epidermal proliferation but also by inducing inflammation.
RATIONALE
The ichthyoses are rare scaling disorders (disorders of cornification) in which a large number of unrelated inherited disorders result is excessive visible scale (4–8). All of these disorders display a prominent permeability barrier abnormality, associated with abnormalities in the architecture of lamellar membranes in the extracellular spaces of the SC, where the barrier resides. Abnormal membrane structure can result directly from abnormalities in lipid metabolism (9–13) (to be discussed in this review) or indirectly from primary abnormalities in the corneocyte that either impede lipid secretion or alter scaffold function (discussed only briefly in this review; see Ref. 13). Initial pathogenic studies classified the inherited ichthyoses as either abnormalities in the structural proteins of the corneocyte “bricks” or as resulting from inborn errors of lipid metabolism (the “mortar”) (11, 12). This approach yielded two key insights: 1) that disorders of lipid metabolism alone can alter the extracellular matrix sufficiently to provoke ichthyotic disorders; and 2) that extracellular lipids contribute to the cohesive properties of normal SC (Table 1). However, it failed to illuminate the functional interdependence of the bricks and mortar components. Moreover, it also failed to predict the epidermal homeo-static responses that occur in response to altered SC function, in which the permeability barrier abnormality leads to epidermal hyperplasia and cytokine signaling of inflammation. This review focuses on the subcellular pathogenesis of ichthyoses as a result of disorders of lipid metabolism, using a function-driven model of disease.
TABLE 1.
Inherited lipid metabolic disorders with ichthyosis
| Metabolic Category | Inheritance Pattern | Multisystem | Affected Protein (Gene) | Normal Function |
|---|---|---|---|---|
| Fatty acid metabolism | ||||
| Refsum disease | Recessive | Yes | Phytanoyl-CoA hydroxylase (PAHX, PHYH ); peroxin 7 receptor (PEX7 ) | α-Hydroxylation of branched chain FFA |
| Sjögren-Larsson syndrome | Recessive | Yes | Fatty aldehyde dehydrogenase (ALDH3A2) | Oxidation of fatty aldehydes to free fatty acids |
| Autosomal recessive congenital ichthyosis | Recessive | No | 12R-Lipoxygenase (ALOX12) | ? Oxygenation of arachidonic acid to12(R)-HPETE |
| Recessive | No | Lipoxygenase-3 (ALOXE3) | ? Hydroxyperoxide isomerization of 12(R)-HPETE to epoxyalcohol metabolites | |
| Recessive | No | Cytochrome P450 4F22(CYP4F22, FLJ39501) | ? ω-Hydroxylation of trioxilins | |
| Recessive | No | Ichthyin (ichthyin) | ? Receptor for hydroxyepoxyalcohol metabolites | |
| Cholesterol metabolism | ||||
| Conradi-Hünermann syndrome | X-linked dominant | Yes | Δ8, Δ7-sterol isomerase emopamil binding protein (EBP) | Distal cholesterol synthetic pathway |
| CHILD syndrome | X-linked dominant | Yes | NAD(P)H steroid dehydrogenase- like protein (NSDHL) | Distal cholesterol synthetic pathway |
| X-linked ichthyosis | X-linked recessive | (Yes) | Ssase | Desulfate cholesterol sulfate |
| Sphingolipid metabolism | ||||
| GD type I | Recessive | Yes | β-Glucocerebrosidase (GBA) | Deglucosylated β-glucocerebrosidase |
| Niemann-Pick disease | Recessive | Yes | Acidic sphingomyelinase (SMPD1) | Generates ceramides from sphingomyelin |
| Triglyceride metabolism | ||||
| Neutral lipid storage disease | Recessive | Yes | CGI-58 acid lipase (ABHD5) | Generates diacylglycerides and FFAs from triacylglycerides |
| Lipid transporter | ||||
| Harlequin ichthyosis | Recessive | No | ATP binding cassette (ABCA12, truncation, deletion) | Transports glucosylceramides into LBs |
| Lamellar ichthyosis (some) | Recessive | No | ATP binding cassette (ABCA12, missense) | Same |
| CEDNIK syndrome | Recessive | Yes | Soluble n-ethylmaleimide-sensitive factor attachment protein receptor (SNAP29) | Facilitates exocytosis of LB contents |
ALOX, arachidonate lipoxygenase; CEDNIK, cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma; CHILD, congenital hemidysplasia with ichthyosiform erythroderma and limb defects; GD, Gaucher disease; LB, lamellar body; 12(R)-HPETE, 12(R)-hydroperoxyeicosa-tetraenoic acid; SSase, steroid sulfatase.
ROLE OF THE PERMEABILITY BARRIER IN DISEASE PATHOGENESIS
All of the ichthyoses studied to date, whether primary disorders of lipid or protein metabolism, have demonstrated a permeability barrier abnormality (9, 10, 13). Because permeability barrier requirements generally drive metabolic responses in the underlying epidermis (15, 16), the clinical phenotypes in the ichthyoses almost certainly reflect a best effort attempt by the epidermis to normalize barrier function (9). These metabolic responses to a flawed barrier, although only partially successful, usually suffice to allow survival in a dry, terrestrial environment. For example, in HI, in which few if any lipids are delivered to the SC interstices (17), the epidermis compensates as well as it can with an intense, hyperplastic response (increased cell proliferation in response to a defective barrier) that generates multiple layers of corneocytes (the “make more cells” imperative) (9) (see below). Even in inherited disorders that affect the structural proteins of the corneocyte bricks, permeability barrier abnormalities result from downstream alterations in the extracellular, lipid-enriched matrix (14), although by divergent mechanisms. For example, transglutaminase 1 (TGM1)-negative lamellar ichthyosis (LI) and loricrin keratoderma represent disorders in which the primary enzyme and its principal substrate, respectively, which both are involved in the formation of the corneocyte, are affected. In both of these disorders, the cornified envelope is attenuated, resulting in an impaired corneocyte scaffold, leading, in turn, to fragmented and foreshortened lamellar membranes (18, 19). These altered membranes lead to an impaired barrier caused by the leakage of water via the extracellular pathway. This link between a defective cornified envelope and the extracellular avenue of increased transepidermal water loss (TEWL) in both LI and loricrin keratoderma provides definitive proof that the corneocyte provides the scaffold necessary for the supramolecular organization of the lipid-enriched, extracellular matrix.
Alternatively, in epidermolytic hyperkeratosis, mutated keratins (either keratin 1 or 10) form abnormal, dominant-negative keratin pairs that disrupt the cytoskeleton, thereby impeding LB exocytosis (20). Once again, the barrier abnormality is provoked via a defect in the extracellular matrix (i.e., a reduction in secreted lipids) (20). Thus, in inherited disorders of corneocyte proteins of diverse etiology, the protein abnormality ultimately provokes a defect in the extracellular lamellar membranes (mortar) (18–20). This secondary defect in the extracellular matrix then allows accelerated, extracellular transcutaneous water movement (i.e., the permeability barrier abnormality), which drives epidermal hyperplasia, resulting in a thickened (ichthyotic) SC.
Finally, and pertinent for the inflammation that accompanies many of the ichthyoses, abnormal permeability barrier function inevitably stimulates signaling mechanisms that attempt to restore barrier function (21), but it also recruits downstream inflammation (i.e., another example of the “outside-inside” pathogenesis of inflammatory dermatoses) (21, 22). Moreover, if this cytokine cascade is sustained, both epidermal hyperplasia with hyperkeratosis and inflammation develop (21, 23), accounting for clinical features of the inflammatory ichthyoses.
DISORDERS OF NONPOLAR LIPID PROCESSING
Neutral lipid storage disease (Chanarin-Dorfman syndrome)
Neutral lipid storage disease with ichthyosis (NLSDI), or Chanarin-Dorfman syndrome [OMIM, Online Mendelian Inheritance in Man (OMIM) 275630], is a rare, recessive disorder often caused by mutations in a gene encoding for a putative lipid hydrolase, ABHD5 (also known as CGI-58) (24–27), that leads to an accumulation of cytosolic triacylglycerides (TAGs). Although the presence of ichthyosis indicates an ABHD5 mutation, kindreds with a lipid storage myopathy but no ichthyosis have been linked instead to mutations in desnutrin (PNPLA2, ATGL, or TTS-22), for which ABHD5 serves as a coactivator largely restricted to adipose tissue (28). CGI-58/ABHD5 is located on chromosome 3p21; it has seven exons and is expressed in many tissues, including skin. Affected patients are almost all homozygous, although a few cases of compound hetero-zygosity have been reported. CGI-58/ABHD5 encodes for a 349 amino acid protein that coactivates a newly identified lipase, ATGL, that hydrolyzes TAGs into diacylglycerides for phospholipid and free fatty acid production (29). However, the pathway that leads to cytosolic TAG accumulation in NLSDI has not been fully characterized. Labeling studies suggest that diacylglyceride is used for phospholipid synthesis, but excess substrate in NLSDI is stored in tissues as TAG (30, 31). Whether the lack of lipase activity results in a lack of phospholipid loading into LBs is likely but not yet investigated (Fig. 1). Alternatively, the protein encoded by CGI-58/ABHD5 may affect epoxide hydroxylation, but this remains to be confirmed experimentally (see below). One important consequence of the lack of phospholipids would be a downstream deficiency of free fatty acids (in epidermis, all secreted phospholipids are hydrolyzed normally into a large pool of nonessential FFAs that is an important constituent of the extracellular lamellar bilayers in SC) (32).
Fig. 1.

Proposed pathogenesis of neutral lipid storage disease. LB, lamellar body; PL, phospholipids; SC, stratum corneum; TAG, triacylglyceride; TEWL, transepidermal water loss.
TAG accumulation in cytosolic droplets in multiple tissues allows for the rapid clinical diagnosis of NLSDI by Oil Red O staining of frozen skin biopsies, which demonstrate diagnostic lipid droplets in both the epidermal basal layer and in appendageal epithelia (33), or by visual inspection of leukocytes on blood smears (33, 34) (Table 2). Although the ichthyosiform phenotype in NLSDI is non-diagnostic, it most closely resembles the nonbullous congenital ichthyosiform erythroderma (CIE) variant of autosomal recessive congenital ichthyosis (ARCI; see below) (33, 35). However, NLSDI patients also often experience pruritus, with or without atopic features (34, 35), an erythrokeratoderma variabilis-like dermatosis (24), or a severe “oily” (seborrheic) type of ichthyosis (36).
TABLE 2.
Ichthyoses that have or could have lamellar phase separation as a basis for barrier abnormality
| Disease | Enzymatic Defect | Barrier Dysfunction | Lamellar/Nonlamellar Phase Separation | Phase-Separated Lipid |
|---|---|---|---|---|
| Neutral lipid storage disease | Neutral lipid hydrolase (CGI-58) | Demonstrated (35) | Demonstrated (35) | Triglycerides |
| Recessive X-linked ichthyosis | SSase | Demonstrated (225) | Demonstrated (225) | Cholesterol sulfate |
| Refsum disease | Phytanoyl-CoA hydroxylase (PhyH); peroxin 7 receptor | Not assessed | Not assessed | Phytanic acid in all glycerolipids (226) |
| Sjögren-Larsson | Fatty aldehyde dehydrogenase | Not assessed | Demonstrated (227) | Not assessed |
| GD | β-Glucocerebrosidase | Demonstrated (228) | Demonstrated (228) | Glucosylceramides |
| CHILD syndrome | NAD(P)H 3β-hydroxysteroid dehydrogenase (NSDHL) | Not assessed | Not assessed | Not assessed |
| Niemann-Pick disease | Acidic sphingomyelinase | Demonstrated (14, 229) | Demonstrated (229) | Not assessed |
Neutral lipid-positive storage vacuoles likely do not account for the ichthyosiform phenotype in NLSDI, because these large, cytosolic inclusions eventually become entombed within corneocytes, where they likely are unavailable to influence extracellular functions, such as permeability barrier homeostasis or desquamation. Moreover, comparable cytosolic lipid droplets occur as a nonspecific response to toxic insults, as in many hyperplastic dermatoses (37–41). More pertinent instead to disease phenotype in NLSDI could be amorphous, lipid microinclusions that occur within epidermal LBs (33). These organelle contents are secreted, along with normal-appearing lamellar membranes, at the stratum granulosum (SG)/SC interface (33). Accordingly, LBs normally encapsulate several types of lipase activity (42–44), including the coactivator encoded by CGI-58/ABHD5 (26, 45). Therefore, the enzyme mutation and the lipid microinclusions in NLSDI are likely linked to disease pathogenesis through their colocalization within LBs.
Barrier function, assessed as abnormalities in TEWL, is markedly abnormal in NLSDI, with basal TEWL levels up to 3-fold higher than in age-matched, normal controls (35). These levels are comparable to those reported for other ichthyoses with a similar phenotype, such as nonbullous CIE and TGM1-negative LI (46, 47).
Recent studies suggest that it is the persistence of secreted, “unprocessed” TAG, coupled with decreased FFA caused by the lack of phospholipid precursors, that likely accounts for the permeability barrier abnormality in NLSDI (35). In normal epidermis, LBs are replete with lamellar membranes that show no evidence of nonlamellar discontinuities. After secretion, secreted lamellar contents transform into “mature” lamellar membrane structures that regulate permeability barrier homeostasis, which again fill the SC interstices (48). Thus, in normal human epidermis, a uniform lamellar phase that completely fills the SC interstices equates with permeability barrier competence (48). In NLSDI, LBs display lipid microinclusions that transform into electron-lucent, lipid-filled “clefts” after secretion (33). To delineate whether these clefts contain phase-separated (nonlamellar) lipid, we assessed tissue samples after ruthenium tetroxide postfixation, a method that allows for the visualization of hydrophobic lipid structures, such as the extracellular lamellar membranes (49), coupled with embedding in a lipid-retaining resin (LR White). Using this method, the clefts did not appear empty but rather filled with an amorphous, electron-dense material adjacent to arrays of lamellar membranes (35). Moreover, these new images provide additional insights into the pathogenesis of NLSDI, which can be ascribed to lamellar/nonlamellar phase separation within the SC interstices (Fig. 1). Phase separation occurs in lipid-based, membrane bilayers when the amount of nonpolar lipid exceeds the capability of this lipid to incorporate into polar lipid-based, lamellar membranes (50–52). These results suggest that ceramide (Cer)-based membrane bilayers of the SC interstices, like their phospholipid-based counterparts, display a limited capacity to incorporate non-polar species, such as triacylglycerols.
To assess whether an inhomogeneous extracellular matrix forms an inherently less effective permeability barrier than interstices that are uniformly replete with lamellar membranes, we perfused the SC of NLSDI with a water-soluble, electron-dense tracer, lanthanum nitrate. Whereas the interstices of normal human SC completely exclude water-soluble molecules, lanthanum permeates through nonlamellar domains in the extracellular spaces at all levels of the SC in NLSDI (35). In summary, these studies show that lamellar/nonlamellar phase separation underlies the permeability barrier abnormality in NLSDI.
ARCI
The ARCIs comprise a group of disorders of cornification with congenital presentation. On clinical, biochemical, and morphological grounds, it has long been recognized that this group is clinically heterogeneous, likely comprising multiple disease entities (53–56). Indeed, these patients reveal a remarkable diversity of underlying genetic mutations (57–65). Before this genetic diversity became known, the LI phenotype, with large dark, plate-like scaling, had been distinguished on clinical grounds from nonbullous congenital erythroderma (CIE or nonbullous CIE), with fine scaling involving flexural sites, and often with prominent erythema (54), but many intermediate phenotypes also have been described (5, 6).
