

Abstract
The significance of intracellular β-amyloid (Aβ42) accumulation is increasingly recognized in Alzheimer's disease (AD) pathogenesis. Aβ removal mechanisms that have attracted attention include IDE/neprilysin degradation and antibody-mediated uptake by immune cells. However, the role of the ubiquitin-proteasome system (UPS) in the disposal of cellular Aβ has not been fully explored. The E3 ubiquitin ligase Parkin targets several proteins for UPS degradation, and Parkin mutations are the major cause of autosomal recessive Parkinson's disease. We tested whether Parkin has cross-function to target misfolded proteins in AD for proteasome-dependent clearance in SH-SY5Y and primary neuronal cells. Wild-type Parkin greatly decreased steady-state levels of intracellular Aβ42, an action abrogated by proteasome inhibitors. Intracellular Aβ42 accumulation decreased cell viability and proteasome activity. Accordingly, Parkin reversed both effects. Changes in mitochondrial ATP production from Aβ or Parkin did not account for their effects on the proteasome. Parkin knock-down led to accumulation of Aβ. In AD brain, Parkin was found to interact with Aβ and its levels were reduced. Thus, Parkin is cytoprotective, partially by increasing the removal of cellular Aβ through a proteasome-dependent pathway.
Keywords: amyloid, ubiquitin ligase, Parkin, Alzheimer's, proteasome
Parkin is a 465-amino-acid protein containing an N-terminal ubiquitin-like (Ubl) domain linked to a C-terminal RING box (Shimura et al., 2000). Parkin functions as an E3 ubiquitin-protein ligase (Imai et al., 2000; Shimura et al., 2000; Zhang et al., 2000), facilitating the proteasomal degradation of misfolded proteins. Several Parkin gene mutations have been linked to autosomal-recessive Parkinsonism with juvenile onset (Kitada et al., 1998; Lucking et al., 2000).
In cell culture systems, Parkin fusion proteins have been shown to interact with several proteins, including the α-synuclein-binding protein synphilin-1 (Chung et al., 2001), actin filaments (Huynh et al., 2000), and α/β tubulin (Ren et al., 2003). Parkin has also been found to be up-regulated during the integrated cellular response to misfolded protein-induced ER stress (Imai et al., 2001). Specific targets of Parkin activity having intrinsic toxic and aggregative properties include Pael-R, the Parkin-associated endothelin-like receptor (Imai et al., 2001), and possibly an O-glycosylated form of α-synuclein (Shimura et al., 2001). Thus, Parkin has been shown to suppress the toxicity of PAEL-R (Imai et al., 2001), mutated α-synuclein A30P (Petrucelli et al., 2002; Lo Bianco et al., 2004), and a poly(Q)-expanded mutant of ataxin-3 (Tsai et al., 2003). Deletions in the Parkin gene result in the accumulation of nonubiquitinated forms of α-synuclein and Pael-R in the brain (Imai et al., 2001; Shimura et al., 2001).
The accumulation of neuronal β-amyloid (Aβ) is increasingly recognized as a critical factor in Alzheimer's disease (AD) and related pathologies (Hartmann, 1999; Wilson et al., 1999; Gouras et al., 2005). Soluble fractions of AD brain Aβ are better correlated with disease severity than the larger insoluble pool (McLean et al., 1999), pointing to a possible contribution from the cellular compartment. There is also evidence to suggest that extracellular, parenchymal Aβ plaques in the brain are derived from degenerating Aβ42-laden neuronal cell bodies (Gouras et al., 2000; D'Andrea et al., 2001).
Expanding data have shown the importance of Aβ degradation or clearance mechanisms. Among these, the endopeptidases insulin-degrading enzyme (IDE) and neprilysin (NEP) have received the most attention (Selkoe, 2001). IDE engages extracellular secreted monomeric and plaque Aβ and, through a cytosolic pool of enzyme, the amyloid intracellular domain (AICD; Qiu et al., 1998; Edbauer et al., 2002; Farris et al., 2003; Leissring et al., 2003). NEP is a major extracellular Aβ-degrading enzyme; it decreases plaque formation and is active against mono- and oligomeric Aβ (Kanemitsu et al., 2003; Marr et al., 2003). Less is known about the clearance of intracellularly generated Aβ. Both IDE and a proteasome-dependent pathway have been shown to degrade endoplasmic reticulum (ER)-localized Aβ in transfected HeLa cells. However, only 30% of expressed Aβ was sensitive to the inhibitor MG132, suggesting limited proteasome involvement in that system (Schmitz et al., 2004). The mechanism behind the proteasome contribution was not further explored. Nevertheless, synthetic Aβ and purified 20S proteasome preparations interact to form complexes (Gregori et al., 1997).
Studies of cells and tissues derived from Parkin knockout mice and flies have indicated an increased sensitivity to cellular stressors of various types (Palacino et al., 2004; Pesah et al., 2004). Our earlier study with Parkin knockout skeletal muscle (Rosen et al., 2006) found enhanced accumulation of fragments of the endogenous β-amyloid precursor protein, including intracellular β-amyloid, and a heightened sensitivity to oxidative stress. Here we examine whether additional manipulations of Parkin expression can influence the accumulation and toxic effects of intracellular β-amyloid in neuronal cells. From its action as a ubiquitin ligase, but likely having other functions, we predicted that Parkin confers cytoprotection against Aβ42 when over-expressed and that this property would be proteasome dependent. Indeed, we found that proteasome inhibition interferes with Parkin's ability to reverse intracellular β-amyloid-induced toxicity and accumulation. These findings were extended to uncover Parkin and β-amyloid associations in the AD brain.
Materials and Methods
Cloning and Viral Preparation
N-terminal myc epitope-tagged, human WT, T240R, and ΔUbl (residues 77–465) Parkin cDNAs in pcDNA3.1 (Shimura et al., 2000) were amplified along with the cytomegalovirus (CMV) promoter [forward primer GAT CAT TGG ACT TAA TTA ACA GAT ATA CGC GTT GAC ATT GAT TA and reverse primer CGC CAC TGT GCT GGA TAT CT] and subcloned into the Pac1 and BamH1 sites of a lentiviral backbone (see Fig. 1A; Lois et al., 2002). The Parkin lentiviral constructs (Lv-WT Parkin; Lv-T240R; Lv-ΔUbl) were packaged into HEK293FT cells using vesicular stomatitis virus (VSVg) as source for envelope protein and the delta 8.9 plasmid, containing genes encoding for core proteins and enzymes to reverse transcribe the lentiviral RNA genome after infection (Lois et al., 2002). The Aβ1–42 cDNA fragment was amplified from a Herpes virus clone using a BamH1-con-taining forward primer (GAG CGG ATC CAT CGC GAT GCT GCC C) and a Not1-containing reverse primer (CAT GCG GCC GCT ACG CTA TGA CAA C) and subcloned into pcDNA3.1 (Invitrogen, Carlsbad, CA). Adenovirus-based constructs harboring a doxycycline-inducible Aβ42 and the positive transactivator (AdTRE-Aβ ± Dox) were prepared as previously described (Magrané et al., 2004). Parkin siRNA duplex (sense strand sequence ugguuuuccagugcaacucc) was purchased from Dharmacon.
Fig. 1.