Although a functional barrier abnormality is present in all ARCI subtypes studied to date (19, 46), structural and biochemical differences between the LI and the CIE phenotypes provided initial clues about the heterogeneity within this group of ichthyoses (54, 56, 66). The major distinguishing feature of the LI phenotype is abnormal cornified envelope cross-linking attributable to TGM1 deficiency, resulting in attenuated cornified envelopes (19). In contrast, the CIE phenotypes (in which cornified envelopes are normal) display prominent abnormalities in LBs and SC extracellular lamellar membranes. Although the number of LBs is increased in CIE, many of these organelles are smaller than normal and show abnormal internal organization (i.e., fragmented lipid lamellae), often giving them a vacuolated appearance (56). The SC of CIE individuals also retains large amounts of exogenous n-alkanes, whereas TAG and FFA levels decrease (54). Together, disorganized lamellar arrays and nonpolar nonlamellar/lamellar phase separation account for the barrier abnormality (56). However, in CIE, several variable ultrastructural features were observed only in subsets of patients. These variable findings include the following: 1) an absence of electron-lucent lamellae; 2) abnormal spacing and interruptions of lamellar structures; and 3) intracellular lipid droplets and vesicular complexes, within both the SC and the SG (10, 46, 56, 66, 67). An alternative classification of the variable ultrastructural findings in ARCI is widely applied in Europe: type I is characterized by abundant lipid droplets within corneocytes; type II shows polygonal clefts within the SC; type III shows vesicular and membranous structures in the SG; and type IV is characterized by lentiform swollen areas within corneocytes and perinuclear accumulation of curved membranes in the SG (55, 66). Because this classification preceded the use of ruthenium tetroxide to evaluate membrane structures, it is of limited utility.
Today, the variability of ARCI morphology can be explained by the genetic heterogeneity that is now becoming apparent, although some ultrastructural findings most likely reflect nonspecific sequelae of disturbed cornification. Subsequently, it became clear that newly discovered gene mutations do not always correlate well with or explain the observed clinical and morphological phenotypes (e.g., the LI phenotype is frequently, but not exclusively, caused by TGM1 deficiency). The same TGM1 mutation can cause both LI and CIE phenotypes, and the LI phenotype can result from mutations other than TGM1 (68–71).
Several of the newly discovered mutations causing ACRI govern the synthesis of enzymes directly involved in the production, transport, or assembly of lipid components of the SC (Table 1). In an intense, ongoing effort, detailed genotype-phenotype relationships, including structural correlations, are being established. Best studied for their consequences for the epidermal permeability barrier are mutations in ichthyin on chromosome 5q33, which putatively encodes for a transmembrane receptor. By electron microscopy, the SG of patients with ichthyin mutations contains many empty or partially filled vacuolar and vesicular structures, which are thought to represent defective LB (71). In one study, 85% of patients with this morphologic pattern were found to have mutations in ichthyin (71). Because ichthyin mutations do not result in decreased mRNA levels, the responsible mutations likely alter the function, rather than the expression levels, of the putative receptor.
It has been proposed that the endogenous ligands for the putative ichthyin receptor are hydroxyepoxyalcohols (69), presumably generated in normal epidermis (72) and reportedly esterified at high rates into phospholipids (73). Epidermal hydroxyepoxyalcohols are metabolic products of 12R-lipoxygenase (LOX) and hydroperoxide isomerase (epoxyalcohol synthase) eLOX3 (74, 75) (Fig. 2). Mutations in ALOX12 (for arachidonate lipoxygenase) and ALOXE3 on chromosome 17p13, which result in the complete loss of enzymatic activity as a result of abnormal protein folding, are relatively common (>10%) among patients with ARCI (60, 61, 70, 76). Thus, these enzymes and the putative ichthyin receptor may function in concert along the same metabolic pathway, catabolizing leukotriene derivatives of arachidonic acid to hydroxyepoxyalcohol end products, specifically 12(R)-hepoxilin A3 and 12(R)-hydroperoxy-eicosatetraenoic acid (75, 76) (Fig. 2). Additional evidence for the relevance of LOX deficiency for the permeability barrier derives from mouse models that display increased TEWL, resulting in death within 3–5 days after birth (77, 78). Ultrastructural examination of the SC of these animals revealed vesicular structures in the upper SG cell layers that are comparable to the structural abnormalities in the SC of human ARCI subjects with ichthyin mutations. In mouse epidermis, these findings also correlate with an increase in protein-bound, ester-linked lipid species (77). Corneocytes isolated from LOX-deficient animals are more fragile and show abnormal filaggrin processing, features that have not yet been identified in affected human skin.
Fig. 2.

Potential disruptions in peroxidated lipid pathways in autosomal recessive congenital ichthyosis. CYP4F22, cytochrome P450, family 4, subfamily F, polypeptide 22; FALDH, fatty aldehyde dehydrogenase; PPAR, peroxisome proliferator-activated receptor; 12R-HPETE, 12(R)-hydro-peroxyeicosatetraenoic acid; 12R-LOX, 12R-lipoxygenase.
The initial transformation of arachidonic acid into epoxyalcohols and the downstream effects on the putative ichthyin receptor have been proposed to provide a framework for several intermediate metabolic steps that could, when disturbed, cause an ARCI phenotype and permeability barrier abnormalities (14, 70) (Fig. 2). First, pedigrees with ARCI linked to the ALOX12/ALOXE3 chromosomal region on 17p13 but lacking mutations in these genes indicate that there may be at least one additional gene in this region coding for a protein within the same pathway (70). Second, in other ARCI kindreds, mutations in cytochrome P450, family 4, subfamily F, polypeptide 22 (Cyp4F22) on chromosome 19p12 encode a putative fatty acid ω-hydroxylase and may perturb a late enzymatic event in the epoxyalcohol oxidation and hydroxylation cascade (79). Yet, information on SC structure and barrier function is lacking in these patients. Third, fatty aldehyde dehy-drogenase, which is deficient in Sjögren-Larsson syndrome (SLS) (see below), could have the ability to oxidize trioxilin products within the above pathway. However, the prominent central nervous system abnormalities that are present in SLS, but lacking in the ARCI phenotypes, indicate that the pathophysiological consequences of blockade at this step are broader in scope. Moreover, differences in the cutaneous phenotype of SLS (a “lichenified” rather than a “scaly” pattern accompanied by prominent pruritus) suggest that other substrates may be affected. Finally, CGI-58/ABHD5, which is mutated in neutral lipid storage disease (see above), has also been proposed to function as an epoxide hydroxylase in the same biochemical pathway (79). However, there is no reason to suspect that the activities of this lipase are restricted to these epoxide metabolites. Indeed, labeling studies suggest a broader effect on acyl-lipid metabolism (see above). Finally, although a unitary pathway hypothesis always is attractive (70), it should be recalled that mutations in disparate genes, such as TGM1 (see above), cause identical phenotypes. More likely is the hypothesis that any derangement of epidermal lipid metabolism can provoke an ichthyosiform phenotype. Finally, it still remains to be seen how many distinct clinical, morphological, and biochemical disease subsets will be distinguished among these patients, or whether most will converge into a common phenotype.
Although a lack of peroxidated lipid metabolites may be the common pathogenetic basis in ARCI phenotypes that are not caused by TGM1 deficiency (Fig. 2), the mechanism whereby this abnormality provokes disease is unknown. The pathogenesis of the barrier abnormality could be related to essential fatty acid deficiency, in which a lack of substrate for the ω-esterification of Cers to acylceramide is known to provoke a barrier abnormality (80–83). Alternatively, some of the accumulating hydroxyepoxyalcohol substrates are potent and selective activators of peroxisome proliferator-activated receptor (PPAR) α (84), a ligand-activated nuclear hormone receptor with prodifferentiating and anti-inflammatory activity in the epidermis (85, 86) (Fig. 2). In addition, Cyp4F22 activity also likely generates potent PPARα agonists, because it is a homolog of the leukotriene B2-ω-hydroxylase and ω-hydroxylation of other eicosanoids enhances PPAR-activating properties (79, 87). Yet, the biological significance of this association remains unclear, because PPARα deficiency results only in transient developmental defects in fetal mouse epidermis (85), presumably as a result of redundancy in other epidermal nuclear hormone receptors. Finally, one or more of these metabolites could mobilize intracellular calcium, thereby altering permeability barrier homeostasis by downregulating LB secretion (88, 89). This last possibility is consistent with the LB secretory defect that has been described in preliminary studies of this group of ichthyoses (e.g., ichthyin mutations; see above).
OTHER DISORDERS OF NONPOLAR LIPID METABOLISM
SLS
Several other primary disorders of nonpolar lipid metabolism display an ichthyotic phenotype with additional systemic abnormalities (Table 2). In at least one of these disorders, SLS, lamellar/nonlamellar phase separation could provoke both a barrier abnormality and the distinctive clinical phenotype. SLS is a recessively inherited disorder of the brain and skin, attributable to the accumulation of free and esterified long-chain aliphatic alcohols (90) as a result of defective peroxisomal oxidation of long-chain aliphatic alcohols. A variety of mutations in the ALDH3A2 gene, encoding the microsomal enzyme fatty aldehyde dehydrogenase, have been described (91, 92). Patients with SLS display a characteristic triad of mental retardation, spastic diplegia or quadradiplegia, and ichthyosis (91, 93). The epidermal phenotype is quite characteristic, exhibiting a ridge or lichenified pattern with fine, brown desquamation and prominent pruritus, which may be caused by coaccumulation of the proinflammatory leukotriene metabolite, leukotriene B4 (91, 94). Although the pathogenesis of the putative barrier abnormality is still unknown, epidermal LB contents are abnormal (95) and extracellular lamellar bilayers exhibit structural abnormalities consistent with lamellar/nonlamellar phase separation (95, 96).
Disorders of peroxisomal fatty acid metabolism
In two recessively inherited, nonpolar disorders of peroxisomal lipid metabolism, Refsum disease (RD) and rhizomelic chondrodysplasia punctata (RCDP; OMIM 21508), similar pathomechanisms to SLS could be operative2 (Table 2). RD (OMIM 266500) is a rare disorder caused by a defect in the first step in peroxisomal β-oxidation of phytanic acid, a C16 saturated fatty acid with four additional methyl groups at C3, C7, C11, and C15 (96). This branched chain fatty acid is enriched in tissues of ruminant animals (97). Accumulation of phytanic acid, although characteristic of RD, is not pathogenic, because increased phytanic acid levels occur in other disorders of peroxisomal biogenesis, including SLS and RCDP (97). In RD, peroxisomal β-oxidation of phytanic acid is blocked by the presence of the methyl group at the 3-position. Mutations in the gene encoding phytanoyl-CoA hydroxylase (PAHX, PHYH) occur in up to 80% of RD patients (98), but some patients do not have PAHX mutations but rather mutations in peroxin 7 receptor (PEX7) (99). PEX7 mutations underlie the more severe phenotype, RCDP, in which severe skeletal defects predominate (99, 100). A mild ichthyosiform phenotype, albeit poorly described, can also be present. In all RD cases, multisystem accumulation of phytols, predominantly phytanic acid, occurs, sometimes in millimolar concentrations (97). Severely affected patients can die in childhood, but most do not become symptomatic until adolescence, from a disease complex that includes retinitis pigmentosum, deafness, cerebellar ataxia, anosmia, and ichthyosis (98). The initial symptom is often night blindness, which can progress to blindness. Death usually results from cardiac arrhythmia, but these as well as other disease symptoms improve with the implementation of a phytol-free diet (97, 98). The pathogenesis of the disease complex in RD could be explained in part by the high affinity of phytanic acid for the retinoid X receptor and PPARα (100, 101). Although purely speculative, the symptoms of RD mimic several features of hypervitaminosis A, which include visual, neurological, and desquamatory abnormalities. In addition, phytanic acid can induce apoptosis in cardiac and neuronal cells and can mobilize Ca2+ from mitochondrial stores (97). The relative roles of these mechanisms in disease pathogenesis remain unknown.
Disorders of distal sterologenesis with ichthyosiform phenotypes
Two multisystem syndromes with ichthyosis, X-linked dominant chondrodysplasia punctata type 2 (CDPX2) (Conradi-Hünermann-Happle syndrome; OMIM 302960) and CHILD syndrome (OMIM 308050), are caused by mutations in genes encoding enzymes of the postsqualene cholesterol biosynthetic pathway. CDPX2 is caused by mutations in the EBP (for emopamil binding protein) gene that encodes 3β-hydroxysteroid-Δ8,Δ7-isomerase, catalyzing the conversion of 8(9)-cholestenol to lathostero1 (102, 103), resulting in diagnostic deviations of the sterol precursors 8-dehydrocholesterol and 8(9)-cholestenol (104). Mutations either in EBP or in NAD(P)H steroid dehydrogenase-like protein, encoding a member of the enzyme complex that removes the C-4 methyl group in the next-most proximal step of the pathways, have been reported to underlie CHILD syndrome [104, 105; D. K. Grange et al., cited in (106)]. Given the close approximation of the metabolic blockages, the striking phenotypic similarities and genetic overlap are not surprising (reviewed in Ref. 106). Both are X-dominant, proposed male-lethal traits associated with asymmetric skeletal malformations and a variety of other deficits. Cutaneous features in CDPX2, the more common of the two conditions, are most striking in the neonate, in which linear bands of scaling are distributed in a morphogenic pattern (the lines of Blaschko), postulated to conform to regions in which the mutant X chromosome is the active X (107, 108), accompanied by a generalized erythroderma. The cutaneous features resolve after infancy, leaving atrophy (follicular atrophoderma and alopecia) and, in some instances, a mild ichthyosis on the extremities (108). Disease severity is dependent on both the specific mutations and the extent to which the mutant X chromosome is active in affected tissues (109–112). The resolution of the cutaneous phenotype presumably reflects the dilution of effects as a consequence of the diminished viability of keratinocytes bearing the mutant X (113). The cutaneous phenotype in CHILD syndrome differs in its distribution, which is typically limited to one side of the body (104, 105). Resolution also occurs, but usually it is only partial. The skeletal defects and internal organ involvement are also restricted to the same, usually right, side.
The pathophysiologic basis for the ichthyosiform phenotype, like the manifestations of the syndromes, is unclear. The multisystem malformations that characterize disorders of postsqualene sterologenesis have been attributed to the following: 1) deficiency of bulk cholesterol in membrane function; 2) toxic effects of accumulated precursors; and/or 3) developmental effects of altered Hedgehog pathway signaling (114). In skin, it seems likely that cholesterol deficiency per se is the major contributor, because a cutaneous phenotype does not occur in either Smith-Lemli-Opitz syndrome (OMIM 270400), attributable to 7-dehydrocholesterol reductase deficiency, or in hairless mice treated with the 7-hydrocholesterol inhibitor AY9944 (115). In contrast, blockade of the Δ24 reductase, converting desmosterol to cholesterol, through the inhibitors triparanol and 20,25-diazocholesterol, is associated with ichthyosis in both rodent models and human (115, 116). It is likely, therefore, that 7-dehydrocholesterol, but not desmosterol, can substitute for cholesterol in the formation of SC lamellar membranes.
Before the identification of primary sterologenesis defects in CDPX2 and CHILD syndromes, these disorders were thought to be related to the peroxisomal biogenesis disorders. Deficient peroxisomal function in cultured fibroblasts has been described in both CDPX2 and CHILD syndromes (113, 117–120) and in the murine homolog of EBP deficiency, the bare patches mouse (113), which displays cutaneous defects that, like the phenotype in CDPX2, resolve over time (113, 121). The clinical phenotypes of the postsqualene sterologenesis and peroxisome biogenesis disorders bear some striking resemblances (121), including skeletal defects (chondrodysplasia punc-tata), central nervous system and/or hepatic involvement, and ichthyosis in the PEX7 disorders (rhizomelic chondrodysplasia punctata) and adult RD. Although their contribution to cellular cholesterol synthesis overall is unclear, the localization of the postsqualene enzymes in the sterol biosynthetic pathway to peroxisomes likely explains these phenotypic convergences.