A–I: Parkin expression systems. A: Schematic representation of the lentiviral construct encoding WT and mutant (T240R and ΔUbl) myc-tagged Parkin. B: Western blot of whole-cell extracts demonstrating expression of lentiviral Parkin constructs WT, T240R, and ΔUbl. Immunofluorescence analysis of SH-SY5Y cells transduced for Parkin (D) (rhodamine fluorescence) and its accompanying myc tag (C) (FITC fluorescence) at ×40. F: Hoechst 33258. G: Hoffman modulation contrast. H: Anti-Parkin labeling reveals expression in both the cytosol and the nucleus of SH-SY5Y cells (confocal 0.1 μm optical section). I: Subcellular fractionation of Adv-Aβ42-infected SH-SY5Y cells to localize β-amyloid and Parkin expression. Equal amounts of protein from each fraction were resolved on 4–12% Bis-Tris NuPAGE gels, so the levels shown would not necessarily reflect their relative absolute contributions to total cellular protein. Endogenous Parkin is found in all compartments, especially the mitochondrial fraction (P2), as characterized by cytochrome C localization. Heterologous Aβ42 is similarly abundant in mitochondria but also resides with Parkin in membrane-associated fractions (P3). Aβ and Parkin levels found in the crude nuclear fraction (P1) likely represent a contribution from either unbroken cells and/or nuclear membrane breakdown, as per cytochrome C contamination.
Cell Culture, Transfection, and Viral Infection
Human neuroblastoma SH-SY5Y cells (seeding density 2 × 105 cells) were grown in 24-well dishes to 70% confluence in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) plus added 10% (vol/vol) heat-inactivated fetal bovine serum (FBS; Invitrogen), penicillin/streptomycin, and 2 mM L-glutamine at 37°C and 5% CO2. Cultures were washed twice in phosphate-buffered saline (PBS), reincubated in DMEM plus 8 μg/ml protamine, and infected at a multiplicity of infection (m.o.i.) of 10 with the myc-Parkin lentiviral constructs for 24 hr. Thereafter, cells were coinfected with 100 m.o.i. of AdTRE-Aβ virus for an additional 24 hr. Finally; Aβ42 expression was induced with 1 μg/ml doxycycline over the next 16–24 hr. Where indicated, cells were finally treated overnight with the proteasome inhibitor Z-Leu-Leu-Leu-al (MG132; 20 μM) before harvest. Transient transfection experiments were performed in DMEM using Lipofectamine 2000 (Invitrogen) under serum-free conditions. Infection with the myc-Parkin lentiviral constructs was performed 24 hr before transfection with Aβ1–42.
Cells were harvested in 1× lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM sodium orthovanadate, 1 μg/ml leupeptin, and 0.1 mM PMSF) and centrifuged at 10,000g for 20 min at 4°C, and the supernatant containing the soluble fraction was collected. The pellet, containing the insoluble fraction, was suspended in 70% formic acid (stock) on ice for 2–3 hr and neutralized with 1 N NaOH. Protein estimation was performed using the microscale Bio-Rad Protein Assay (Bio-Rad, Hercules, CA). Subcellular fractionation began with cell lysis in hypotonic buffer (10 mM Tris-HCl, pH 7.5, 10 mM KCl, 1 mM MgCl2). After addition of sucrose (0.25 M final), differential centrifugation of the whole-cell lysate (WCE) produced fractions P1 (mostly nuclei, 1,200g, 10 min), P2 (mitochondria, 5,000g), P3 (vesicular, 22,000g, 30 min), and S3 (residual, including soluble proteins).
Primary Mouse Cortical Neuron Cultures
Mouse cortex was isolated from 2-day-old pups, and the cells were dissociated by limited trypsinization, followed by trituration through a Pasteur pipette. Approximately 1,500 cells/mm2 were seeded onto glass coverslips precoated with poly-L-ornithine and laminin (5 μg/ml each in PBS; Sigma, St. Louis, MO). Cortical cultures were grown at 37°C and 5% CO2 for 10 days in Neurobasal medium (Life Technologies, Grand Island, NY) supplemented with 2% (v/v) B-27 supplement (Invitrogen) and 50 μM of β-mercaptoethanol (Sigma). Culture media were changed every second day. Neurons were infected with myc-Parkin lentiviral and/or AdTRE-Aβ viral constructs as described above and treated with 10 μM MG132.
Antibodies, Immunoprecipitation, and Immunoblotting
Immunoprecipitation of Aβ42 from cell lysates was performed in 1× STEN lysis buffer (50 mM Tris, pH 7.6, 150 mM NaCl, 2 mM EDTA, 0.2% NP-40, 0.2% BSA, 20 mM PMSF, and 1× protease cocktail inhibitor) with anti-Aβ rabbit polyclonal R1282 (1:100). Immunostaining of myc-tagged Parkin was performed with anti-Parkin rabbit polyclonal (1:300) antibody (Cell Signaling Technology, Beverly, MA) and goat anti-rabbit Cy3 or anti-myc-tag mouse monoclonal clone 9E10 antibody (1:200; Upstate, Lake Placid, NY) and goat anti-mouse Cy2. Anti-20S proteasome (Calbiochem, La Jolla, CA) recognizes various 27–30-kD core subunits (1:100 immunoprecipitation, 1:1,000 Western blot). Whole-cell lysates or soluble/insoluble fractions (20 μg protein) were separated by SDS-PAGE on NuPAGE 4–12% Bis-Tris gels (Invitrogen) and then blotted onto PVDF membrane (Immobilon-P; Millipore, Bedford, MA). Membranes were blocked in TBS containing 5% (w/v) nonfat dry milk. Immunoblot signal detection was performed using anti-Aβ42 mouse monoclonal 6E10 (1:600) and anti-Parkin mouse monoclonal PRK8 (1:500; Signet, Dedham, MA). After incubation with primary antibodies for 18 hr at 4°C, blots were washed, incubated in HRP-conjugated secondary antibodies (1:2,000 dilution; Dako, Carpinteria, CA), and washed again, and the signals were visualized by using enhanced chemiluminescence reagents and film from GE Healthcare.
Human and Transgenic Mouse Brain Samples
AD and age-matched control human brain samples were obtained from the Harvard Brain Tissue Resource Center at McLean Hospital (Belmont, MA). All AD samples were Braak stage V, characterized by moderately abundant neuronal loss and NFT formation in limbic and medial temporal structures. Rapid autopsy brain samples were frozen at post-mortem intervals ranging from 7 to 21 hr. Cortical tissues from Brodmann areas 10 (frontal pole) and 20/36 (inf./med. temporal) were pulverized at −80°C and homogenized in lysis buffer (Tris-HCl (pH 7.4, 20 mM), NaCl (150 mM), Na4P2O7 (10 mM), Na3VO4 (2 mM), protease inhibitor cocktail (Complete; Roche Biochemicals, Indianapolis, IN), phenylmethylsulfonyl fluoride (PMSF; 44 mg/ml), and Triton X-100:1% v/v) on ice before centrifugation at 10,000g for 15 min. The whole-brain lysate supernatant was loaded directly for Western blot analysis (20 μg of total protein) or used for immunoprecipitation (IP; 100–300 μg of protein). After a preclearing incubation with protein A/G-Sepharose (Sigma, St. Louis, MO), extracts were incubated for 3–4 hr at 4°C with 3 μg primary antibody and protein A/G-Sepharose for an additional 1 hr. Immunoprecipitates were harvested by centrifugation at 14,000g for 5 min at 4°C and washed several times in 4°C, 1× phosphate-buffered saline (PBS) containing protease inhibitor cocktail (Roche Biochemical, Indianapolis, IN) and PMSF before elution and electrophoresis. Six-month-old Parkin knockout and littermate control mousse brains (Palacino et al., 2004) were kindly provided by Dr. Jie Shen, Department of Neurology, Harvard Medical School, and extracts were prepared in the same way as for the human samples.
Cell Viability Assays
To measure cell viability, cells were washed twice in warm D-PBS and incubated in 1 ml DMEM (no serum) containing 0.5 mg (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT; Molecular Probes, Eugene, OR) for 2–3 hr at 37°C and 5% CO2. 4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate (WST-1) was alternatively used with equal results. The medium was aspirated, and the cells were washed twice with warm D-PBS. The formazan salts were dissolved in 1 ml pure ethanol. Cells were homogenized by repetitive pipetting and centrifuged for 5 min at 4,500 rpm, and the supernatant was collected. Absorbance was read against an ethanol blank at 564 nm.