DISORDERS OF POLAR LIPID PROCESSING
RXLI
Molecular genetics and biochemistry
RXLI is caused by null mutations in the gene encoding the microsomal enzyme, steroid sulfatase (SSase) (122, 123). Because of its location on the distal tip of the short arm of the X chromosome (124–128), the SSase gene has been the subject of considerable research. The gene mutations/deletions in RXLI (129–133) provoke ichthyosiform skin changes, with occasional extracutaneous organ system involvement, as a result of contiguous gene syndromes (134–136).
SSase is a classic microsomal enzyme that further localizes to coated pits in the plasma membrane (137, 138), where it hydrolyzes the 3β-sulfate esters from both CSO4 and sulfated steroid hormones. In epidermis, SSase activity is low in the basal and spinous layers, whereas enzyme levels peak in the SG (10–20 times higher) and persist into the SC, where it is concentrated in membrane domains (2) (Fig. 3). In cytochemical studies, SSase activity localizes not only within the cytosol (i.e., microsomes) but also within LBs, and SSase is delivered to the interstices of the lower SC by LB secretion (2). Thus, SSase, like other lipid hydrolases that are involved in the extracellular processing of secreted polar lipids (48), uses the LB secretory system to reach sites where it participates in the regulation of permeability barrier homeostasis and desquamation.
Fig. 3.

How steroid sulfatase (SSase) deficiency leads to recessive X-linked ichthyosis. CE, cornified envelope; CLE, cornified lipid envelope; PKC, protein kinase C; TGM1, transglutaminase 1.
As a result of enzyme deficiency in RXLI, CSO4 accumulates in the epidermis (139–141), in erythrocyte cell membranes (140, 142), and in the LDL (β-lipoprotein) fraction of plasma, where it produces diagnostic alterations in electrophoretic mobility (140, 143). But CSO4 levels in epidermis/SC are 1 order of magnitude higher than the levels in blood (140, 142, 144), likely explaining the prominence of skin versus other organ involvement in RXLI. Normally, CSO4 constitutes ~5% of the total lipid of human SG, declining to ~1% of lipid mass in the outer SC, through ongoing hydrolysis of CSO4 by SSase during SC transit (145, 146) (Fig. 3). However, as a result of absent SSase activity, the SC typically contains 10–12% CSO4 (by dry weight) in RXLI (144). Like other SC lipids, CSO4 is concentrated in the SC interstices, but in contrast to other barrier lipid precursors, it is not delivered by LB secretion (147). Rather, its mode of delivery to the SC interstices can be explained by its amphilicity, which allows it to diffuse readily across the cell membrane (148). Thus, in the absence of a lipid milieu within corneocytes, CSO4 likely partitions preferentially to the highly hydrophobic, extracellular domains of the SC.
CSO4 “cycle” and its regulatory significance
Because cholesterol sulfotransferase activity, which generates CSO4, predominates in the lower nucleated cell layers of the epidermis, whereas SSase peaks in the outer epidermis, Epstein et al. (149) proposed that an “epidermal CSO4” exists in the epidermis, in which cholesterol is first sulfated in the lower epidermis and then desulfated back to cholesterol in outer epidermal layers (Fig. 4). Disruption of this CSO4 cycle accounts for both the abnormal desquamation and the permeability barrier abnormality in RXLI (see below).
Fig. 4.

Consequences of the epidermal cholesterol sulfate cycle for normal skin. Chol, cholesterol; SULT2B1b, cholesterol sulfotransferase.
Sulfation of cholesterol by cholesterol sulfotransferase is a step that is intimately linked to keratinization (150–152), including cornification in the epidermis (153–155). For example, CSO4 levels are several orders of magnitude higher in keratinizing than in mucosal epithelia (150), and reversal of keratinization, through the induction of mucous metaplasia in keratinizing epithelia (e.g., by the application of exogenous retinoids), dramatically reduces tissue CSO4 levels (156, 157). Moreover, cholesterol sulfotransferase expression is linked to epidermal development in utero (158, 159), and CSO4 levels increase late in fetal development (160).
CSO4 is a potent regulatory molecule in many extracutaneous tissues (161, 162). For example, whereas retinoic acid inhibits cholesterol sulfotransferase expression, PPARα and liver X receptor activators stimulate its expression (163). CSO4 stimulates epidermal differentiation by at least two related mechanisms (Fig. 4): 1) it activates the η isoform of protein kinase C (164–166), which in turn stimulates the phosphorylation of differentiation-linked proteins, assessed as increased cornified envelope formation (167); and 2) it is also a transcriptional regulator of proteins involved in cornified envelope formation, such as TGM1 and involucrin, operating through an activator protein-1 binding site in the promoter region (168, 169). It is likely that these two mechanisms are linked, as shown in Fig. 4: protein kinase C activation by CSO4 could phosphorylate activator protein-1, leading to enhanced transcriptional regulation of TGM1 and involucrin. Together, these observations provide an explanation for the biological significance of the CSO4 cycle.
Basis for the permeability barrier abnormality in RXLI
Patients with RXLI display only a minimal basal barrier abnormality (170, 171) but a pronounced delay in recovery kinetics after acute perturbations (171), suggesting that excess CSO4 destabilizes permeability barrier homeostasis. To assess this hypothesis, we performed both in vitro and in vivo studies, showing, first, that CSO4 fails to form eutectic mixtures with other SC lipids, with excess CSO4 apparently segregating within nonlamellar domains in model lipid mixtures (51) and in diseased SC (52). Accordingly, ultrastructural images of SC in RXLI show frequent but focal nonlamellar domains, with disruption of the extracellular lamellae (2, 172). Yet, the barrier abnormality could be attributable not only to excess CSO4 but also to decreased cholesterol (the cholesterol content of the SC in RXLI is reduced by ~50%) (144), and a discrete decrease in cholesterol produces abnormal extracellular lamellar membranes (173). To varying extents, the decrease in cholesterol in RXLI could be caused by (Fig. 3) the following: 1) reduced generation of cholesterol from CSO4 as a result of the enzyme deficiency (172, 174); and/or 2) CSO4-mediated inhibition of HMG-CoA reductase, the rate-limiting enzyme of cholesterol synthesis (174). CSO4, like oxygenated sterols, is a potent inhibitor of cholesterol synthesis (174), consistent with the reduced levels of cholesterol in the SC of RXLI (144). Finally, CSO4 inhibits the TGM1-mediated attachment of ω-hydroxyceramides to the cornified envelope in vitro, a step that forms the corneocyte lipid envelope (175). Yet, the cornified envelope/corneocyte-bound lipid envelope scaffold does not appear abnormal in RXLI (P. M. Elias et al., unpublished observations). Thus, the dominant mechanisms that account for the barrier abnormality in RXLI appear to be lamellar/nonlamellar phase separation of excess CSO4 and reduced cholesterol content of the SC lamellar membranes (2).
Mechanisms proposed to cause abnormal desquamation in RXLI
Kinetic studies have demonstrated that the hyperkeratosis in RXLI reflects delayed desquamation (12, 176). The basis for this classic, retention type of ichthyosis is the persistence of corneodesmosomes at all levels of the SC (cited in Ref. 2). Two key serine proteases, kallikrein (KLK) 7 [stratum corneum chymotryptic enzyme (SCCE)] and KLK5 [stratum corneum tryptic enzyme (SCTE)], are primary mediators of desquamation that degrade corneo-desmosomes in vitro (177). CSO4 may increase SC retention through the known ability of this lipid to function as a serine protease inhibitor (2, 12). Moreover, although the activities of these enzymes are restricted by the acidic pH of normal SC (SCCE and SCTE exhibit neutral pH optima), the pH of the SC in RXLI is even lower than that of normal SC (178). As a result, serine protease activity in RXLI is reduced below the levels in normal SC (2). An unrelated mechanism, which could contribute to increased SC cohesion in RXLI, posits that Ca2+, if present in sufficient quantities, could stabilize highly anionic SO4 groups (from the persistence of CSO4) on adjacent lamellar bilayers (179). Indeed, CSO4-containing lipo-somes aggregate avidly in the presence of calcium (180, 181). Moreover, the lower SC in RXLI demonstrates abundant Ca2+ in extracellular domains, which preferentially localize along the external faces of opposing corneodesmosomes (2). Thus, the delayed degradation of corneodesmosomes in RXLI could be attributable in part to leakage of Ca2+ into the lower SC (caused by the barrier defect), with the formation of Ca2+ bridges between adjacent corneodesmosomes (2).
Sphingolipidoses
In its most severe form, the sphingolipidosis, type 2 GD can present with a neonatal “collodion baby” phenotype ichthyosis (182–184). Studies in patients, transgenic murine analogs, and inhibitor-based models have shown that substantial reductions in lysosomal β-glucocerebrosidase (EC 3.2.1.45) can provoke GD, a profound barrier abnormality (3, 183); in contrast, diminished activity of another key Cer-generating enzyme, acidic sphingomyelinase, the causative enzyme of Niemann-Pick disease, although also provoking barrier abnormalities (4, 185), rarely, if ever, causes ichthyosiform skin changes. The distinct cutaneous phenotypes may reflect the generation of the full spectrum of all nine human SC Cer species from glucosylceramides, whereas acidic sphingomyelinase generates only two SC Cers from corresponding sphingomyelin precursors (186). Cers constitute 50% of the extracellular lamellar membranes in SC; as such, they are absolutely required for normal barrier function (186). When β-glucocerebrosidase levels are very low (<90% of normal), ichthyotic signs can emerge (182–184, 187), attributable to both hyperplasia consequent to a severe permeability barrier abnormality (188) and direct mitogenic activity of excess glucosylceramides (189) (Fig. 5). The persistence of glucosylceramides in extracellular lamellar membranes also imparts an “immature” appearance that is quite distinctive and potentially diagnostic of GD (3, 187). Decreased Cer in relation to cholesterol and FFA in GD (185, 188) also likely results again in lamellar/nonlamellar phase separation caused by altered molar ratios of the three key lipids, analogous to the effects of excess CSO4 in RXLI (2) (see above). This conclusion is based upon the observation that topical Cers normalize barrier function in severe glucocerebrosidase deficiency (4), but the cause of the barrier abnormality in GD is more complex, because topical Cers do not normalize barrier function in the face of enzymatic blockade (183). Hence, it is likely that both decreased Cers and excess glucosylceramides contribute to the barrier abnormality in GD (Fig. 5). The metabolic production and fate of epidermal Cer is the topic of a subsequent review in this series by Drs. W. Holleran and Y. Uchida.
Fig. 5.
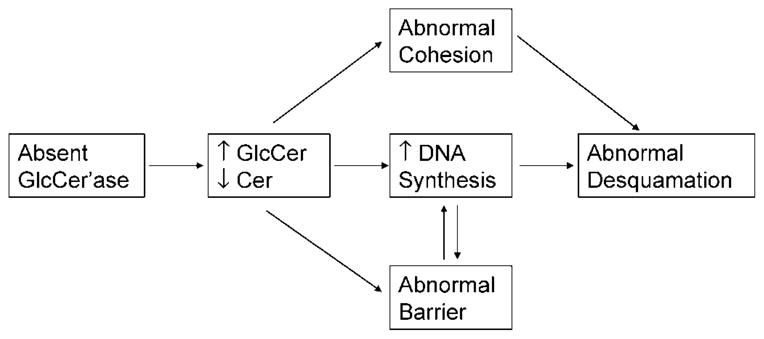
Potential pathogenic mechanisms in Gaucher disease. Cer, ceramide; GlcCer, glucocerebroside; GlcCer’ase, glucocerebrosidase.
FAILURE OF LB ASSEMBLY OR SECRETION
HI
HI is a rare, recessively inherited disorder that presents at birth with a thick, plate-like encasement of the entire skin surface that has life-threatening consequences. In neonates who survive the perinatal period, the plate-like encasement is shed, and the phenotype shifts to a severe ichthyosiform erythroderma (9, 190). Transcutaneous water loss rates remain extremely high (47), explaining at least in part the prominent epidermal hyperplasia and hyperkeratosis that are presumably driven by the permeability barrier abnormality, as described above. The barrier defect in HI is a primary disorder of the LB secretory system (191). Specifically, LBs with replete lamellar contents are found only rarely in HI. Instead, the cytosol of the SG layer contains numerous, small vesicular structures (192), which presumably represent nascent LBs that lack internal contents. It is likely that these nascent organelles undergo exocytosis, because the cornified lipid envelope, a structure thought to derive from the fusion of LB with the plasma membrane, is normal in HI (192). Nevertheless, the extracellular spaces of the SC are largely devoid of lamellar membranes (192).
ABCs are a large group of proteins that mediate the transport of a variety of different substrates across cellular membranes. These proteins contain two transmembrane sequences and two ATP binding domains, which undergo conformational changes that facilitate first the binding and then the dissociation of attached lipids (193). To date, 48 ABC genes have been identified, which have been further divided into seven subfamilies, based on sequence homology and supramolecular organization of the nucleotide binding folds (194–197). The ABCA subfamily comprises 12 functional transporters that all mediate lipid transport (198), with the exception of one pseudogene (ABCA11). ABCA transporters function as components of highly specialized cellular lipid-transporting organelles in major physiological systems, in which defects cause severe inherited diseases in the cardiovascular, visual, and respiratory systems. Accordingly, gene mutations of ABCA proteins are linked to several recessive disorders of lipid metabolism, including ABCA1 [Tangier disease (156, 157)], ABCA4 [Stargardt disease (158–160)], and surfactant deficiency in newborns, which has been linked to ABCA3 deficiency (161, 162). Further study has revealed that ABCA3 regulates lipid transport into the LBs of alveolar type 2 cells (199, 200).
ABCA12 is a recently characterized member of the ABC transporter superfamily, which serves as a putative transporter for glucosylceramides from the Golgi apparatus (201) into epidermal LBs. In HI [and in some cases of ARCI with a LI phenotype (202)], truncation or deletion mutations in both alleles of the gene that encodes ABCA12 (203) result in a failure to deliver newly synthesized glucosylceramides into nascent LBs (194, 204). As a result, few if any lamellar lipids are delivered to the SC interstices (192), and as noted above, a profound barrier abnormality results (205). Recent studies in HI keratinocytes have demonstrated not only defective glucosylceramide transport into LBs but also that corrective transfer of the ABCA12 gene into HI keratinocytes restores normal glucosylceramide loading into LBs (194). In those HI patients with residual ABCA12 expression, it is possible that topical treatment with either PPARγ or PPARδ activators could be beneficial, because our recent studies show that these two nuclear hormone receptors upregulate ABCA12 expression in normal keratinocytes (198). Mutations in the lipid transporter ABCA12 place HI into a disease spectrum with ARCI (202). Although in HI the genetic ABCA12 abnormalities are truncations or deletions, the ABCA12 mutations reported in ARCI to date have been solely missense mutations (202).
Because HI is characterized not only by a profound barrier abnormality but also by striking hyperkeratosis (thickening of the SC), the lipid transporter defect likely underlies the desquamation abnormality as well, but by an indirect mechanism. Because lipid delivery to LBs is required for the subsequent or concurrent importation of proteins into these organelles (206), it is likely that a failure of lipid delivery also impairs the delivery of hydrolytic enzymes to LBs in HI; therefore, little or no enzymes are delivered to the SC interstices (Fig. 6). Because an array of LB-derived proteases is required for normal desquamation (207–209), the failure of protease delivery could result in corneodesmosome retention, explaining (along with the intense hyperplastic response to the barrier abnormality) the extreme hyperkeratosis in neonates with HI.
Fig. 6.
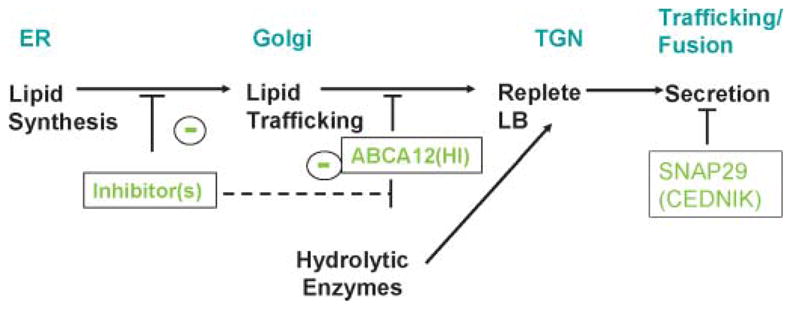
Protein delivery to lamellar bodies is dependent upon prior lipid deposition: sites of blockade in harlequin ichthyosis and cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma syndrome. ER, endoplasmic reticulum; TGN, trans-Golgi network.
Cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma syndrome
A noncongenital neurocutaneous syndrome with microcephaly, mental retardation, generalized ichthyosis, and palmoplantar keratoderma was recently ascribed to mutations in the SNAP29 gene, encoding for the SNARE29 protein involved in intracellular vesicle fusion (210). Aside from normal-appearing LBs, the epidermis of these patients exhibits numerous vesicular structures of varying size in the SG and SC layers that contain glucosylceramide, KLK5, and KLK7 (210). As the SNARE proteins are known to mediate neuromediator secretion (211), SNAP29 mutations could cause a permeability barrier abnormality by impairing LB secretion. The defect is possibly limited to a subset of LBs in the skin, explaining the selective enrichment of glucosylceramide, KLK5, and KLK7 in the retained vesicular structures seen by electron microscopy.
SYSTEMIC CONSEQUENCES OF BARRIER ABNORMALITIES IN THE ICHTHYOSES
The importance of calories lost through evaporation has been long recognized in the treatment of children with thermal burns and in premature infants who have immature skin barriers (212), but this factor has not been addressed previously in children with extensive inflammatory or genetic skin diseases. Short stature has been reported in some ichthyoses, such as Netherton syndrome (213), HI (214), and trichothiodystrophy (214–216), but growth failure is present at times in other forms of ichthyosis, suggesting that common pathogenic mechanism(s) could be operative. Although epidermal inflammation and hyperproliferation have been proposed to explain growth failure in adults with exfoliative erythroderma (217), negative nitrogen balance does not occur until losses exceed 17 g/m2/day (218). Therefore, nutrient drain from a hyperplastic epidermis alone is unlikely to account for the growth failure in these children. Alternatively, recent studies have shown that caloric losses from an impaired permeability barrier is the most likely cause of growth failure in severe ichthyosis phenotypes (47). Because transcutaneous evaporation is necessarily accompanied by a loss of heat (0.58 kcal/ml) (219), excessive rates of TEWL can constitute a significant caloric drain. All of these pediatric subjects displayed impaired barrier function with a marked increase in TEWL rates, resulting in large daily volumes of evaporative water loss, but TEWL rates ranged widely among study patients, as may be expected in view of the genetic heterogeneity included under the umbrella term “ichthyosis” (220).
The number of kilocalories lost from daily total TEWL ranged from 84 to 1,015 kcal (8–42 kcal/kg/day, with a mean of 433 ± 272 kcal/day) in this cohort, in contrast to expected rates of 41 to 132 kcal/day for age-matched children with competent barriers. In patients with moderate to severe barrier abnormalities, the consequent caloric drain from heat evaporation appeared sufficient to account for growth failure. Moreover, and alternatively, evaporative caloric losses could be compounded by caloric expenditures from cutaneous hyperplasia, chronic inflammation, which would be expected to increase metabolic rates, and/or anorexia accompanying systemic inflammation. Indeed, in the subject in whom resting energy expenditure was measured, it was 19% or higher than predicted; patients with the highest rates of TEWL also displayed the highest resting expenditure, suggesting that the severity of the barrier defect correlates with increased metabolic demands. Some patients were in positive caloric balance at the time of study, but they had dropped below normal growth patterns early in life (221). Hence, their current positive caloric balance likely reflected that they had now reached a steady state of growth, but they remained below normal body weights. A significant number of these patients were in negative energy balance, suggesting how precariously these patients maintain energy balance.
As noted above, both possession of the correct type and quantity of lipid and their organization into lamellar sheets are required for the formation of a competent barrier (222–224). Indeed, ultrastructural assessment of permeability barrier-related structures predicted the severity of the functional defect in ichthyosis patients with growth failure (47). The most severe barrier defects were observed in patients with HI and Netherton syndrome, and assessment of the LB secretory system and evaluation of cutaneous barrier function by measurement of TEWL correlated well with the magnitude of caloric drain from cutaneous water losses in these patients.
Acknowledgments
Our work on the pathogenesis of the ichthyoses has been supported by National Institutes of Health Grants AR-19098 and AR-39448 and by the Medical Research Service, Department of Veterans Affairs. Ms. Joan Wakefield provided superb editorial assistance, and Dr. Kenneth R. Feingold critically reviewed the manuscript.
Abbreviations
- ALOX
arachidonate lipoxygenase
- ARCI
autosomal recessive congenital ichthyosis
- CDPX2
X-linked dominant chondrodysplasia punctata type 2
- Cer
ceramide
- CHILD
congenital hemidysplasia with ichthyosiform erythroderma and limb defects
- CIE
congenital ichthyosiform erythroderma
- CSO4
cholesterol sulfate
- GD
Gaucher disease
- HI
harlequin ichthyosis
- KLK
kallikrein
- LB
lamellar body
- LI
lamellar ichthyosis
- LOX
12R-lipoxygenase
- NLSDI
neutral lipid storage disease with ichthyosis
- OMIM
Online Mendelian Inheritance in Man
- PPAR
peroxisome proliferator-activated receptor
- RCDP
rhizomelic chondrodysplasia punctata
- RD
Refsum disease
- RXLI
recessive X-linked ichthyosis
- SSase
steroid sulfatase
- SC
stratum corneum
- SCCE
stratum corneum chymotryptic enzyme
- SCTE
stratum corneum tryptic enzyme
- SG
stratum granulosum
- SLS
Sjögren-Larsson syndrome
- TAG
triacylglyceride
- TEWL
transepidermal water loss
- TGM1
transglutaminase 1
Footnotes
An additional pathomechanism could also be operative in CHILD (for congenital hemidysplasia with ichthyosiform erythroderma and limb defects) and Conradi-Hünermann syndromes; that is, an accumulation of distal sterol precursors (7-dehydrocholesterol/zymosterol) could result in lamellar membranes that are deficient in cholesterol. Cholesterol is one of the key lipids (with Cers and free fatty acids) that are required to form mature lamellar membranes, and such cholesterol-deficient membranes provide a suboptimal barrier (Ref. 1; p. 74).
References
- 1.Feingold KR. The regulation and role of epidermal lipid synthesis. Adv Lipid Res. 1991;24:57–82. doi: 10.1016/b978-0-12-024924-4.50007-9. [DOI] [PubMed] [Google Scholar]
- 2.Elias PM, Crumrine D, Rassner U, Hachem JP, Menon GK, Man W, Choy MH, Leypoldt L, Feingold KR, Williams ML. Basis for abnormal desquamation and permeability barrier dysfunction in RXLI. J Invest Dermatol. 2004;122:314–319. doi: 10.1046/j.1523-1747.2003.22258.x. [DOI] [PubMed] [Google Scholar]
- 3.Holleran WM, Ziegler SG, Goker-Alpan O, Eblan MJ, Elias PM, Schiffmann R, Sidransky E. Skin abnormalities as an early predictor of neurologic outcome in Gaucher disease. Clin Genet. 2006;69:355–357. doi: 10.1111/j.1399-0004.2006.00589.x. [DOI] [PubMed] [Google Scholar]
- 4.Schmuth M, Man MQ, Weber F, Gao W, Feingold KR, Fritsch P, Elias PM, Holleran WM. Permeability barrier disorder in Niemann-Pick disease: sphingomyelin-ceramide processing required for normal barrier homeostasis. J Invest Dermatol. 2000;115:459–466. doi: 10.1046/j.1523-1747.2000.00081.x. [DOI] [PubMed] [Google Scholar]
- 5.Williams ML. Ichthyosis: mechanisms of disease. Pediatr Dermatol. 1992;9:365–368. doi: 10.1111/j.1525-1470.1992.tb00632.x. [DOI] [PubMed] [Google Scholar]
- 6.Williams ML. Epidermal lipids and scaling diseases of the skin. Semin Dermatol. 1992;11:169–175. [PubMed] [Google Scholar]
- 7.DiGiovanna JJ, Robinson-Bostom L. Ichthyosis: etiology, diagnosis, and management. Am J Clin Dermatol. 2003;4:81–95. doi: 10.2165/00128071-200304020-00002. [DOI] [PubMed] [Google Scholar]
- 8.Oji V, Traupe H. Ichthyoses: differential diagnosis and molecular genetics. Eur J Dermatol. 2006;16:349–359. [PubMed] [Google Scholar]
- 9.Williams ML, Elias PM. From basketweave to barrier. Unifying concepts for the pathogenesis of the disorders of cornification. Arch Dermatol. 1993;129:626–629. doi: 10.1001/archderm.129.5.626. [DOI] [PubMed] [Google Scholar]
- 10.Bouwstra JA, Ponec M. The skin barrier in healthy and diseased state. Biochim Biophys Acta. 2006;1758:2080–2095. doi: 10.1016/j.bbamem.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 11.Williams ML, Elias PM. The extracellular matrix of stratum corneum: role of lipids in normal and pathological function. Crit Rev Ther Drug Carrier Syst. 1987;3:95–122. [PubMed] [Google Scholar]
- 12.Williams ML. Lipids in normal and pathological desquamation. Adv Lipid Res. 1991;24:211–262. doi: 10.1016/b978-0-12-024924-4.50012-2. [DOI] [PubMed] [Google Scholar]
- 13.Schmuth M, Gruber R, Elias PM, Williams ML. Ichthyosis update: towards a function-driven model of pathogenesis of the disorders of cornification and the role of corneocyte proteins in these disorders. Adv Dermatol. 2007;23:231–256. doi: 10.1016/j.yadr.2007.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bouwstra JA, Ponec M. The skin barrier in healthy and diseased state. Biochim Biophys Acta. 2006;1758:2080–2095. doi: 10.1016/j.bbamem.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 15.Elias PM. Stratum corneum defensive functions: an integrated view. J Invest Dermatol. 2005;125:183–200. doi: 10.1111/j.0022-202X.2005.23668.x. [DOI] [PubMed] [Google Scholar]
- 16.Elias PM, Feingold KR. Permeability barrier homeostasis. In: Elias PM, Feingold KR, editors. Skin Barrier. Taylor & Francis; New York: 2006. pp. 337–362. [Google Scholar]
- 17.Akiyama M. Pathomechanisms of harlequin ichthyosis and ABCA transporters in human diseases. Arch Dermatol. 2006;142:914–918. doi: 10.1001/archderm.142.7.914. [DOI] [PubMed] [Google Scholar]
- 18.Schmuth M, Fluhr JW, Crumrine DC, Uchida Y, Hachem JP, Behne M, Moskowitz DG, Christiano AM, Feingold KR, Elias PM. Structural and functional consequences of loricrin mutations in human loricrin keratoderma (Vohwinkel syndrome with ichthyosis) J Invest Dermatol. 2004;122:909–922. doi: 10.1111/j.0022-202X.2004.22431.x. [DOI] [PubMed] [Google Scholar]
- 19.Elias PM, Schmuth M, Uchida Y, Rice RH, Behne M, Crumrine D, Feingold KR, Holleran WM. Basis for the permeability barrier abnormality in lamellar ichthyosis. Exp Dermatol. 2002;11:248–256. doi: 10.1034/j.1600-0625.2001.110308.x. [DOI] [PubMed] [Google Scholar]
- 20.Schmuth M, Yosipovitch G, Williams ML, Weber F, Hintner H, Ortiz-Urda S, Rappersberger K, Crumrine D, Feingold KR, Elias PM. Pathogenesis of the permeability barrier abnormality in epidermolytic hyperkeratosis. J Invest Dermatol. 2001;117:837–847. doi: 10.1046/j.0022-202x.2001.01471.x. [DOI] [PubMed] [Google Scholar]
- 21.Elias PM, Feingold KR. Does the tail wag the dog? Role of the barrier in the pathogenesis of inflammatory dermatoses and therapeutic implications. Arch Dermatol. 2001;137:1079–1081. [PubMed] [Google Scholar]
- 22.Elias PM. Stratum corneum architecture, metabolic activity and interactivity with subjacent cell layers. Exp Dermatol. 1996;5:191–201. doi: 10.1111/j.1600-0625.1996.tb00117.x. [DOI] [PubMed] [Google Scholar]
- 23.Elias PM, Wood LC, Feingold KR. Epidermal pathogenesis of inflammatory dermatoses. Am J Contact Dermat. 1999;10:119–126. [PubMed] [Google Scholar]
- 24.Pujol RM, Gilaberte M, Toll A, Florensa L, Lloreta J, Gonzalez-Ensenat MA, Fischer J, Azon A. Erythrokeratoderma variabilis-like ichthyosis in Chanarin-Dorfman syndrome. Br J Dermatol. 2005;153:838–841. doi: 10.1111/j.1365-2133.2005.06828.x. [DOI] [PubMed] [Google Scholar]
- 25.Lefevre C, Jobard F, Caux F, Bouadjar B, Karaduman A, Heilig R, Lakhdar H, Wollenberg A, Verret JL, Weissenbach J, et al. Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. Am J Hum Genet. 2001;69:1002–1012. doi: 10.1086/324121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akiyama M, Sawamura D, Nomura Y, Sugawara M, Shimizu H. Truncation of CGI-58 protein causes malformation of lamellar granules resulting in ichthyosis in Dorfman-Chanarin syndrome. J Invest Dermatol. 2003;121:1029–1034. doi: 10.1046/j.1523-1747.2003.12520.x. [DOI] [PubMed] [Google Scholar]
- 27.Ben Selma Z, Yilmaz S, Schischmanoff PO, Blom A, Ozogul C, Laroche L, Caux F. A novel S115G mutation of CGI-58 in a Turkish patient with Dorfman-Chanarin syndrome. J Invest Dermatol. 2007;127:2273–2276. doi: 10.1038/sj.jid.5700860. [DOI] [PubMed] [Google Scholar]