20S Proteasome Activity Assay
Human neuroblastoma SH-SY5Y cells were washed twice in PBS and incubated with the fluorescent 20S proteasome-specific substrate succinyl-LLVY-AMC (250 μM) at 37°C for 2 hr. This assay reflects chymotrypsin-like activity of the proteasome. The medium was discarded, and cells were lysed in 50 mM HEPES, pH 7.5, 5 mM EDTA, 150 mM NaCl, and 1% Triton X-100, containing 2 mM ATP. The AMC fluorophore, which is released after cleavage from succinyl-LLVY-AMC (Chemicon, Temecula, CA), is detected using a 380/460-nm filter set in a fluorometer (excitation at 351 nm and emission at 430 nm).
ATP Measurement
Mitochondrial ATP production was determined as described elsewhere (Veereshwarayya et al., 2006). Mitochondria were freshly isolated from cells (Manfredi et al., 2001) exposed to virus encoding the transgenes. Graphs and statistical analyses were performed in GraphPad Prism (GraphPad, San Diego, CA).
Results
To study the affect of Parkin on the biology of intracellular β-amyloid in neuronal cell types, we generated recombinant lentiviral constructs expressing myc epitope-tagged forms of either WT or mutant Parkin (Fig. 1A). Human neuroblastoma SH-SY5Y cells were infected for 24 hr with 10 m.o.i. of myc-tagged Parkin lentivirus prior to extraction. Western blot analyses of total protein from infected cells identified proteins with the expected molecular weight for the WT and T240R (∼54 kD) and the ΔUbl (∼42 kD) forms of Parkin (Fig. 1B). Immunofluorescent staining analysis of infected SH-SY5Y cells (Fig. 1C–H) revealed ubiquitous expression of Parkin in cytosolic as well as nuclear compartments (confocal image, Fig. 1H). Myc epitope tag expression correlated exactly with transfected levels of Parkin. Myc expression was not observed in uninfected control cells, which otherwise exhibited low levels of endogenous Parkin (not shown). Although Parkin is considered an ER-associated and cytosolic protein, endogenous nuclear localization is also observed (Stichel et al., 2000), contributing to the nuclear immunoreactivity in our virally mediated overexpression system. With a subcellular fractionation method (Fig. 1I), Parkin expression was readily detected in microsomal (P3) and cytosolic (S3) fractions. Consistent with the staining studies, some nuclear Parkin is also seen (P1). We also observed enrichment in the mitochondrial fraction (P2; confirmed by cytochrome c detection). These data are consistent with results in Parkin-expressing PC-12 cells (Darios et al., 2003). Adenovirally-expressing Aβ shares microsomal and mitochondrial compartments with Parkin.
To begin to examine the influence of Parkin on intracellular β-amyloid, we utilized a previously described doxycycline-inducible, adenoviral-mediated system for Aβ expression (Magrané et al., 2004) in SH-SY5Y cells and primary cortical neurons. Extraction of SH-SY5Y cells induced for 24 hr with doxycycline shows that intracellular Aβ accumulates in both detergent-soluble and -insoluble fractions of these cells (Fig. 2A). Knowing from our previous work that the absence of Parkin can increase Aβ levels, we tested the ability of the lentivirally expressed wild-type Parkin to mitigate accumulation of intracellular Aβ (Fig. 2B). Overexpression of wild-type Parkin, but neither mutant (T240R or ΔUbl) molecule, led to a substantial decrease in levels of intracellular β-amyloid in both detergent-soluble and -insoluble compartments (Fig. 2, lanes 4, 5) compared with control cells (Fig. 2, lanes 2, 3). Interestingly, an increase in both monomeric and oligomeric Aβ levels is seen in the soluble and insoluble fractions when either of the mutant Parkin constructs was overexpressed compared with control. The relative levels of oligomer to total Aβ species increased only slightly (Supp. Info. Fig. 1). Next, we tested the effect of proteasome inhibition on Aβ levels and, in turn, on the action of Parkin to curtail Aβ accumulation. In Figure 2C, MG132 or lactacystin treatment aggravates Aβ build-up. With the proteasome inhibited, Parkin however, has no ability to reduce these levels (Fig. 2D,E). Thus Parkin action is proteasome dependent. Neither MG132 nor Aβ, alone or in combination, significantly affects endogenous or expressed Parkin levels to account for these results. Similar experiments in cultures of primary mouse cortical neurons yielded the same striking results (Fig. 2F,G).
Fig. 2.

Aβ42 and Parkin levels in SH-SY5Y cells and primary cortical neurons in the presence of proteasome inhibitors. A: Cellular expression of β-amyloid under the inducible control of an adenovirus-based vector system shows equal partitioning into detergent (TX-100)-soluble and -insoluble fractions. SH-SY5Y cells infected at 100 m.o.i. and induced with 1 μg/ml doxycycline for 24 hr. The pellet was solubilized in 25% formic acid. B: Western blot analysis showing the effects of Parkin on steady-state levels of soluble (S) and insoluble (I) Aβ42 in SH-SY5Y cells. Aβ42 levels (top gel) detected with 6E10 antibody and Parkin levels (bottom gel) after stripping and reprobing with anti-Parkin antibody (PRK8). Wild-type but not mutant Parkin greatly reduces levels of Aβ42 in both compartments. Equal amounts of each soluble fraction sample (lanes 2, 4, 6, 8) were loaded, as shown in the actin blot below. C: Proteasome inhibition with MG132 (M) or lactacystin (L) increases steady-state levels of Aβ42. D: Parkin expression mitigates whole-cell Aβ42 levels, except where MG132 is present. E: Densitometric analysis of Parkin activity to lower Aβ levels and abrogation by inhibition of the proteasome (P < 0.02 compared with Parkin alone, n = 2 experiments). F, G: Parkin expression decreases steady-state levels of Aβ42 in whole-cell soluble (S) and insoluble (I) fractions prepared from primary neuronal cultures. An equal amount of protein (30 μg) corresponding to soluble fractions was loaded into each well on a 4–12% NuPAGE Bis-Tris gel. The insoluble fraction contained the entire cell pellet. Note that proteasome inhibition does not affect Parkin levels.
To address whether the observed changes in Aβ levels with Parkin and proteasome inhibitors are reflected in neuronal viability, SH-SY5Y cells were infected with adenoviral Aβ and the various Parkin expression constructs, and survival was tested utilizing either MTT or WST assays. The expression and accumulation of intracellular Aβ42 in human SH-SY5Y neuroblastoma cells caused a significant decrease in cell viability (∼35%; Fig. 3A, bar 3 compared with uninfected or no added doxycycline controls, bars 1, 2; P < 0.05). Overexpression of WT Parkin (bar 5) nearly fully reversed the effects of intracellular Aβ42 on cell death (P < 0.05). Neither T240R nor ΔUbl Parkin was able to reverse Aβ42-induced cell death (bars 6, 7). Inhibition of the proteasome with MG132 resulted in a minor decrease of viability (bar 8 vs. bar 1). However, exposure of Aβ42-stressed cells to MG132 completely abolished the ability of WT Parkin to protect against Aβ42-induced cell death (bar 10 vs. bar 5). This result is in agreement with the Aβ assays, suggesting that the mechanism of protection conferred by Parkin against cellular Aβ42 is, in part, dependent on the proteasome. Moreover, the effects of Parkin on amyloid were limited to intracellular accumulation, in that Parkin shows no activity to neutralize the toxicity associated with extracellularly applied synthetic Aβ in this assay (Fig. 3B). We also controlled for the possibility that serum could be a contributing source of the intracellular Aβ that is reduced by Parkin or provide some other cofactor to degrade Aβ. In Figure 3C, Parkin significantly decreased intracellular Aβ in either serum-containing or serum-free conditions.
Fig. 3.

Cell death in Aβ42-stressed neuroblastoma cells: rescue by WT Parkin. A: MTT reduction is inhibited in Aβ-expressing SH-SY5Y cells in the presence of doxycycline (bar 3) and reversed only by WT Parkin (bar 5) and not its mutant forms. No reversal is obtained in the presence of MG132 (bar 10). Mean ± SD, n = 5, P < 0.05, Mann-Whitney test. *Significantly different from control (bar 1), #significantly different from Aβ42-stressed cells (bar 3). All values expressed as percentage of control. B: Synthetic Aβ, when added to SH-SY5Y cell medium (10 μM), is equally toxic but not rescued through Parkin expression; n = 4. C: In the absence of serum components, Parkin similarly reduces levels of intracellular Aβ. The lower amount of cellular Aβ under serum-free conditions likely reflects both stress and reduced uptake of exogenous Aβ present in the serum.
To test directly the involvement of proteasome function in the effect of Parkin of eliminating steady-state levels of intracellular Aβ42, proteasome activity was analyzed in lysates from SH-SY5Y cells exposed to combinations of Parkin and Aβ constructs and treatment with proteasome inhibitors. Expression of intracellular Aβ42 significantly decreased (P < 0.05) the activity level of the 20S proteasome compared with control (Fig. 4A, bar 5 vs. bar 1 or 4). This result was confirmed in an in vitro assay with synthetic Aβ peptide (not shown). WT Parkin significantly reversed (P < 0.05) these effects (bar 7), whereras both T240R and ΔUbl failed to reverse the inhibition of the 20S proteasome by intracellular Aβ42 (bars 8, 9). Accumulation of intracellular Aβ accordingly caused a slight increase in the overall level of ubiquitinated cellular proteins, also reversed by Parkin. However, this was difficult to quantify owing to the appearance of a generalized smear of such proteins on Western blots of antiubiquitin immunoprecipitations (not shown). Furthermore, inhibition of the proteasome with either MG132 or lactacystin (bars 2, 3) was not reversed by WT Parkin in control (bar 11) or Aβ42-stressed (bar 14) cells. Importantly, WT Parkin has an intrinsic capacity to increase substrate throughput significantly (bar 10). In further support of the hypothesis that intracellular Aβ is both toxic to and degraded by the proteasome, we show in Figure 4B that immunoprecipitation of 20S proteasome also precipitates Aβ42 (lane 6). Their interaction in Aβ-expressing SH-SY5Y cells is dissociable under standard denaturing and nonreducing (without β-mercaptoethanol) conditions (lane 5).
Fig. 4.

Aβ42 impairs 20S proteasome activity while associating with it and is rescued by Parkin. A: The activity of the 20S proteasome is inhibited in Aβ42-stressed SH-SY5Y cells (bar 5 vs. bar 4, *P < 0.05) and rescued by WT Parkin (bar 7 vs. bar 5, #P < 0.05) but not up to stimulated levels afforded by Parkin alone (bar 10 vs. bar 4, *P < 0.05). WT Parkin cannot overcome MG132 or lactacystin toxicities, under conditions with or without Aβ42 (bars 11 and 14 vs. 2, 3, and 6). Triplicate of n = 2 experiments. B: Aβ association with the proteasome. SH-SY5Y cells were induced to express β-amyloid, and the 20S proteasome was immunoprecipitated with an antibody recognizing multiple α and β subunits in the 27–30-kD range. The IPs were solubilized in sample buffer lacking (−) or containing (+) the reducing agent β-mercaptoethanol (β-ME). More Aβ immuno-reactivity is released from association with the anti-20S precipitate in the presence of β-ME (lane 6 vs. 5). C: Left: Effects of Parkin on simultaneous Aβ-affected and mitochondria-/proteasome-inhibited cells. The mitochondrial inhibitors rotenone and CCCP do not affect chymotrypsin-like proteasome activity, in contradistinction to Aβ. However, in their presence, Parkin stimulation is reduced. [Mean ± 1 SD, n = 5: vs. ctrl (bar 1) *P < 0.05, vs. Aβ alone (bar 4), #P < 0.05, Mann-Whitney test.] Note that bars 1, 2, 3, 4 correspond to bars 1, 2, 10, 5 in A. Right: Parkin reverses ATP loss in Aβ42- and mitochondrial poison-stressed cells (bars 3, 4 and bars 5–8, respectively). [Mean ± 1SD, n = 3, vs. Aβ, rotenone, or CCCP alone *P < 0.05, Mann-Whitney test.]
The inhibition of intact cell-based proteasome function, an energy-requiring activity, by Aβ and its rescue by Parkin could be explained through local and/or indirect effects on the mitochondrial production of ATP. Parkin and Aβ each (Figs. 1I, 4B) and individually (Gregori et al., 1997; Ardley et al., 2003; Darios et al., 2003; Lopez-Salon et al., 2003; Schmitz et al., 2004; Caspersen et al., 2005) localize to both structures. We tested the dependence of proteasome activity on mitochondrial ATP production in the context of Aβ and/or Parkin expression in Figure 4C. In the presence of mitochondrial poisons, rotenone or CCCP, cellular proteasome activity is not significantly affected. However, there is also no added boost to activity in the presence of Parkin, as there is when Parkin alone is added (Fig. 4C, left).
When ATP levels are examined, Parkin alone has no effect but is shown to restore the moderate ATP depletion caused by rotenone/CCCP and Aβ treatments (Fig. 4C, right). Thus, although Parkin is protective of mitochondrial activity, its action at the proteasome level is direct and is not limited by the sort of reductions in mitochondrial ATP caused by Aβ. Accordingly, oligomycin, which inhibits mitochondrial and proteasomal ATPase activities, severely limits mitochondrial ATP production and begins to inhibit proteasome activity, despite Parkin expression (data not shown). Therefore, the amyloid effect on the proteasome is not through moderate declines in ATP. Moreover, our in vitro assays show inhibition of 20S activity by Aβ when ATP levels are not limiting (data not shown).
The impressive effect of Parkin to reduce resting cellular Aβ levels most likely reflects an increase in proteasome throughput (degradation). Indeed, we tested whether Aβ can be targeted to the proteasome by ubiquitination, in cells made to overexpress Parkin and ubiquitin and exposed to proteasome inhibitor. We find under these conditions that a moderate fraction of Aβ is covalently modified with one or two ubiquitin molecules, arguing again for an interaction between β-amyloid and the proteasomal pathway (Supp. Info. Fig. 2). To test whether interfering with endogenous Parkin expression would influence intracellular Aβ accumulation in the opposite direction, we utilized siRNA against Parkin to decrease endogenous expression in SH-SY5Y cells. As shown in Figure 5A (left panel), Parkin siRNA led to decreased endogenous Parkin expression and a corresponding increase in the levels of induced intracellular Aβ. Moreover, we confirmed earlier results in parkin null skeletal muscle cells (Rosen et al., 2006) by testing for endogenous Aβ levels in the brains from these knockout mice to show a similar up-regulation (Fig. 5A, right).
Fig. 5.