- 28.Fischer J, Lefevre C, Morava E, Mussini JM, Laforet P, Negre-Salvayre A, Lathrop M, Salvayre R. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat Genet. 2007;39:28–30. doi: 10.1038/ng1951. [DOI] [PubMed] [Google Scholar]
- 29.Lass A, Zimmermann R, Haemmerle G, Riederer M, Schoiswohl G, Schweiger M, Kienesberger P, Strauss JG, Gorkiewicz G, Zechner R. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman syndrome. Cell Metab. 2006;3:309–319. doi: 10.1016/j.cmet.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 30.Williams ML, Coleman RA, Placezk D, Grunfeld C. Neutral lipid storage disease: a possible functional defect in phospholipid-linked triacylglycerol metabolism. Biochim Biophys Acta. 1991;1096:162–169. doi: 10.1016/0925-4439(91)90055-e. [DOI] [PubMed] [Google Scholar]
- 31.Igal RA, Coleman RA. Neutral lipid storage disease: a genetic disorder with abnormalities in the regulation of phospholipid metabolism. J Lipid Res. 1998;39:31–43. [PubMed] [Google Scholar]
- 32.Mao-Qiang M, Feingold KR, Jain M, Elias PM. Extracellular processing of phospholipids is required for permeability barrier homeostasis. J Lipid Res. 1995;36:1925–1935. [PubMed] [Google Scholar]
- 33.Elias PM, Williams ML. Neutral lipid storage disease with ichthyosis. Defective lamellar body contents and intracellular dispersion. Arch Dermatol. 1985;121:1000–1008. [PubMed] [Google Scholar]
- 34.Williams ML, Koch TK, McDonnell JJ, Frost P, Epstein LB, Grizzard WS, Epstein CH, Opitz JM, Reynolds JF. Ichthyosis and neutral-lipid storage disease. Am J Med Genet. 1985;20:711–726. doi: 10.1002/ajmg.1320200417. [DOI] [PubMed] [Google Scholar]
- 35.Demerjian M, Crumrine DA, Milstone LM, Williams ML, Elias PM. Barrier dysfunction and pathogenesis of neutral lipid storage disease with ichthyosis (Chanarin-Dorfman syndrome) J Invest Dermatol. 2006;126:2032–2038. doi: 10.1038/sj.jid.5700332. [DOI] [PubMed] [Google Scholar]
- 36.Solomon C, Bernier L, Germain L, Dufour R, Davignon J. Severe oily ichthyosis in monozygotic twins mimicking Chanarin-Dorfman syndrome but not associated with a mutation of the CGI58 gene. Arch Dermatol. 2006;142:402–403. doi: 10.1001/archderm.142.3.402. [DOI] [PubMed] [Google Scholar]
- 37.Zaynoun ST, Aftimos BG, Tenekjian KK, Bahuth N, Kurban AK. Extensive pityriasis alba: a histological histochemical and ultrastructural study. Br J Dermatol. 1983;108:83–90. doi: 10.1111/j.1365-2133.1983.tb04582.x. [DOI] [PubMed] [Google Scholar]
- 38.Johnson BL, Kramer EM, Lavker RM. The keratotic tumors of Cowden’s disease: an electron microscopic study. J Cutan Pathol. 1987;14:291–298. doi: 10.1111/j.1600-0560.1987.tb00502.x. [DOI] [PubMed] [Google Scholar]
- 39.Kanerva L. Electron microscopy of the effects of dithranol on healthy and on psoriatic skin. Am J Dermatopathol. 1990;12:51–62. doi: 10.1097/00000372-199002000-00008. [DOI] [PubMed] [Google Scholar]
- 40.el-Shoura SM, Tallab TM. Richner-Hanhart’s syndrome: new ultrastructural observations on skin lesions of two cases. Ultrastruct Pathol. 1997;21:51–56. doi: 10.3109/01913129709023247. [DOI] [PubMed] [Google Scholar]
- 41.Monteiro-Riviere NA, Inman AO, Riviere JE. Skin toxicity of jet fuels: ultrastructural studies and the effects of substance P. Toxicol Appl Pharmacol. 2004;195:339–347. doi: 10.1016/j.taap.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 42.Menon GK, Ghadially R, Williams ML, Elias PM. Lamellar bodies as delivery systems of hydrolytic enzymes: implications for normal and abnormal desquamation. Br J Dermatol. 1992;126:337–345. doi: 10.1111/j.1365-2133.1992.tb00675.x. [DOI] [PubMed] [Google Scholar]
- 43.Elias PM, Cullander C, Mauro T, Rassner U, Komuves L, Brown BE, Menon GK. The secretory granular cell: the outermost granular cell as a specialized secretory cell. J Investig Dermatol Symp Proc. 1998;3:87–100. doi: 10.1038/jidsymp.1998.20. [DOI] [PubMed] [Google Scholar]
- 44.Rassner U, Feingold KR, Crumrine DA, Elias PM. Coordinate assembly of lipids and enzyme proteins into epidermal lamellar bodies. Tissue Cell. 1999;31:489–498. doi: 10.1054/tice.1999.0050. [DOI] [PubMed] [Google Scholar]
- 45.Yamaguchi T, Omatsu N, Matsushita S, Osumi T. CGI-58 interacts with perilipin and is localized to lipid droplets. Possible involvement of CGI-58 mislocalization in Chanarin-Dorfman syndrome. J Biol Chem. 2004;279:30490–30497. doi: 10.1074/jbc.M403920200. [DOI] [PubMed] [Google Scholar]
- 46.Lavrijsen AP, Bouwstra JA, Gooris GS, Weerheim A, Bodde HE, Ponec M. Reduced skin barrier function parallels abnormal stratum corneum lipid organization in patients with lamellar ichthyosis. J Invest Dermatol. 1995;105:619–624. doi: 10.1111/1523-1747.ep12323752. [DOI] [PubMed] [Google Scholar]
- 47.Moskowitz DG, Fowler AJ, Heyman MB, Cohen SP, Crumrine D, Elias PM, Williams ML. Pathophysiologic basis for growth failure in children with ichthyosis: an evaluation of cutaneous ultrastructure, epidermal permeability barrier function, and energy expenditure. J Pediatr. 2004;145:82–92. doi: 10.1016/j.jpeds.2004.03.052. [DOI] [PubMed] [Google Scholar]
- 48.Elias PM, Menon GK. Structural and lipid biochemical correlates of the epidermal permeability barrier. Adv Lipid Res. 1991;24:1–26. doi: 10.1016/b978-0-12-024924-4.50005-5. [DOI] [PubMed] [Google Scholar]
- 49.Swartzendruber DC, I, Burnett H, Wertz PW, Madison KC, Squier CA. Osmium tetroxide and ruthenium tetroxide are complementary reagents for the preparation of epidermal samples for transmission electron microscopy. J Invest Dermatol. 1995;104:417–420. doi: 10.1111/1523-1747.ep12665909. [DOI] [PubMed] [Google Scholar]
- 50.Bangham AD. Lipid bilayers and biomembranes. Annu Rev Biochem. 1972;41:753–776. doi: 10.1146/annurev.bi.41.070172.003541. [DOI] [PubMed] [Google Scholar]
- 51.Rehfeld SJ, Williams ML, Elias PM. Interactions of cholesterol and cholesterol sulfate with free fatty acids: possible relevance for the pathogenesis of recessive X-linked ichthyosis. Arch Dermatol Res. 1986;278:259–263. doi: 10.1007/BF00407734. [DOI] [PubMed] [Google Scholar]
- 52.Rehfeld SJ, Plachy WZ, Williams ML, Elias PM. Calorimetric and electron spin resonance examination of lipid phase transitions in human stratum corneum: molecular basis for normal cohesion and abnormal desquamation in recessive X-linked ichthyosis. J Invest Dermatol. 1988;91:499–505. doi: 10.1111/1523-1747.ep12476654. [DOI] [PubMed] [Google Scholar]
- 53.Swanbeck G. The ichthyosis. Acta Derm Venereol Suppl (Stockh) 1981;95:88–90. [PubMed] [Google Scholar]
- 54.Williams ML, Elias PM. Heterogeneity in autosomal recessive ichthyosis. Clinical and biochemical differentiation of lamellar ichthyosis and nonbullous congenital ichthyosiform erythroderma. Arch Dermatol. 1985;121:477–488. doi: 10.1001/archderm.121.4.477. [DOI] [PubMed] [Google Scholar]
- 55.Arnold ML, Anton-Lamprecht I, Melz-Rothfuss B, Hartschuh W. Ichthyosis congenita type III. Clinical and ultrastructural characteristics and distinction within the heterogeneous ichthyosis congenita group. Arch Dermatol Res. 1988;280:268–278. doi: 10.1007/BF00440599. [DOI] [PubMed] [Google Scholar]
- 56.Ghadially R, Williams ML, Hou SY, Elias PM. Membrane structural abnormalities in the stratum corneum of the autosomal recessive ichthyoses. J Invest Dermatol. 1992;99:755–763. doi: 10.1111/1523-1747.ep12614489. [DOI] [PubMed] [Google Scholar]
- 57.Russell LJ, DiGiovanna JJ, Rogers GR, Steinert PM, Hashem N, Compton JG, Bale SJ. Mutations in the gene for transglutaminase 1 in autosomal recessive lamellar ichthyosis. Nat Genet. 1995;9:279–283. doi: 10.1038/ng0395-279. [DOI] [PubMed] [Google Scholar]
- 58.Huber M, Rettler I, Bernasconi K, Frenk E, Lavrijsen SP, Ponec M, Bon A, Lautenschlager S, Schorderet DF, Hohl D. Mutations of keratinocyte transglutaminase in lamellar ichthyosis. Science. 1995;267:525–528. doi: 10.1126/science.7824952. [DOI] [PubMed] [Google Scholar]
- 59.Hennies HC, Kuster W, Wiebe V, Krebsova A, Reis A. Genotype/phenotype correlation in autosomal recessive lamellar ichthyosis. Am J Hum Genet. 1998;62:1052–1061. doi: 10.1086/301818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jobard F, Lefevre C, Karaduman A, Blanchet-Bardon C, Emre S, Weissenbach J, Ozguc M, Lathrop M, Prud’homme JF, Fischer J. Lipoxygenase-3 (ALOXE3) and 12(R)-lipoxygenase (ALOX12B) are mutated in non-bullous congenital ichthyosiform erythroderma (NCIE) linked to chromosome 17p13.1. Hum Mol Genet. 2002;11:107–113. doi: 10.1093/hmg/11.1.107. [DOI] [PubMed] [Google Scholar]
- 61.Akiyama M, Sawamura D, Shimizu H. The clinical spectrum of nonbullous congenital ichthyosiform erythroderma and lamellar ichthyosis. Clin Exp Dermatol. 2003;28:235–240. doi: 10.1046/j.1365-2230.2003.01295.x. [DOI] [PubMed] [Google Scholar]
- 62.Vahlquist A, Ganemo A, Pigg M, Virtanen M, Westermark P. The clinical spectrum of congenital ichthyosis in Sweden: a review of 127 cases. Acta Derm Venereol Suppl (Stockh) 2003;83:34–47. [PubMed] [Google Scholar]
- 63.Eckl KM, Krieg P, Kuster W, Traupe H, Andre F, Wittstruck N, Furstenberger G, Hennies HC. Mutation spectrum and functional analysis of epidermis-type lipoxygenases in patients with autosomal recessive congenital ichthyosis. Hum Mutat. 2005;26:351–361. doi: 10.1002/humu.20236. [DOI] [PubMed] [Google Scholar]
- 64.Fischer J, Faure A, Bouadjar B, Blanchet-Bardon C, Karaduman A, Thomas I, Emre S, Cure S, Ozguc M, Weissenbach J, et al. Two new loci for autosomal recessive ichthyosis on chromosomes 3p21 and 19p12-q12 and evidence for further genetic heterogeneity. Am J Hum Genet. 2000;66:904–913. doi: 10.1086/302814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Richard G. Molecular genetics of the ichthyoses. Am J Med Genet C Semin Med Genet. 2004;131C:32–44. doi: 10.1002/ajmg.c.30032. [DOI] [PubMed] [Google Scholar]
- 66.Anton-Lamprecht I. Ultrastructural identification of basic abnormalities as clues to genetic disorders of the epidermis. J Invest Dermatol. 1994;103(Suppl):6–12. doi: 10.1111/1523-1747.ep12398887. [DOI] [PubMed] [Google Scholar]
- 67.Ganemo A, Pigg M, Virtanen M, Kukk T, Raudsepp H, Rossman-Ringdahl I, Westermark P, Niemi KM, Dahl N, Vahlquist A. Autosomal recessive congenital ichthyosis in Sweden and Estonia: clinical, genetic and ultrastructural findings in eighty-three patients. Acta Derm Venereol. 2003;83:24–30. doi: 10.1080/00015550310002666. [DOI] [PubMed] [Google Scholar]
- 68.Lawlor F. Progress of a harlequin fetus to nonbullous ichthyosiform erythroderma. Pediatrics. 1988;82:870–873. [PubMed] [Google Scholar]
- 69.Lefevre C, Bouadjar B, Karaduman A, Jobard F, Saker S, Ozguc M, Lathrop M, Prud’homme JF, Fischer J. Mutations in ichthyin a new gene on chromosome 5q33 in a new form of autosomal recessive congenital ichthyosis. Hum Mol Genet. 2004;13:2473–2482. doi: 10.1093/hmg/ddh263. [DOI] [PubMed] [Google Scholar]
- 70.Lesueur F, Bouadjar B, Lefevre C, Jobard F, Audebert S, Lakhdar H, Martin L, Tadini G, Karaduman A, Emre S, et al. Novel mutations in ALOX12B in patients with autosomal recessive congenital ichthyosis and evidence for genetic heterogeneity on chromosome 17p13. J Invest Dermatol. 2007;127:829–834. doi: 10.1038/sj.jid.5700640. [DOI] [PubMed] [Google Scholar]
- 71.Dahlqvist J, Klar J, Hausser I, Anton-Lamprecht I, Pigg MH, Gedde-Dahl T, Jr, Ganemo A, Vahlquist A, Dahl N. Congenital ichthyosis: mutations in ichthyin are associated with specific structural abnormalities in the granular layer of epidermis. J Med Genet. 2007;44:615–620. doi: 10.1136/jmg.2007.050542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Anton R, Vila L. Stereoselective biosynthesis of hepoxilin B3 in human epidermis. J Invest Dermatol. 2000;114:554–559. doi: 10.1046/j.1523-1747.2000.00903.x. [DOI] [PubMed] [Google Scholar]
- 73.Anton R, Camacho M, Puig L, Vila L. Hepoxilin B3 and its enzymatically formed derivative trioxilin B3 are incorporated into phospholipids in psoriatic lesions. J Invest Dermatol. 2002;118:139–146. doi: 10.1046/j.0022-202x.2001.01593.x. [DOI] [PubMed] [Google Scholar]
- 74.Krieg P, Marks F, Furstenberger G. A gene cluster encoding human epidermis-type lipoxygenases at chromosome 17p13.1: cloning, physical mapping, and expression. Genomics. 2001;73:323–330. doi: 10.1006/geno.2001.6519. [DOI] [PubMed] [Google Scholar]
- 75.Yu Z, Schneider C, Boeglin WE, Marnett LJ, Brash AR. The lipoxygenase gene ALOXE3 implicated in skin differentiation encodes a hydroperoxide isomerase. Proc Natl Acad Sci USA. 2003;100:9162–9167. doi: 10.1073/pnas.1633612100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yu Z, Schneider C, Boeglin WE, Brash AR. Mutations associated with a congenital form of ichthyosis (NCIE) inactivate the epidermal lipoxygenases 12R-LOX and eLOX3. Biochim Biophys Acta. 2005;1686:238–247. doi: 10.1016/j.bbalip.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 77.Epp N, Furstenberger G, Muller K, de Juanes S, Leitges M, Hausser I, Thieme F, Liebisch G, Schmitz G, Krieg P. 12R-Lipoxygenase deficiency disrupts epidermal barrier function. J Cell Biol. 2007;177:173–182. doi: 10.1083/jcb.200612116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moran JL, Qiu H, Turbe-Doan A, Yun Y, Boeglin WE, Brash AR, Beier DR. A mouse mutation in the 12R-lipoxygenase, Alox12b, disrupts formation of the epidermal permeability barrier. J Invest Dermatol. 2007;127:1893–1897. doi: 10.1038/sj.jid.5700825. [DOI] [PubMed] [Google Scholar]
- 79.Lefevre C, Bouadjar B, Ferrand V, Tadini G, Megarbane A, Lathrop M, Prud’homme JF, Fischer J. Mutations in a new cytochrome P450 gene in lamellar ichthyosis type 3. Hum Mol Genet. 2006;15:767–776. doi: 10.1093/hmg/ddi491. [DOI] [PubMed] [Google Scholar]
- 80.Elias PM, Brown BE. The mammalian cutaneous permeability barrier: defective barrier function in essential fatty acid deficiency correlates with abnormal intercellular lipid deposition. Lab Invest. 1978;39:574–583. [PubMed] [Google Scholar]
- 81.Elias PM, Brown BE, Ziboh VA. The permeability barrier in essential fatty acid deficiency: evidence for a direct role for linoleic acid in barrier function. J Invest Dermatol. 1980;74:230–233. doi: 10.1111/1523-1747.ep12541775. [DOI] [PubMed] [Google Scholar]
- 82.Nugteren DH, Christ-Hazelhof E, van der Beek A, Houtsmuller UM. Metabolism of linoleic acid and other essential fatty acids in the epidermis of the rat. Biochim Biophys Acta. 1985;834:429–436. doi: 10.1016/0005-2760(85)90017-7. [DOI] [PubMed] [Google Scholar]
- 83.Hou SY, Mitra AK, White SH, Menon GK, Ghadially R, Elias PM. Membrane structures in normal and essential fatty acid-deficient stratum corneum: characterization by ruthenium tetroxide staining and x-ray diffraction. J Invest Dermatol. 1991;96:215–223. doi: 10.1111/1523-1747.ep12461361. [DOI] [PubMed] [Google Scholar]
- 84.Yu Z, Schneider C, Boeglin WE, Brash AR. Epidermal lipoxygenase products of the hepoxilin pathway selectively activate the nuclear receptor PPARalpha. Lipids. 2007;42:491–497. doi: 10.1007/s11745-007-3054-4. [DOI] [PubMed] [Google Scholar]
- 85.Schmuth M, Schoonjans K, Yu QC, Fluhr JW, Crumrine D, Hachem JP, Lau P, Auwerx J, Elias PM, Feingold KR. Role of peroxisome proliferator-activated receptor alpha in epidermal development in utero. J Invest Dermatol. 2002;119:1298–1303. doi: 10.1046/j.1523-1747.2002.19605.x. [DOI] [PubMed] [Google Scholar]
- 86.Dubrac S, Stoitzner P, Pirkebner D, Elentner A, Schoonjans K, Auwerx J, Saeland S, Hengster P, Fritsch P, Romani N, et al. Peroxisome proliferator-activated receptor-alpha activation inhibits Langerhans cell function. J Immunol. 2007;178:4362–4372. doi: 10.4049/jimmunol.178.7.4362. [DOI] [PubMed] [Google Scholar]
- 87.Cowart LA, Wei S, Hsu MH, Johnson EF, Krishna MU, Falck JR, Capdevila JH. The CYP4A isoforms hydroxylate epoxyeicosatrienoic acids to form high affinity peroxisome proliferator-activated receptor ligands. J Biol Chem. 2002;277:35105–35112. doi: 10.1074/jbc.M201575200. [DOI] [PubMed] [Google Scholar]
- 88.Rivier M, Safonova I, Lebrun P, Griffiths CE, Ailhaud G, Michel S. Differential expression of peroxisome proliferator-activated receptor subtypes during the differentiation of human keratinocytes. J Invest Dermatol. 1998;111:1116–1121. doi: 10.1046/j.1523-1747.1998.00439.x. [DOI] [PubMed] [Google Scholar]
- 89.Reynaud D, Demin PM, Sutherland M, Nigam S, Pace-Asciak CR. Hepoxilin signaling in intact human neutrophils: biphasic elevation of intracellular calcium by unesterified hepoxilin A3. FEBS Lett. 1999;446:236–238. doi: 10.1016/s0014-5793(99)00225-2. [DOI] [PubMed] [Google Scholar]
- 90.Rizzo WB, Craft DA. Sjogren-Larsson syndrome: accumulation of free fatty alcohols in cultured fibroblasts and plasma. J Lipid Res. 2000;41:1077–1081. [PubMed] [Google Scholar]
- 91.Rizzo WB. Sjogren-Larsson syndrome: molecular genetics and biochemical pathogenesis of fatty aldehyde dehydrogenase deficiency. Mol Genet Metab. 2007;90:1–9. doi: 10.1016/j.ymgme.2006.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gordon N. Sjogren-Larsson syndrome. Dev Med Child Neurol. 2007;49:152–154. doi: 10.1111/j.1469-8749.2007.00152.x. [DOI] [PubMed] [Google Scholar]
- 93.Willemsen MA, LIJ, Steijlen PM, Rotteveel JJ, de Jong JG, van Domburg PH, Mayatepek E, Gabreels FJ, Wanders RJ. Clinical, biochemical and molecular genetic characteristics of 19 patients with the Sjogren-Larsson syndrome. Brain. 2001;124:1426–1437. doi: 10.1093/brain/124.7.1426. [DOI] [PubMed] [Google Scholar]
- 94.Wedi B, Kapp A. Pathophysiological role of leuko-trienes in dermatological diseases: potential therapeutic implications. BioDrugs. 2001;15:729–743. doi: 10.2165/00063030-200115110-00004. [DOI] [PubMed] [Google Scholar]
- 95.Shibaki A, Akiyama M, Shimizu H. Novel ALDH3A2 heterozygous mutations are associated with defective lamellar granule formation in a Japanese family of Sjogren-Larsson syndrome. J Invest Dermatol. 2004;123:1197–1199. doi: 10.1111/j.0022-202X.2004.23505.x. [DOI] [PubMed] [Google Scholar]
- 96.Koone MD, Rizzo WB, Elias PM, Williams ML, Lightner V, Pinnell SR. Ichthyosis, mental retardation, and asymptomatic spasticity. A new neurocutaneous syndrome with normal fatty alcohol:NAD+ oxidoreductase activity. Arch Dermatol. 1990;126:1485–1490. doi: 10.1001/archderm.126.11.1485. [DOI] [PubMed] [Google Scholar]
- 97.van den Brink DM, Wanders RJ. Phytanic acid: production from phytol, its breakdown and role in human disease. Cell Mol Life Sci. 2006;63:1752–1765. doi: 10.1007/s00018-005-5463-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jansen GA, Ferdinandusse S, Hogenhout EM, Verhoeven NM, Jakobs C, Wanders RJ. Phytanoyl-CoA hydroxylase deficiency. Enzymological and molecular basis of classical Refsum disease. Adv Exp Med Biol. 1999;466:371–376. [PubMed] [Google Scholar]
- 99.van den Brink DM, Brites P, Haasjes J, Wierzbicki AS, Mitchell J, Lambert-Hamill M, de Belleroche J, Jansen GA, Waterham HR, Wanders RJ. Identification of PEX7 as the second gene involved in Refsum disease. Am J Hum Genet. 2003;72:471–477. doi: 10.1086/346093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pahan K, Khan M, Singh I. Phytanic acid oxidation: normal activation and transport yet defective alpha-hydroxylation of phytanic acid in peroxisomes from Refsum disease and rhizomelic chondrodysplasia punctata. J Lipid Res. 1996;37:1137–1143. [PubMed] [Google Scholar]
- 101.Verhoeven NM, Jakobs C, Carney G, Somers MP, Wanders RJ, Rizzo WB. Involvement of microsomal fatty aldehyde dehydrogenase in the alpha-oxidation of phytanic acid. FEBS Lett. 1998;429:225–228. doi: 10.1016/s0014-5793(98)00574-2. [DOI] [PubMed] [Google Scholar]
- 102.Derry JM, Gormally E, Means GD, Zhao W, Meindl A, Kelley RI, Boyd Y, Herman GE. Mutations in a delta 8-delta 7 sterol isomerase in the tattered mouse and X-linked dominant chondrodysplasia punctata. Nat Genet. 1999;22:286–290. doi: 10.1038/10350. [DOI] [PubMed] [Google Scholar]
- 103.Braverman N, Lin P, Moebius FF, Obie C, Moser A, Glossmann H, Wilcox WR, Rimoin DL, Smith M, Kratz L, et al. Mutations in the gene encoding 3 beta-hydroxysteroid-delta 8, delta 7-isomerase cause X-linked dominant Conradi-Hunermann syndrome. Nat Genet. 1999;22:291–294. doi: 10.1038/10357. [DOI] [PubMed] [Google Scholar]
- 104.Grange DK, Kratz LE, Braverman NE, Kelley RI. CHILD syndrome caused by deficiency of 3beta-hydroxysteroid-delta8, delta7-isomerase. Am J Med Genet. 2000;90:328–335. doi: 10.1002/(sici)1096-8628(20000214)90:4<328::aid-ajmg13>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 105.Konig A, Happle R, Bornholdt D, Engel H, Grzeschik KH. Mutations in the NSDHL gene, encoding a 3beta-hydroxysteroid dehydrogenase, cause CHILD syndrome. Am J Med Genet. 2000;90:339–346. [PubMed] [Google Scholar]
- 106.Kelley RI, Herman GE. Inborn errors of sterol biosynthesis. Annu Rev Genomics Hum Genet. 2001;2:299–341. doi: 10.1146/annurev.genom.2.1.299. [DOI] [PubMed] [Google Scholar]
- 107.Happle R. Lyonization and the lines of Blaschko. Hum Genet. 1985;70:200–206. doi: 10.1007/BF00273442. [DOI] [PubMed] [Google Scholar]
- 108.Happle R. X-linked dominant chondrodysplasia punc-tata. Review of literature and report of a case. Hum Genet. 1979;53:65–73. doi: 10.1007/BF00289453. [DOI] [PubMed] [Google Scholar]
- 109.Ikegawa S, Ohashi H, Ogata T, Honda A, Tsukahara M, Kubo T, Kimizuka M, Shimode M, Hasegawa T, Nishimura G, et al. Novel and recurrent EBP mutations in X-linked dominant chondrodysplasia punctata. Am J Med Genet. 2000;94:300–305. doi: 10.1002/1096-8628(20001002)94:4<300::aid-ajmg7>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 110.Steijlen PM, van Geel M, Vreeburg M, Marcus-Soekarman D, Spaapen LJ, Castelijns FC, Willemsen M, van Steensel MA. Novel EBP gene mutations in Conradi-Hunermann-Happle syndrome. Br J Dermatol. 2007;157:1225–1229. doi: 10.1111/j.1365-2133.2007.08254.x. [DOI] [PubMed] [Google Scholar]
- 111.Feldmeyer L, Mevorah B, Grzeschik KH, Huber M, Hohl D. Clinical variation in X-linked dominant chondrodysplasia punctata (X-linked dominant ichthyosis) Br J Dermatol. 2006;154:766–769. doi: 10.1111/j.1365-2133.2006.07137.x. [DOI] [PubMed] [Google Scholar]
- 112.Herman GE, Kelley RI, Pureza V, Smith D, Kopacz K, Pitt J, Sutphen R, Sheffield LJ, Metzenberg AB. Characterization of mutations in 22 females with X-linked dominant chondrodysplasia punctata (Happle syndrome) Genet Med. 2002;4:434–438. doi: 10.1097/00125817-200211000-00006. [DOI] [PubMed] [Google Scholar]
- 113.Emami S, Hanley KP, Esterly NB, Daniallinia N, Williams ML. X-linked dominant ichthyosis with peroxisomal deficiency. An ultrastructural and ultracytochemical study of the Conradi-Hunermann syndrome and its murine homologue, the bare patches mouse. Arch Dermatol. 1994;130:325–336. doi: 10.1001/archderm.130.3.325. [DOI] [PubMed] [Google Scholar]
- 114.Cooper MK, Wassif CA, Krakowiak PA, Taipale J, Gong R, Kelley RI, Porter FD, Beachy PA. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat Genet. 2003;33:508–513. doi: 10.1038/ng1134. [DOI] [PubMed] [Google Scholar]
- 115.Elias P, Williams ML, Maloney M, Fritsch P, Chung J-C. Applications of the diazacholesterol animal model of ichthyosis. In: Marks R, Plewig G, editors. Skin Models. Springer; Berlin: 1986. pp. 122–135. [Google Scholar]
- 116.Elias PM, Lampe MA, Chung JC, Williams ML. Diazacholesterol-induced ichthyosis in the hairless mouse. I. Morphologic, histochemical, and lipid biochemical characterization of a new animal model. Lab Invest. 1983;48:565–577. [PubMed] [Google Scholar]
- 117.Clayton PT, Kalter DC, Atherton DJ, Besley GT, Broadhead DM. Peroxisomal enzyme deficiency in X-linked dominant Conradi-Hunermann syndrome. J Inherit Metab Dis. 1989;12(Suppl 2):358–360. doi: 10.1007/BF03335422. [DOI] [PubMed] [Google Scholar]
- 118.Holmes RD, Wilson GN, Hajra AK. Peroxisomal enzyme deficiency in the Conradi-Hunerman form of chondrodysplasia punctata. N Engl J Med. 1987;316:1608. doi: 10.1056/NEJM198706183162517. [DOI] [PubMed] [Google Scholar]
- 119.Emami S, Rizzo WB, Hanley KP, Taylor JM, Goldyne ME, Williams ML. Peroxisomal abnormality in fibroblasts from involved skin of CHILD syndrome. Case study and review of peroxisomal disorders in relation to skin disease. Arch Dermatol. 1992;128:1213–1222. [PubMed] [Google Scholar]
- 120.Wilson CJ, Aftimos S. X-linked dominant chondrodysplasia punctata: a peroxisomal disorder? Am J Med Genet. 1998;78:300–302. doi: 10.1002/(sici)1096-8628(19980707)78:3<300::aid-ajmg19>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 121.Steinberg SJ, Dodt G, Raymond GV, Braverman NE, Moser AB, Moser HW. Peroxisome biogenesis disorders. Biochim Biophys Acta. 2006;1763:1733–1748. doi: 10.1016/j.bbamcr.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 122.Koppe G, Marinkovic-Ilsen A, Rijken Y, De Groot WP, Jobsis AC. X-linked icthyosis. A sulphatase deficiency. Arch Dis Child. 1978;53:803–806. doi: 10.1136/adc.53.10.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Webster D, France JT, Shapiro LJ, Weiss R. X-linked ichthyosis due to steroid-sulphatase deficiency. Lancet. 1978;1:70–72. doi: 10.1016/s0140-6736(78)90005-3. [DOI] [PubMed] [Google Scholar]
- 124.Tiepolo L, Zuffardi O, Rodewald A. Nullisomy for the distal portion of Xp in a male child with a X/Y translocation. Hum Genet. 1977;39:277–281. doi: 10.1007/BF00295420. [DOI] [PubMed] [Google Scholar]
- 125.Mohandas T, Shapiro LJ, Sparkes RS, Sparkes MC. Regional assignment of the steroid sulfatase-X-linked ichthyosis locus: implications for a noninactivated region on the short arm of human X chromosome. Proc Natl Acad Sci USA. 1979;76:5779–5783. doi: 10.1073/pnas.76.11.5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Li XM, Yen P, Mohandas T, Shapiro LJ. A long range restriction map of the distal human X chromosome short arm around the steroid sulfatase locus. Nucleic Acids Res. 1990;18:2783–2788. doi: 10.1093/nar/18.9.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shapiro LJ. Steroid sulfatase deficiency and the genetics of the short arm of the human X chromosome. Adv Hum Genet. 1985;14:331–381. 388–399. doi: 10.1007/978-1-4615-9400-0_5. [DOI] [PubMed] [Google Scholar]