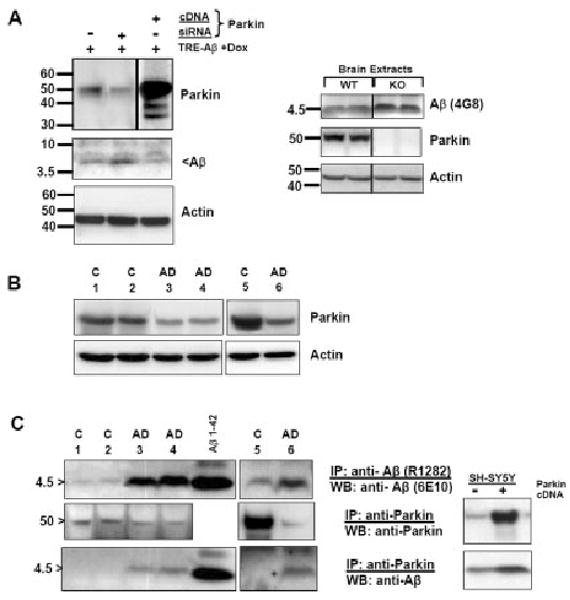
Parkin accelerates Aβ degradation. A: Left: Knock-down of Parkin in cell culture increases accumulation of cellular Aβ. SH-SY5Y cells were transfected with 80 pM Parkin siRNA for 48 hr simultaneously with inducible Aβ cDNA transfection. A companion transfection was performed with Parkin cDNA (a shorter exposure for this lane is shown). Duplicate experiments were performed with identical results. Right: Aβ expression in Parkin knockout mice. Whole-brain lysate from a pair of 6–7-month-old Parkin null mice and control littermates were immunoprecipitated with anti-Aβ (4G8) and immunoblotted with an anti-rodent Aβ-specific antibody (upper panel). Parkin immunoblot is shown below. A younger pair of mice gave similar results (not shown). B: Human autopsy brain samples from sporadic AD patients are deficient in Parkin. Lysates (20 μg protein) were fractionated by Western and probed for Parkin (top) or actin (loading control, bottom). Cortical samples corresponding to control cases 1 and 2 and AD cases 3 and 4 were from frontal pole (BA 10). In an additional experiment, a different control (case 5) and AD (case 6) sample were obtained from inferomedial temporal cortex (BA 20/36). All AD cases were Braak stage V. C: Immunoprecipitates (IP) of Aβ (top) or Parkin (middle and bottom) were prepared from lysates (200 μg protein) corresponding to the same cases as in B. The lower levels of Parkin were nonetheless associated with larger quantities of Aβ in all AD cases compared with control (bottom panel). Inset: Parkin was immunoprecipitated from SH-SY5Y extracts prepared from cells transfected with either control (−) or Parkin (+) vector and containing Aβ42. Western blots were probed for either Parkin (top) or β-amyloid (bottom). Increased Parkin leads to increased coimmunoprecipitable Aβ. Forty micrograms of total protein; higher exposure reveals oligomers in the pull down (results not shown).
The significance of Parkin's expanded function to eliminate Aβ in cell culture through UPS action was further probed in human brain. We tested for differences in the predicted interaction between Parkin and Aβ in two regions of cortex from AD (Braak V) and age-matched control cases. In frontal cortex lysates from two AD cases, endogenous Parkin levels were depressed (Fig. 5B; lanes 3, 4) relative to levels in unaffected brain (lanes 1, 2). We confirmed this difference in the temporal cortex from a third case compared with its control (lanes 5, 6). Immunoprecipitable Aβ levels were much higher in all cases compared with control, as expected (Fig. 5C, top row). The previous finding of reduced Parkin in AD brain was confirmed through immunoprecipitation (Fig. 5C, second row). In spite of the relative reduction in Parkin levels, Aβ peptide was clearly pulled down in anti-Parkin IPs performed on AD brains (Fig. 5C, third row). By utilizing the SH-SY5Y cell culture model, we confirmed that IP of endogenous Parkin pulls down detectable levels of cellular Aβ. Parkin expression enhances precipitable levels of Aβ (Fig. 5C, inset). An immunohistochemical comparison of samples from AD brain and age-matched controls revealed that β-amyloid deposition in extracellular plaques was not associated with Parkin, as expected. These samples also confirmed the Western data in demonstrating the deficiency of cellular Parkin in AD neurons (Supp. Info. Fig. 3). The results support a model in which Parkin can directly participate in Aβ degradation.
Discussion
For Parkinson's disease, a large body of evidence exists to support proteasome dysfunction as a major contributor to disease pathogenesis (McNaught et al., 2003; Petrucelli and Dawson, 2004; Ross and Pickart, 2004; Snyder and Wolozin, 2004). Our experiments with Parkin and β-amyloid removal build on prior studies that point to an expanded involvement of the UPS in AD brain. For instance, damage to proteasome activities is readily detected in AD brain (Keller et al., 2000b; Lopez Salon et al., 2000), wherein ubiquitin (Ub) and its conjugates are deposited in neuritic plaque and PHF/NFT structures (Perry et al., 1987; Morishima-Kawashima et al., 1993). In this regard, it was shown experimentally that synthetic β-amyloid directly binds to (Gregori et al., 1997) and inhibits substrate degradation by the 20S proteasome (Gregori et al., 1995; Lopez Salon et al., 2003). Additionally, phospho-tau is directly conjugated to Ub in AD brain and in vitro, thereby targeting it for UPS degradation (Perry et al., 1989; Morishima-Kawashima et al., 1993; Shimura et al., 2004). Recently, a mouse strain expressing mutated human tau on a Parkin null background displayed elevated levels of hyperphosphorylated tau and amyloid deposits in the hippocampus and in other sites, along with reduced antioxidant capacity and alterations in chaperone expression (Rodríguez-Navarro et al., 2008). Our findings on β-amyloid degradation and Parkin, already linked to early-onset Parkinson's disease, suggest that Parkin may have pluri-functional action across other neurodegenerative disorders, including AD. For instance, Parkin has been shown to eliminate polyglutamine-expanded proteins, reducing their aggregation and toxicity, and to restore proteasome function (Tsai et al., 2003).
This study demonstrates that Parkin expression facilitates the removal of intracellular β-amyloid. Conversely, the absence of Parkin activity tends to raise resting Aβ levels. Inhibition of the proteasome also results in the accumulation of Aβ42, further implicating proteasome-mediated degradation. This latter aspect confirms other studies that employ synthetic amyloid applications or nonneuronal cell lines (Lopez Salon et al., 2003; Schmitz et al., 2004), but we utilized a system to regulate the expression of Aβ42 in the cytosol of neuronal cells. By focusing on intracellular Aβ42, extracellular mechanisms for Aβ42 disposal such as through the endocytosis/lysosomal pathway or cell surface IDE are isolated aside. Furthermore, Parkin does not influence the toxicity of extracellular amyloid, nor does the latter appear to be a major source of cellular Aβ through internalization. The same finding under serum-free conditions also argues against the existence of extracellular trophic factors or enzymes having clearance properties to confound the Parkin effect. Our results point to Parkin as an important chaperone for the proteasomal-based removal of both soluble and insoluble cytosolic Aβ42.
In accordance with these findings, Parkin has the expected function of protecting cell viability from the toxic effects of Aβ42 buildup. The mechanism for this is likely the direct removal of Aβ42 through proteasomal degradation. However, additional possibilities may account for the cytoprotection. First, Parkin may indirectly reduce sensitivity to amyloid-induced ROS and buildup of peroxidized products while acting at the level of the mitochondrion (Darios et al., 2003; Palacino et al., 2004; Jiang et al., 2004; Kuroda et al., 2006). Second, Parkin has direct action on the function of isolated mitochondria, insofar as we give evidence for recovered ATP production under Aβ42 and mitochondrial inhibitor conditions. However, these levels of ATP reduction per se (and their restoration) were not found to drive the proteasome dysfunction (and subsequent recovery) in the presence of Aβ42 and Parkin, respectively. Next, we demonstrated that intracellular Aβ42 inhibits proteasomal activity in vivo and in vitro (not shown). We found that Aβ in association with immunoprecipitated proteasomal subunits provides additional support for such an interaction, with damaging consequences to proteasomal function. An indirect mechanism involving oxidative damage may also play a role (Ding and Keller, 2001). In any case, althoughh Parkin is shown to preserve proteasome function under Aβ42 stress, we also show that Parkin intrinsically stimulates proteasomal activity as a third cytoprotective mechanism. Previous findings support the idea that overexpression of Parkin stimulates proteasome activity and reduces levels of carbonyl- and 3-nitrotyrosine-modified proteins (Hyun et al., 2002). Thus, the failures of the inactive T240R (RING 1 domain) and ΔUbl mutants (Shimura et al., 2000) to confer cytoprotection from Aβ toxicity, together with our observation that neither mutant changes proteasome activity, are consistent with loss of function (Darios et al., 2003; von Coelln et al., 2004; Kuroda et al., 2006; Hampe et al., 2006). However, the small increase in Aβ monomer and oligomer levels in their presence compared with control may indicate a dominant negative action in this regard.