- 128.Tiepolo L, Zuffardi O, Fraccaro M, di Natale D, Gargantini L, Muller CR, Ropers HH. Assignment by deletion mapping of the steroid sulfatase X-linked ichthyosis locus to Xp223. Hum Genet. 1980;54:205–206. doi: 10.1007/BF00278973. [DOI] [PubMed] [Google Scholar]
- 129.Ballabio A, Parenti G, Carrozzo R, Sebastio G, Andria G, Buckle V, Fraser N, Craig I, Rocchi M, Romeo G, et al. Isolation and characterization of a steroid sulfatase cDNA clone: genomic deletions in patients with X-chromosome-linked ichthyosis. Proc Natl Acad Sci USA. 1987;84:4519–4523. doi: 10.1073/pnas.84.13.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bonifas JM, Morley BJ, Oakey RE, Kan YW, Epstein EH., Jr Cloning of a cDNA for steroid sulfatase: frequent occurrence of gene deletions in patients with recessive X chromosome-linked ichthyosis. Proc Natl Acad Sci USA. 1987;84:9248–9251. doi: 10.1073/pnas.84.24.9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Conary JT, Lorkowski G, Schmidt B, Pohlmann R, Nagel G, Meyer HE, Krentler C, Cully J, Hasilik A, von Figura K. Genetic heterogeneity of steroid sulfatase deficiency revealed with cDNA for human steroid sulfatase. Biochem Biophys Res Commun. 1987;144:1010–1017. doi: 10.1016/s0006-291x(87)80064-5. [DOI] [PubMed] [Google Scholar]
- 132.Gillard EF, Affara NA, Yates JR, Goudie DR, Lambert J, Aitken DA, Ferguson-Smith MA. Deletion of a DNA sequence in eight of nine families with X-linked ichthyosis (steroid sulphatase deficiency) Nucleic Acids Res. 1987;15:3977–3985. doi: 10.1093/nar/15.10.3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shapiro LJ, Yen P, Pomerantz D, Martin E, Rolewic L, Mohandas T. Molecular studies of deletions at the human steroid sulfatase locus. Proc Natl Acad Sci USA. 1989;86:8477–8481. doi: 10.1073/pnas.86.21.8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Schmickel RD. Contiguous gene syndromes: a component of recognizable syndromes. J Pediatr. 1986;109:231–241. doi: 10.1016/s0022-3476(86)80377-8. [DOI] [PubMed] [Google Scholar]
- 135.Schnur RE, Trask BJ, van den Engh G, Punnett HH, Kistenmacher M, Tomeo MA, Naids RE, Nussbaum RL. An Xp22 microdeletion associated with ocular albinism and ichthyosis: approximation of breakpoints and estimation of deletion size by using cloned DNA probes and flow cytometry. Am J Hum Genet. 1989;45:706–720. [PMC free article] [PubMed] [Google Scholar]
- 136.de Vries B, Eussen B, van Diggelen O, van der Heide A, Deelen W, Govaerts L, Lindhout D, Wouters C, Van Hemel J. Submicroscopic Xpter deletion in a boy with growth and mental retardation caused by a familial t(X;14) Am J Med Genet A. 1999;87:189–194. doi: 10.1002/(sici)1096-8628(19991119)87:2<189::aid-ajmg12>3.3.co;2-h. [DOI] [PubMed] [Google Scholar]
- 137.Willemsen R, Kroos M, Hoogeveen AT, van Dongen JM, Parenti G, van der Loos CM, Reuser AJ. Ultra-structural localization of steroid sulphatase in cultured human fibroblasts by immunocytochemistry: a comparative study with lysosomal enzymes and the mannose 6-phosphate receptor. Histochem J. 1988;20:41–51. doi: 10.1007/BF01745968. [DOI] [PubMed] [Google Scholar]
- 138.Dibbelt L, Herzog V, Kuss E. Human placental sterylsulfatase: immunocytochemical and biochemical localization. Biol Chem Hoppe Seyler. 1989;370:1093–1102. doi: 10.1515/bchm3.1989.370.2.1093. [DOI] [PubMed] [Google Scholar]
- 139.Kubilus J, Tarascio AJ, Baden HP. Steroid-sulfatase deficiency in sex-linked ichthyosis. Am J Hum Genet. 1979;31:50–53. [PMC free article] [PubMed] [Google Scholar]
- 140.Epstein EH, Jr, Krauss RM, Shackleton CH. X-linked ichthyosis: increased blood cholesterol sulfate and electrophoretic mobility of low-density lipoprotein. Science. 1981;214:659–660. doi: 10.1126/science.6945674. [DOI] [PubMed] [Google Scholar]
- 141.Elias PM, Williams ML, Maloney ME, Bonifas JA, Brown BE, Grayson S, Epstein EHJ. Stratum corneum lipids in disorders of cornification. Steroid sulfatase and cholesterol sulfate in normal desquamation and the pathogenesis of recessive X-linked ichthyosis. J Clin Invest. 1984;74:1414–1421. doi: 10.1172/JCI111552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bergner EA, Shapiro LJ. Metabolism of 3H-dehydroepiandrosterone sulphate by subjects with steroid sulphatase deficiency. J Inherit Metab Dis. 1988;11:403–415. doi: 10.1007/BF01800429. [DOI] [PubMed] [Google Scholar]
- 143.Ibsen HH, Brandrup F, Secher B. Topical cholesterol treatment of recessive X-linked ichthyosis. Lancet. 1984;2:645. doi: 10.1016/s0140-6736(84)90642-1. [DOI] [PubMed] [Google Scholar]
- 144.Williams ML, Elias PM. Stratum corneum lipids in disorders of cornification: increased cholesterol sulfate content of stratum corneum in recessive X-linked ichthyosis. J Clin Invest. 1981;68:1404–1410. doi: 10.1172/JCI110391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Long SA, Wertz PW, Strauss JS, Downing DT. Human stratum corneum polar lipids and desquamation. Arch Dermatol Res. 1985;277:284–287. doi: 10.1007/BF00509081. [DOI] [PubMed] [Google Scholar]
- 146.Ranasinghe AW, Wertz PW, Downing DT, Mackenzie IC. Lipid composition of cohesive and desquamated corneocytes from mouse ear skin. J Invest Dermatol. 1986;86:187–190. doi: 10.1111/1523-1747.ep12284246. [DOI] [PubMed] [Google Scholar]
- 147.Grayson S, Johnson-Winegar AG, Wintroub BU, Isseroff RR, Epstein EH, Jr, Elias PM. Lamellar body-enriched fractions from neonatal mice: preparative techniques and partial characterization. J Invest Dermatol. 1985;85:289–294. doi: 10.1111/1523-1747.ep12276826. [DOI] [PubMed] [Google Scholar]
- 148.Ponec M, Williams ML. Cholesterol sulfate uptake and outflux in cultured human keratinocytes. Arch Dermatol Res. 1986;279:32–36. doi: 10.1007/BF00404355. [DOI] [PubMed] [Google Scholar]
- 149.Epstein EH, Williams ML, Elias PM. The epidermal cholesterol sulfate cycle. J Am Acad Dermatol. 1984;10:866–868. doi: 10.1016/s0190-9622(84)80144-9. [DOI] [PubMed] [Google Scholar]
- 150.Rearick JI, Jetten AM. Accumulation of cholesterol 3-sulfate during in vitro squamous differentiation of rabbit tracheal epithelial cells and its regulation by retinoids. J Biol Chem. 1986;261:13898–13904. [PubMed] [Google Scholar]
- 151.Rearick JI, Albro PW, Jetten AM. Increase in cholesterol sulfotransferase activity during in vitro squamous differentiation of rabbit tracheal epithelial cells and its inhibition by retinoic acid. J Biol Chem. 1987;262:13069–13074. [PubMed] [Google Scholar]
- 152.Rearick JI, Hesterberg TW, Jetten AM. Human bronchial epithelial cells synthesize cholesterol sulfate during squamous differentiation in vitro. J Cell Physiol. 1987;133:573–578. doi: 10.1002/jcp.1041330320. [DOI] [PubMed] [Google Scholar]
- 153.Williams ML, Rutherford SL, Ponec M, Hincenbergs M, Placzek DR, Elias PM. Density-dependent variations in the lipid content and metabolism of cultured human keratinocytes. J Invest Dermatol. 1988;91:86–91. doi: 10.1111/1523-1747.ep12463297. [DOI] [PubMed] [Google Scholar]
- 154.Jetten AM, George MA, Nervi C, Boone LR, Rearick JI. Increased cholesterol sulfate and cholesterol sulfotransferase activity in relation to the multi-step process of differentiation in human epidermal keratinocytes. J Invest Dermatol. 1989;92:203–209. doi: 10.1111/1523-1747.ep12276731. [DOI] [PubMed] [Google Scholar]
- 155.Higashi Y, Fuda H, Yanai H, Lee Y, Fukushige T, Kanzaki T, Strott CA. Expression of cholesterol sulfotransferase (SULT2B1b) in human skin and primary cultures of human epidermal keratinocytes. J Invest Dermatol. 2004;122:1207–1213. doi: 10.1111/j.0022-202X.2004.22416.x. [DOI] [PubMed] [Google Scholar]
- 156.Jetten AM, George MA, Pettit GR, Herald CL, Rearick JI. Action of phorbol esters, bryostatins, and retinoic acid on cholesterol sulfate synthesis: relation to the multistep process of differentiation in human epidermal keratinocytes. J Invest Dermatol. 1989;93:108–115. doi: 10.1111/1523-1747.ep12277374. [DOI] [PubMed] [Google Scholar]
- 157.Jetten AM, Brody AR, Deas MA, Hook GE, Rearick JI, Thacher SM. Retinoic acid and substratum regulate the differentiation of rabbit tracheal epithelial cells into squamous and secretory phenotype. Morphological and biochemical characterization. Lab Invest. 1987;56:654–664. [PubMed] [Google Scholar]
- 158.Kagehara M, Tachi M, Harii K, Iwamori M. Programmed expression of cholesterol sulfotransferase and transglutaminase during epidermal differentiation of murine skin development. Biochim Biophys Acta. 1994;1215:183–189. doi: 10.1016/0005-2760(94)90109-0. [DOI] [PubMed] [Google Scholar]
- 159.Hanley K, Jiang Y, Katagiri C, Feingold KR, Williams ML. Epidermal steroid sulfatase and cholesterol sulfotransferase are regulated during late gestation in the fetal rat. J Invest Dermatol. 1997;108:871–875. doi: 10.1111/1523-1747.ep12292586. [DOI] [PubMed] [Google Scholar]
- 160.Komuves LG, Hanley K, Jiang Y, Katagiri C, Elias PM, Williams ML, Feingold KR. Induction of selected lipid metabolic enzymes and differentiation-linked structural proteins by air exposure in fetal rat skin explants. J Invest Dermatol. 1999;112:303–309. doi: 10.1046/j.1523-1747.1999.00511.x. [DOI] [PubMed] [Google Scholar]
- 161.Strott CA. Sulfonation and molecular action. Endocr Rev. 2002;23:703–732. doi: 10.1210/er.2001-0040. [DOI] [PubMed] [Google Scholar]
- 162.Strott CA, Higashi Y. Cholesterol sulfate in human physiology: what’s it all about? J Lipid Res. 2003;44:1268–1278. doi: 10.1194/jlr.R300005-JLR200. [DOI] [PubMed] [Google Scholar]
- 163.Jiang YJ, Kim P, Elias PM, Feingold KR. LXR and PPAR activators stimulate cholesterol sulfotransferase type 2 isoform 1b in human keratinocytes. J Lipid Res. 2005;46:2657–2666. doi: 10.1194/jlr.M500235-JLR200. [DOI] [PubMed] [Google Scholar]
- 164.Kashiwagi M, Ohba M, Chida K, Kuroki T. Protein kinase C eta (PKC eta): its involvement in keratinocyte differentiation. J Biochem (Tokyo) 2002;132:853–857. doi: 10.1093/oxfordjournals.jbchem.a003297. [DOI] [PubMed] [Google Scholar]
- 165.Denning MF, Dlugosz AA, Williams EK, Szallasi Z, Blumberg PM, Yuspa SH. Specific protein kinase C isozymes mediate the induction of keratinocyte differentiation markers by calcium. Cell Growth Differ. 1995;6:149–157. [PubMed] [Google Scholar]
- 166.Ikuta T, Chida K, Tajima O, Matsuura Y, Iwamori M, Ueda Y, Mizuno K, Ohno S, Kuroki T. Cholesterol sulfate, a novel activator for the eta isoform of protein kinase C. Cell Growth Differ. 1994;5:943–947. [PubMed] [Google Scholar]
- 167.Kuroki T, Ikuta T, Kashiwagi M, Kawabe S, Ohba M, Huh N, Mizuno K, Ohno S, Yamada E, Chida K. Cholesterol sulfate, an activator of protein kinase C mediating squamous cell differentiation: a review. Mutat Res. 2000;462:189–195. doi: 10.1016/s1383-5742(00)00036-3. [DOI] [PubMed] [Google Scholar]
- 168.Kawabe S, Ikuta T, Ohba M, Chida K, Ueda E, Yamanishi K, Kuroki T. Cholesterol sulfate activates transcription of transglutaminase 1 gene in normal human keratinocytes. J Invest Dermatol. 1998;111:1098–1102. doi: 10.1046/j.1523-1747.1998.00441.x. [DOI] [PubMed] [Google Scholar]
- 169.Hanley K, Wood L, Ng DC, He SS, Lau P, Moser A, Elias PM, Bikle DD, Williams ML, Feingold KR. Cholesterol sulfate stimulates involucrin transcription in keratinocytes by increasing Fra-1, Fra-2, and Jun D. J Lipid Res. 2001;42:390–398. [PubMed] [Google Scholar]
- 170.Lavrijsen AP, Oestmann E, Hermans J, Boddé HE, Vermeer BJ, Ponec M. Barrier function parameters in various keratinization disorders: transepidermal water loss and vascular response to hexyl nicotinate. Br J Dermatol. 1993;129:547–553. doi: 10.1111/j.1365-2133.1993.tb00482.x. [DOI] [PubMed] [Google Scholar]
- 171.Johansen JD, Ramsing D, Vejlsgaard G, Agner T. Skin barrier properties in patients with recessive X-linked ichthyosis. Acta Derm Venereol. 1995;75:202–204. doi: 10.2340/0001555575198201. [DOI] [PubMed] [Google Scholar]
- 172.Zettersten E, Man MQ, Sato J, Denda M, Farrell A, Ghadially R, Williams ML, Feingold KR, Elias PM. Recessive X-linked ichthyosis: role of cholesterol-sulfate accumulation in the barrier abnormality. J Invest Dermatol. 1998;111:784–790. doi: 10.1046/j.1523-1747.1998.00386.x. [DOI] [PubMed] [Google Scholar]
- 173.Feingold KR, Man MQ, Menon GK, Cho SS, Brown BE, Elias PM. Cholesterol synthesis is required for cutaneous barrier function in mice. J Clin Invest. 1990;86:1738–1745. doi: 10.1172/JCI114899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Williams ML, Rutherford SL, Feingold KR. Effects of cholesterol sulfate on lipid metabolism in cultured human keratinocytes and fibroblasts. J Lipid Res. 1987;28:955–967. [PubMed] [Google Scholar]
- 175.Nemes Z, Demeny M, Marekov LN, Fesus L, Steinert PM. Cholesterol 3-sulfate interferes with cornified envelope assembly by diverting transglutaminase 1 activity from the formation of cross-links and esters to the hydrolysis of glutamine. J Biol Chem. 2000;275:2636–2646. doi: 10.1074/jbc.275.4.2636. [DOI] [PubMed] [Google Scholar]
- 176.Frost P, Weinstein GD, Van Scott EJ. The ichthyosiform dermatoses. II. Autoradiographic studies of epidermal proliferation. J Invest Dermatol. 1966;47:561–567. doi: 10.1038/jid.1966.185. [DOI] [PubMed] [Google Scholar]