Our main finding that Parkin accelerates intracellular Aβ degradation in a proteasome-dependent manner (Fig. 2C) raises the next question to be addressed through detailed, further study. Is ubiquitination of Aβ required for the delivery of Aβ to the proteasome? Is it necessary for Aβ to be retrotranslocated through a cytosolic phase, or can it proceed directly from the ER membrane? The primary mechanism to dispose of retained abnormal proteins in the ER is via ERAD, and, in the majority of cases, chaperones escort and assist in the polyubiquitination of soluble substrates to ensure efficient and rapid transit to the cytoplasmic 26S proteasome. However, a group of ERAD substrates does not require ubiquitin (for review see Hershko and Ciechan-over; 1998; Nakatsukasa and Brodsky, 2008), notably hydrophobic peptides (Kisselev et al., 2002; Orlowski and Wilk, 2003). Moreover, a subpopulation of proteasomes is directly in contact and functionally coupled with the cytoplasmic ER face (Bonifacino and Weissman, 1998; Lee et al., 2004). It is therefore possible that a membrane-bound, small peptide such as Aβ can be directly escorted by membrane-associated Parkin or one of its cochaperone binding partners (e.g., CHIP; Imai et al., 2002) to the 19S proteasome cap, without ubiquitination or retrotranslocation.
In preliminary studies under conditions of proteasome inhibition and Parkin and ubiquitin expressions, we give evidence that a moderate fraction of Aβ42 can be conjugated to either mono- or diubiquitin. These findings warrant further work to define the exact nature of the Aβ-substrate and Parkin-E3 ubiquitin ligase relationship.
A fourth mechanism, still compatible with the others, is that Parkin may stimulate or be part of the ER unfolded protein response (UPR) to Aβ. Parkin has been shown to protect cells from chemical and unfolded PAEL-R-induced ER stress and to participate in the UPR (Takahashi and Imai, 2003). The accumulation of intracellular Aβ has previously been shown to sensitize or induce the ER stress response (Ghribi et al., 2003; Esposito et al., 2004; Hoshino et al., 2007; Chafekar et al., 2008). We found that intracellular Aβ modestly increases some markers of ER stress (Grp94/KDEL and ERP72: 1.53 ± 0.16-fold, n = 3; P < .05) but that added Parkin does not further boost this response (1.36 ± 0.05-fold; data not shown). Interestingly, whereas endogenous Parkin levels exhibited some induction to acute Aβ expression (Fig. 2D), others report that general ER stressors do not up-regulate Parkin in SHSY5Y cells (Ledesma et al., 2002; West et al., 2003). Although these effects may be minimal, another possibility, not tested here, is that Aβ may modify Parkin solubility. Oxidative stress-induced declines in the solubility of Parkin have recently been noted by others (Lavoie et al., 2007; Wong et al., 2007).
To extend the importance of these cell culture findings, we tested the association between Parkin and Aβ in brain tissue from AD patients. The reduction of Parkin levels in AD brain (Fig. 5; Supp. Info. Fig. 3) signifies that, along with the general impairment of the UPS machinery in AD (Keller et al., 2000a), this component is additively inadequate to handle the chronic load of intracellular Aβ. We speculate that the interaction of the remaining available Parkin with intracellular Aβ in vivo (Fig. 5) indicates a direct substrate-ligase relationship that is likely enhanced in AD pathogenesis.
The absence of Parkin colocalization with extracellular plaque Aβ is explained by the confinement of Parkin to the intracellular compartment and its lower abundance there in AD. Another possibility, not tested here, is that Parkin binds exclusively to soluble monomeric and oligomeric Aβ and not to the insoluble, fibrillar Aβ species deposited in advanced plaques. These findings also raise the interesting question why the few Parkin-mutant patients who have come to brain autopsy do not display β-amyloid deposition. It is likely that the other Aβ-clearance pathways (Schmitz et al., 2004) are sufficiently intact in them to prevent accumulation. Among UPR-based mechanisms, these could be proteasome-dependent (ERAD) and still bypass a deficiency in Parkin, e.g., the transmembrane E3 ligase HRD1, which shares a Parkin substrate, PAEL-R (Omura et al., 2006), or be independent and involve autophagy (Kruse et al., 2006). Our results recommend therapeutic stimulation of Parkin as a strategy to limit Aβ levels when they exceed a certain threshold and other degradative systems are exhausted in AD.
Supplementary Material
Acknowledgments
The authors thank Dr. Michael Schlossmacher for Parkin cDNAs and helpful discussion, Dr. Dennis Selkoe for the R1282 antibody, Dr. Jie Shen for Parkin null brain samples, the Harvard Brain Tissue Resource Center for the rapid autopsy frozen AD and control samples, and the Bennett Charitable Foundation for partial funding of this work.
Contract grant sponsor: NIH; Contract grant number: NS41373 (to H.W.Q.).
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- Ardley HC, Scott GB, Rose SA, Tan NG, Markham AF, Robinson PA. Inhibition of proteasomal activity causes inclusion formation in neuronal and nonneuronal cells overexpressing Parkin. Mol Biol Cell. 2003;14:4541–4556. doi: 10.1091/mbc.E03-02-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Weissman AM. Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu Rev Cell Dev Biol. 1998;14:19–57. doi: 10.1146/annurev.cellbio.14.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- Chafekar SM, Zwart R, Veerhuis R, Vanderstichele H, Baas F, Scheper W. Increased Abeta1–42 production sensitizes neuroblastoma cells for ER stress toxicity. Curr Alzheimer Res. 2008;5:469–474. doi: 10.2174/156720508785908883. [DOI] [PubMed] [Google Scholar]
- Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM. Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat Med. 2001;7:1144–1150. doi: 10.1038/nm1001-1144. [DOI] [PubMed] [Google Scholar]
- D'Andrea MR, Nagele RG, Wang HY, Peterson PA, Lee DH. Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer's disease. Histopathology. 2001;38:120–134. doi: 10.1046/j.1365-2559.2001.01082.x. [DOI] [PubMed] [Google Scholar]
- Darios F, Corti O, Lucking CB, Hampe C, Muriel MP, Abbas N, Gu WJ, Hirsch EC, Rooney T, Ruberg M, Brice A. Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum Mol Genet. 2003;12:517–526. doi: 10.1093/hmg/ddg044. [DOI] [PubMed] [Google Scholar]
- Ding Q, Keller JN. Proteasome inhibition in oxidative stress neurotoxicity: implications for heat shock proteins. J Neurochem. 2001;77:1010–1017. doi: 10.1046/j.1471-4159.2001.00302.x. [DOI] [PubMed] [Google Scholar]
- Edbauer D, Willem M, Lammich S, Steiner H, Haass C. Insulin-degrading enzyme rapidly removes the beta-amyloid precursor protein intracellular domain (AICD) J Biol Chem. 2002;277:13389–13393. doi: 10.1074/jbc.M111571200. [DOI] [PubMed] [Google Scholar]
- Esposito L, Gan L, Yu GQ, Essrich C, Mucke L. Intracellularly generated amyloid-beta peptide counteracts the antiapoptotic function of its precursor protein and primes proapoptotic pathways for activation by other insults in neuroblastoma cells. J Neurochem. 2004;91:1260–1274. doi: 10.1111/j.1471-4159.2004.02816.x. [DOI] [PubMed] [Google Scholar]
- Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guenette S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003;100:4162–4167. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghribi O, Herman MM, Savory J. Lithium inhibits Abeta-induced stress in endoplasmic reticulum of rabbit hippocampus but does not prevent oxidative damage and tau phosphorylation. J Neurosci Res. 2003;71:853–862. doi: 10.1002/jnr.10511. [DOI] [PubMed] [Google Scholar]
- Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouras GK, Almeida CG, Takahashi RH. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer's disease. Neurobiol Aging. 2005;26:1235–1244. doi: 10.1016/j.neurobiolaging.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Gregori L, Fuchs C, Figueiredo-Pereira ME, Van Nostrand WE, Goldgaber D. Amyloid beta-protein inhibits ubiquitin-dependent protein degradation in vitro. J Biol Chem. 1995;270:19702–19708. doi: 10.1074/jbc.270.34.19702. [DOI] [PubMed] [Google Scholar]
- Gregori L, Hainfeld JF, Simon MN, Goldgaber D. Binding of amyloid beta protein to the 20 S proteasome. J Biol Chem. 1997;272:58–62. doi: 10.1074/jbc.272.1.58. [DOI] [PubMed] [Google Scholar]
- Hampe C, Ardila-Osorio H, Fournier M, Brice A, Corti O. Biochemical analysis of Parkinson's disease-causing variants of Parkin, an E3 ubiquitin-protein ligase with monoubiquitylation capacity. Hum Mol Genet. 2006;15:2059–2075. doi: 10.1093/hmg/ddl131. [DOI] [PubMed] [Google Scholar]
- Hartmann T. Intracellular biology of Alzheimer's disease amyloid beta peptide. Eur Arch Psychiatry Clin Neurosci. 1999;249:291–298. doi: 10.1007/s004060050102. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Hoshino T, Nakaya T, Araki W, Suzuki K, Suzuki T, Mizushima T. Endoplasmic reticulum chaperones inhibit the production of amyloid-beta peptides. Biochem J. 2007;402:581–589. doi: 10.1042/BJ20061318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun DH, Lee M, Hattori N, Kubo S, Mizuno Y, Halliwell B, Jenner P. Effect of wild-type or mutant Parkin on oxidative damage, nitric oxide, antioxidant defenses, and the proteasome. J Biol Chem. 2002;277:28572–28577. doi: 10.1074/jbc.M200666200. [DOI] [PubMed] [Google Scholar]
- Huynh DP, Scoles DR, Ho TH, Del Bigio MR, Pulst SM. Parkin is associated with actin filaments in neuronal and nonneural cells. Ann Neurol. 2000;48:737–744. [PubMed] [Google Scholar]
- Imai Y, Soda M, Takahashi R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J Biol Chem. 2000;275:35661–35664. doi: 10.1074/jbc.C000447200. [DOI] [PubMed] [Google Scholar]
- Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001;105:891–902. doi: 10.1016/s0092-8674(01)00407-x. [DOI] [PubMed] [Google Scholar]
- Imai Y, Soda M, Hatakeyama S, Akagi T, Hashikawa T, Nakayama KI, Takahashi R. CHIP is associated with Parkin, a gene responsible for familial Parkinson's disease, and enhances its ubiquitin ligase activity. Mol Cell. 2002;10:55–67. doi: 10.1016/s1097-2765(02)00583-x. [DOI] [PubMed] [Google Scholar]
- Jiang H, Ren Y, Zhao J, Feng J. Parkin protects human dopaminergic neuroblastoma cells against dopamine-induced apoptosis. Hum Mol Genet. 2004;13:1745–1754. doi: 10.1093/hmg/ddh180. [DOI] [PubMed] [Google Scholar]
- Kanemitsu H, Tomiyama T, Mori H. Human neprilysin is capable of degrading amyloid beta peptide not only in the monomeric form but also the pathological oligomeric form. Neurosci Lett. 2003;350:113–116. doi: 10.1016/s0304-3940(03)00898-x. [DOI] [PubMed] [Google Scholar]
- Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer's disease. J Neurochem. 2000a;75:436–439. doi: 10.1046/j.1471-4159.2000.0750436.x. [DOI] [PubMed] [Google Scholar]
- Keller JN, Lauderback CM, Butterfield DA, Kindy MS, Yu J, Markesbery WR. Amyloid beta-peptide effects on synaptosomes from apolipoprotein E-deficient mice. J Neurochem. 2000b;74:1579–1586. doi: 10.1046/j.1471-4159.2000.0741579.x. [DOI] [PubMed] [Google Scholar]
- Kisselev AF, Kaganovich D, Goldberg AL. Binding of hydrophobic peptides to several noncatalytic sites promotes peptide hydrolysis by all active sites of 20 S proteasomes. Evidence for peptide-induced channel opening in the alpha-rings. J Biol Chem. 2002;277:22260–22270. doi: 10.1074/jbc.M112360200. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Kruse KB, Brodsky JL, McCracken AA. Autophagy: an ER protein quality control process. Autophagy. 2006;2:135–137. doi: 10.4161/auto.2.2.2388. [DOI] [PubMed] [Google Scholar]
- Kuroda Y, Mitsui T, Kunishige M, Matsumoto T. Parkin affects mitochondrial function and apoptosis in neuronal and myogenic cells. Biochem Biophys Res Commun. 2006;348:787–793. doi: 10.1016/j.bbrc.2006.06.201. [DOI] [PubMed] [Google Scholar]
- Lavoie M, Cortese G, Ostaszewski B, Schlossmacher M. The effects of oxidative stress on parkin and other E3 ligases. J Neurochem. 2007;103:2354–2368. doi: 10.1111/j.1471-4159.2007.04911.x. [DOI] [PubMed] [Google Scholar]
- Ledesma MD, Galvan C, Hellias B, Dotti C, Jensen PH. Astrocytic but not neuronal increased expression and redistribution of parkin during unfolded protein stress. J Neurochem. 2002;83:1431–1440. doi: 10.1046/j.1471-4159.2002.01253.x. [DOI] [PubMed] [Google Scholar]
- Lee RJ, Liu CW, Harty C, McCracken AA, Latterich M, Römisch K, DeMartino GN, Thomas PJ, Brodsky JL. Uncoupling retro-translocation and degradation in the ER-associated degradation of a soluble protein. EMBO J. 2004;23:2206–2215. doi: 10.1038/sj.emboj.7600232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, Frosch MP, Selkoe DJ. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40:1087–1093. doi: 10.1016/s0896-6273(03)00787-6. [DOI] [PubMed] [Google Scholar]
- Lo Bianco C, Schneider BL, Bauer M, Sajadi A, Brice A, Iwatsubo T, Aebischer P. Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha-synuclein rat model of Parkinson's disease. Proc Natl Acad Sci U S A. 2004;101:17510–17515. doi: 10.1073/pnas.0405313101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- Lopez Salon M, Morelli L, Castano E, Soto E, Pasquini J. Defective ubiquitination of cerebral proteins in Alzheimer's disease. J Neurosci Res. 2000;62:302–310. doi: 10.1002/1097-4547(20001015)62:2<302::AID-JNR15>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Lopez Salon M, Pasquini L, Besio Moreno M, Pasquini JM, Soto E. Relationship between beta-amyloid degradation and the 26S proteasome in neural cells. Exp Neurol. 2003;180:131–143. doi: 10.1016/s0014-4886(02)00060-2. [DOI] [PubMed] [Google Scholar]
- Lucking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW, Agid Y, Brice A. Association between early-onset Parkinson's disease and mutations in the parkin gene. French Parkinson's Disease Genetics Study Group. N Engl J Med. 2000;342:1560–1567. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- Magrané J, Smith RC, Walsh K, Querfurth HW. Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed beta-amyloid in neurons. J Neurosci. 2004;24:1700–1706. doi: 10.1523/JNEUROSCI.4330-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredi G, Spinazzola A, Checcarelli N, Naini A. Assay of mitochondrial ATP synthesis in animal cells. Methods Cell Biol. 2001;65:133–145. doi: 10.1016/s0091-679x(01)65008-8. [DOI] [PubMed] [Google Scholar]
- Marr RA, Rockenstein E, Mukherjee A, Kindy MS, Hersh LB, Gage FH, Verma IM, Masliah E. Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J Neurosci. 2003;23:1992–1996. doi: 10.1523/JNEUROSCI.23-06-01992.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Belizaire R, Isacson O, Jenner P, Olanow CW. Altered proteasomal function in sporadic Parkinson's disease. Exp Neurol. 2003;179:38–46. doi: 10.1006/exnr.2002.8050. [DOI] [PubMed] [Google Scholar]
- Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Titani K, Ihara Y. Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron. 1993;10:1151–1160. doi: 10.1016/0896-6273(93)90063-w. [DOI] [PubMed] [Google Scholar]
- Nakatsukasa K, Brodsky JL. The recognition and retrotranslocation of misfolded proteins from the endoplasmic reticulum. Traffic. 2008;9:861–870. doi: 10.1111/j.1600-0854.2008.00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura T, Kaneko M, Okuma Y, Orba Y, Nagashima K, Takahashi R, Fujitani N, Matsumura S, Hata A, Kubota K, Murahashi K, Uehara T, Nomura Y. A ubiquitin ligase HRD1 promotes the degradation of Pael receptor, a substrate of Parkin. J Neurochem. 2006;99:1456–1469. doi: 10.1111/j.1471-4159.2006.04155.x. [DOI] [PubMed] [Google Scholar]
- Orlowski M, Wilk S. Ubiquitin-independent proteolytic functions of the proteasome. Arch Biochem Biophys. 2003;415:1–5. doi: 10.1016/s0003-9861(03)00197-8. [DOI] [PubMed] [Google Scholar]
- Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- Perry G, Friedman R, Shaw G, Chau V. Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer disease brains. Proc Natl Acad Sci U S A. 1987;84:3033–3036. doi: 10.1073/pnas.84.9.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry G, Mulvihill P, Fried VA, Smith HT, Grundke-Iqbal I, Iqbal K. Immunochemical properties of ubiquitin conjugates in the paired helical filaments of Alzheimer disease. J Neurochem. 1989;52:1523–1528. doi: 10.1111/j.1471-4159.1989.tb09203.x. [DOI] [PubMed] [Google Scholar]
- Pesah Y, Pham T, Burgess H, Middlebrooks B, Verstreken P, Zhou Y, Harding M, Bellen H, Mardon G. Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development. 2004;131:2183–2194. doi: 10.1242/dev.01095. [DOI] [PubMed] [Google Scholar]
- Petrucelli L, Dawson T. Mechanism of neurodegenerative disease: role of the ubiquitin proteasome system. Ann Med. 2004;36:315–320. doi: 10.1080/07853890410031948. [DOI] [PubMed] [Google Scholar]
- Petrucelli L, O'Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Hardy J, Cookson MR. Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron. 2002;36:1007–1019. doi: 10.1016/s0896-6273(02)01125-x. [DOI] [PubMed] [Google Scholar]
- Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB, Rosner MR, Safavi A, Hersh LB, Selkoe DJ. Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J Biol Chem. 1998;273:32730–32738. doi: 10.1074/jbc.273.49.32730. [DOI] [PubMed] [Google Scholar]
- Ren Y, Zhao J, Feng J. Parkin binds to alpha/beta tubulin and increases their ubiquitination and degradation. J Neurosci. 2003;23:3316–3324. doi: 10.1523/JNEUROSCI.23-08-03316.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Navarro JA, Gòmez A, Rodal I, Perucho J, Martinez A, Furiò V, Ampuero I, Casarejos MJ, Solano RM, de Yébenes JG, Mena MA. Parkin deletion causes cerebral and systemic amyloidosis in human mutated tau over-expressing mice. Hum Mol Genet. 2008;17:3128–3143. doi: 10.1093/hmg/ddn210. [DOI] [PubMed] [Google Scholar]
- Rosen KM, Veereshwarayya V, Moussa CE, Fu Q, Goldberg MS, Schlossmacher MG, Shen J, Querfurth HW. Parkin protects against mitochondrial toxins and beta-amyloid accumulation in skeletal muscle cells. J Biol Chem. 2006;281:12809–12816. doi: 10.1074/jbc.M512649200. [DOI] [PubMed] [Google Scholar]
- Ross C, Pickart C. The ubiquitin–proteasome pathway in Parkinson's disease and other neurodegenerative diseases. Trends Cell Biol. 2004;14:703–711. doi: 10.1016/j.tcb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Schmitz A, Schneider A, Kummer MP, Herzog V. Endoplasmic reticulum-localized amyloid beta-peptide is degraded in the cytosol by two distinct degradation pathways. Traffic. 2004;5:89–101. doi: 10.1111/j.1600-0854.2004.00159.x. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Clearing the brain's amyloid cobwebs. Neuron. 2001;32:177–180. doi: 10.1016/s0896-6273(01)00475-5. [DOI] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson's disease. Science. 2001;293:263–269. doi: 10.1126/science.1060627. [DOI] [PubMed] [Google Scholar]
- Shimura H, Schwartz D, Gygi SP, Kosik KS. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J Biol Chem. 2004;279:4869–4876. doi: 10.1074/jbc.M305838200. [DOI] [PubMed] [Google Scholar]
- Snyder H, Wolozin B. Pathological proteins in Parkinson's disease: focus on the proteasome. J Mol Neurosci. 2004;24:425–442. doi: 10.1385/JMN:24:3:425. [DOI] [PubMed] [Google Scholar]
- Stichel CC, Augustin M, Kuhn K, Zhu XR, Engels P, Ullmer C, Lubbert H. Parkin expression in the adult mouse brain. Eur J Neurosci. 2000;12:4181–4194. [PubMed] [Google Scholar]
- Takahashi R, Imai Y. Pael receptor, endoplasmic reticulum stress, and Parkinson's disease. J Neurol. 2003;250(Suppl 3):III25–III29. doi: 10.1007/s00415-003-1305-8. [DOI] [PubMed] [Google Scholar]
- Tsai YC, Fishman PS, Thakor NV, Oyler GA. Parkin facilitates the elimination of expanded polyglutamine proteins and leads to preservation of proteasome function. J Biol Chem. 2003;278:22044–22055. doi: 10.1074/jbc.M212235200. [DOI] [PubMed] [Google Scholar]
- Veereshwarayya V, Kumar P, Rosen KM, Mestril R, Querfurth H. Differential effects of mitochondrial heat shock protein 60 and related molecular chaperones to prevent intracellular β-amyloid-induced inhibition of complex IV and limit apoptosis. J Biol Chem. 2006;281:29468–29478. doi: 10.1074/jbc.M602533200. [DOI] [PubMed] [Google Scholar]
- von Coelln R, Dawson VL, Dawson TM. Parkin-associated Parkinson's disease. Cell Tissue Res. 2004;318:175–184. doi: 10.1007/s00441-004-0924-4. [DOI] [PubMed] [Google Scholar]
- West AB, Gonzalez-de-Chavez F, Wilkes K, O'Farrell C, Farrer MJ. Parkin is not regulated by the unfolded protein response in human neuroblastoma cells. Neurosci Lett. 2003;341:139–142. doi: 10.1016/s0304-3940(03)00188-5. [DOI] [PubMed] [Google Scholar]
- Wilson CA, Doms RW, Lee VM. Intracellular APP processing and A beta production in Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:787–794. doi: 10.1097/00005072-199908000-00001. [DOI] [PubMed] [Google Scholar]
- Wong E, Tan J, Wang C, Zhang Z, Tay S, Zaiden N, Ko H, Dawson V, Dawson T, Lim K. Relative sensitivity of parkin and other cysteine-containing enzymes to stress-induced solubility alterations. J Biol Chem. 2007;282:12310–12318. doi: 10.1074/jbc.M609466200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, McLaughlin R, Goodyer C, LeBlanc A. Selective cytotoxicity of intracellular amyloid beta peptide1–42 through p53 and Bax in cultured primary human neurons. J Cell Biol. 2002;156:519–529. doi: 10.1083/jcb.200110119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.