- 177.Ekholm IE, Brattsand M, Egelrud T. Stratum corneum tryptic enzyme in normal epidermis: a missing link in the desquamation process? J Invest Dermatol. 2000;114:56–63. doi: 10.1046/j.1523-1747.2000.00820.x. [DOI] [PubMed] [Google Scholar]
- 178.Ohman H, Vahlquist A. The pH gradient over the stratum corneum differs in X-linked recessive and autosomal dominant ichthyosis: a clue to the molecular origin of the “acid skin mantle”? J Invest Dermatol. 1998;111:674–677. doi: 10.1046/j.1523-1747.1998.00356.x. [DOI] [PubMed] [Google Scholar]
- 179.Epstein EH, Williams ML, Elias PM. The epidermal cholesterol sulfate cycle. J Am Acad Dermatol. 1984;10:866–868. doi: 10.1016/s0190-9622(84)80144-9. [DOI] [PubMed] [Google Scholar]
- 180.Abraham W, Wertz PW, Landmann L, Downing DT. Stratum corneum lipid liposomes: calcium-induced transformation into lamellar sheets. J Invest Dermatol. 1987;88:212–214. doi: 10.1111/1523-1747.ep12525375. [DOI] [PubMed] [Google Scholar]
- 181.Hatfield RM, Fung LW. A new model system for lipid interactions in stratum corneum vesicles: effects of lipid composition, calcium, and pH. Biochemistry. 1999;38:784–791. doi: 10.1021/bi981421k. [DOI] [PubMed] [Google Scholar]
- 182.Stone DL, Carey WF, Christodoulou J, Sillence D, Nelson P, Callahan M, Tayebi N, Sidransky E. Type 2 Gaucher disease: the collodion baby phenotype revisited. Arch Dis Child Fetal Neonatal Ed. 2000;82:F163–F166. doi: 10.1136/fn.82.2.F163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Holleran WM, Ginns EI, Menon GK, Grundmann JU, Fartasch M, McKinney CE, Elias PM, Sidransky E. Consequences of beta-glucocerebrosidase deficiency in epidermis. Ultrastructure and permeability barrier alterations in Gaucher disease. J Clin Invest. 1994;93:1756–1764. doi: 10.1172/JCI117160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sidransky E. Gaucher disease: complexity in a “simple” disorder. Mol Genet Metab. 2004;83:6–15. doi: 10.1016/j.ymgme.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 185.Schmuth M, Weber F, Paschke E, Sepp N, Fritsch P. Delayed epidermal permeability repair in patients with sphingomyelinase deficiency (Niemann-Pick disease) J Invest Dermatol. 1999;112:542. [Google Scholar]
- 186.Uchida Y, Hara M, Nishio H, Sidransky E, Inoue S, Otsuka F, Suzuki A, Elias PM, Holleran WM, Hamanaka S. Epidermal sphingomyelins are precursors for selected stratum corneum ceramides. J Lipid Res. 2000;41:2071–2082. [PubMed] [Google Scholar]
- 187.Sidransky E, Fartasch M, Lee RE, Metlay LA, Abella S, Zimran A, Gao W, Elias PM, Ginns EI, Holleran WM. Epidermal abnormalities may distinguish type 2 from type 1 and type 3 of Gaucher disease. Pediatr Res. 1996;39:134–141. doi: 10.1203/00006450-199601000-00020. [DOI] [PubMed] [Google Scholar]
- 188.Jensen JM, Schutze S, Forl M, Kronke M, Proksch E. Roles for tumor necrosis factor receptor p55 and sphingomyelinase in repairing the cutaneous permeability barrier. J Clin Invest. 1999;104:1761–1770. doi: 10.1172/JCI5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Marsh NL, Elias PM, Holleran WM. Glucosylceramides stimulate murine epidermal hyperproliferation. J Clin Invest. 1995;95:2903–2909. doi: 10.1172/JCI117997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Akiyama M. The pathogenesis of severe congenital ichthyosis of the neonate. J Dermatol Sci. 1999;21:96–104. doi: 10.1016/s0923-1811(99)00024-9. [DOI] [PubMed] [Google Scholar]
- 191.Milner ME, O’Guin WM, Holbrook KA, Dale BA. Abnormal lamellar granules in harlequin ichthyosis. J Invest Dermatol. 1992;99:824–829. doi: 10.1111/1523-1747.ep12614791. [DOI] [PubMed] [Google Scholar]
- 192.Elias PM, Fartasch M, Crumrine D, Behne M, Uchida Y, Holleran WM. Origin of the corneocyte lipid envelope (CLE): observations in harlequin ichthyosis and cultured human keratinocytes. J Invest Dermatol. 2000;115:765–769. doi: 10.1046/j.1523-1747.2000.00124-5.x. [DOI] [PubMed] [Google Scholar]
- 193.Hollenstein K, Frei D, Locher K. Structure of an ABC transporter in complex with its binding protein. Nature. 2007;446:213–216. doi: 10.1038/nature05626. [DOI] [PubMed] [Google Scholar]
- 194.Akiyama M, Sugiyama-Nakagiri Y, Sakai K, McMillan JR, Goto M, Arita K, Tsuji-Abe Y, Tabata N, Matsuoka K, Sasaki R, et al. Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J Clin Invest. 2005;115:1777–1784. doi: 10.1172/JCI24834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Allikmets R, Gerrard B, Hutchinson A, Dean M. Characterization of the human ABC superfamily: isolation and mapping of 21 new genes using the expressed sequence tags database. Hum Mol Genet. 1996;5:1649–1655. doi: 10.1093/hmg/5.10.1649. [DOI] [PubMed] [Google Scholar]
- 196.Borst P, Elferink RO. Mammalian ABC transporters in health and disease. Annu Rev Biochem. 2002;71:537–592. doi: 10.1146/annurev.biochem.71.102301.093055. [DOI] [PubMed] [Google Scholar]
- 197.Dean M, Rzhetsky A, Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001;11:1156–1166. doi: 10.1101/gr.184901. [DOI] [PubMed] [Google Scholar]
- 198.Jiang YJ, Lu B, Kim P, Paragh G, Schmitz G, Elias PM, Feingold KR. PPAR and LXR activators regulate ABCA12 expression in human keratinocytes. J Invest Dermatol. 2008;128:104–109. doi: 10.1038/sj.jid.5700944. [DOI] [PubMed] [Google Scholar]
- 199.Mulugeta S, Gray JM, Notarfrancesco KL, Gonzales LW, Koval M, Feinstein SI, Ballard PL, Fisher AB, Shuman H. Identification of LBM180, a lamellar body limiting membrane protein of alveolar type II cells, as the ABC transporter protein ABCA3. J Biol Chem. 2002;277:22147–22155. doi: 10.1074/jbc.M201812200. [DOI] [PubMed] [Google Scholar]
- 200.Yamano G, Funahashi H, Kawanami O, Zhao LX, Ban N, Uchida Y, Morohoshi T, Ogawa J, Shioda S, Inagaki N. ABCA3 is a lamellar body membrane protein in human lung alveolar type II cells. FEBS Lett. 2001;508:221–225. doi: 10.1016/s0014-5793(01)03056-3. [DOI] [PubMed] [Google Scholar]
- 201.Sakai K, Akiyama M, Sugiyama-Nakagiri Y, McMillan JR, Sawamura D, Shimizu H. Localization of ABCA12 from Golgi apparatus to lamellar granules in human upper epidermal keratinocytes. Exp Dermatol. 2007;16:920–926. doi: 10.1111/j.1600-0625.2007.00614.x. [DOI] [PubMed] [Google Scholar]
- 202.Lefevre C, Audebert S, Jobard F, Bouadjar B, Lakhdar H, Boughdene-Stambouli O, Blanchet-Bardon C, Heilig R, Foglio M, Weissenbach J, et al. Mutations in the transporter ABCA12 are associated with lamellar ichthyosis type 2. Hum Mol Genet. 2003;12:2369–2378. doi: 10.1093/hmg/ddg235. [DOI] [PubMed] [Google Scholar]
- 203.Thomas AC, Cullup T, Norgett EE, Hill T, Barton S, Dale BA, Sprecher E, Sheridan E, Taylor AE, Wilroy RS, et al. ABCA12 is the major harlequin ichthyosis gene. J Invest Dermatol. 2006;126:2408–2413. doi: 10.1038/sj.jid.5700455. [DOI] [PubMed] [Google Scholar]
- 204.Kelsell DP, Norgett EE, Unsworth H, Teh MT, Cullup T, Mein CA, Dopping-Hepenstal PJ, Dale BA, Tadini G, Fleckman P, et al. Mutations in ABCA12 underlie the severe congenital skin disease harlequin ichthyosis. Am J Hum Genet. 2005;76:794–803. doi: 10.1086/429844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Moskowitz DG, Fowler AJ, Heyman MB, Cohen SP, Crumrine D, Elias PM, Williams ML. Pathophysiological basis for growth failure in children with ichthyosis: an evaluation of cutaneous ultrastructure, epidermal permeability barrier function, and energy expenditure. J Pediatr. 2004;145:82–92. doi: 10.1016/j.jpeds.2004.03.052. [DOI] [PubMed] [Google Scholar]
- 206.Rassner UA, Crumrine DA, Nau P, Elias PM. Microwave incubation improves lipolytic enzyme preservation for ultrastructural cytochemistry. Histochem J. 1997;29:387–392. doi: 10.1023/a:1026438917856. [DOI] [PubMed] [Google Scholar]
- 207.Brattsand M, Stefansson K, Lundh C, Haasum Y, Egelrud T. A proteolytic cascade of kallikreins in the stratum corneum. J Invest Dermatol. 2005;124:198–203. doi: 10.1111/j.0022-202X.2004.23547.x. [DOI] [PubMed] [Google Scholar]
- 208.Caubet C, Jonca N, Brattsand M, Guerrin M, Bernard D, Schmidt R, Egelrud T, Simon M, Serre G. Degradation of corneodesmosome proteins by two serine proteases of the kallikrein family, SCTE/KLK5/hK5 and SCCE/KLK7/hK7. J Invest Dermatol. 2004;122:1235–1244. doi: 10.1111/j.0022-202X.2004.22512.x. [DOI] [PubMed] [Google Scholar]
- 209.Horikoshi T, Igarashi S, Uchiwa H, Brysk H, Brysk MM. Role of endogenous cathepsin D-like and chymotrypsin-like proteolysis in human epidermal desquamation. Br J Dermatol. 1999;141:453–459. doi: 10.1046/j.1365-2133.1999.03038.x. [DOI] [PubMed] [Google Scholar]
- 210.Sprecher E, Ishida-Yamamoto A, Mizrahi-Koren M, Rapaport D, Goldsher D, Indelman M, Topaz O, Chefetz I, Keren H, O’Brien TJ, et al. A mutation in SNAP29, coding for a SNARE protein involved in intracellular trafficking, causes a novel neurocutaneous syndrome characterized by cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma. Am J Hum Genet. 2005;77:242–251. doi: 10.1086/432556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Hepp R, Langley K. SNAREs during development. Cell Tissue Res. 2001;305:247–253. doi: 10.1007/s004410100359. [DOI] [PubMed] [Google Scholar]
- 212.Cartlidge P, Rutter N. Skin barrier function. In: Polin R, Fox W, editors. Fetal and Neonatal Physiology. WB Saunders; Philadelphia, PA: 1998. pp. 771–788. [Google Scholar]
- 213.Greene SL, Muller SA. Netherton’s syndrome. Report of a case and review of the literature. J Am Acad Dermatol. 1985;13:329–337. [PubMed] [Google Scholar]
- 214.Sybert VP. Genetic Skin Disorders. 13–16. Oxford University Press; Oxford, UK: 1997. pp. 205–206. [Google Scholar]
- 215.Traupe H. A Guide to Clinical Diagnosis, Genetic Counseling, and Therapy. Springer Verlag; New York: 1989. Ichthyosis; p. 253. [Google Scholar]
- 216.Williams M, Shwayder T. Pediatric Dermatology. Churchill Livingstone; New York: 1995. Ichthyosis and disorders of cornification; pp. 413–454. [Google Scholar]
- 217.Judge MR, Morgan G, Harper JI. A clinical and immunological study of Netherton’s syndrome. Br J Dermatol. 1994;131:615–621. doi: 10.1111/j.1365-2133.1994.tb04971.x. [DOI] [PubMed] [Google Scholar]
- 218.Freedberg IM, Baden HP. The metabolic response to exfoliation. J Invest Dermatol. 1962;38:277–284. doi: 10.1038/jid.1962.50. [DOI] [PubMed] [Google Scholar]
- 219.Perlstein P. Physical environment. In: Fanaroff A, Martin R, editors. Neonatal-Perinatal Medicine. Mosby-Year Book; St. Louis, MO: 1997. pp. 481–501. [Google Scholar]
- 220.Williams ML, Elias PM. Genetically transmitted, generalized disorders of cornification. The ichthyoses. Dermatol Clin. 1987;5:155–178. [PubMed] [Google Scholar]
- 221.Fowler AJ, Moskowitz DG, Wong A, Cohen SP, Williams ML, Heyman MB. Nutritional status and gastrointestinal structure and function in children with ichthyosis and growth failure. J Pediatr Gastroenterol Nutr. 2004;38:164–169. doi: 10.1097/00005176-200402000-00012. [DOI] [PubMed] [Google Scholar]
- 222.Williams ML, Hanley K, Elias PM, Feingold KR. Ontogeny of the epidermal permeability barrier. J Investig Dermatol Symp Proc. 1998;3:75–79. doi: 10.1038/jidsymp.1998.18. [DOI] [PubMed] [Google Scholar]
- 223.Lampe MA, Burlingame AL, Whitney J, Williams ML, Brown BE, Roitman E, Elias PM. Human stratum corneum lipids: characterization and regional variations. J Lipid Res. 1983;24:120–130. [PubMed] [Google Scholar]
- 224.Elias PM, Cooper ER, Korc A, Brown BE. Percutaneous transport in relation to stratum corneum structure and lipid composition. J Invest Dermatol. 1981;76:297–301. doi: 10.1111/1523-1747.ep12526137. [DOI] [PubMed] [Google Scholar]
- 225.Elias PM, Crumrine D, Rassner U, Hachem JP, Menon GK, Man W, Choy MH, Leypoldt L, Feingold KR, Williams ML. Basis for abnormal desquamation and permeability barrier dysfunction in RXLI. J Invest Dermatol. 2004;122:314–319. doi: 10.1046/j.1523-1747.2003.22258.x. [DOI] [PubMed] [Google Scholar]
- 226.van den Brink DM, Wanders RJ. Phytanic acid: production from phytol, its breakdown and role in human disease. Cell Mol Life Sci. 2006;63:1752–1765. doi: 10.1007/s00018-005-5463-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Shibaki A, Akiyama M, Shimizu H. Novel ALDH3A2 heterozygous mutations are associated with defective lamellar granule formation in a Japanese family of Sjogren-Larsson syndrome. J Invest Dermatol. 2004;123:1197–1199. doi: 10.1111/j.0022-202X.2004.23505.x. [DOI] [PubMed] [Google Scholar]
- 228.Holleran WM, Ginns EI, Menon GK, Grundmann JU, Fartasch M, McKinney CE, Elias PM, Sidransky E. Consequences of beta-glucocerebrosidase deficiency in epidermis. Ultrastructure and permeability barrier alterations in Gaucher disease. J Clin Invest. 1994;93:1756–1764. doi: 10.1172/JCI117160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Schmuth M, Man MQ, Weber F, Gao W, Feingold KR, Fritsch P, Elias PM, Holleran WM. Permeability barrier disorder in Niemann-Pick disease: sphingomyelin-ceramide processing required for normal barrier homeostasis. J Invest Dermatol. 2000;115:459–466. doi: 10.1046/j.1523-1747.2000.00081.x. [DOI] [PubMed] [Google Scholar]