

Abstract
Background
Matrix metalloproteinases (MMPs) are implicated in the pathogenesis of varicose veins. We have shown that MMP-2 causes relaxation of venous segments and suggested a role of venous smooth muscle (VSM) hyperpolarization; however, the downstream mechanisms are unclear. We tested whether MMP-2 induced venous relaxation involves inhibition of the Ca2+ mobilization mechanisms of VSM contraction due to generation of Arg-Gly-Asp (RGD)-containing peptides.
Methods
Circular segments of inferior vena cava (IVC) were isolated from male Sprague-Dawley rats, suspended between two wires in a tissue bath, and isometric contraction was measured. Contraction data in mg/mg tissue were presented as means±SEM.
Results
In IVC incubated in normal Krebs (2.5 mM Ca2+), the α-adrenergic agonist phenylephrine (Phe, 10-5 M) caused initial peak (133.2±17.5) followed by a maintained contraction (73.4±11.6), that was inhibited by MMP-2 (1 μg/mL) to 32.4±12.8 in 30 min. The inhibitory effects of MMP-2 were reversible by washing the tissue with Krebs or in the presence of the MMP inhibitors TIMP-1 (1 μg/ml), Ro 28-2653 and BB-94 (10-6 M), and were not associated with changes in IVC structure, demonstrating specificity. Angiotensin II (AngII, 10-6 M) caused a monophasic contraction (114.2±12.2), that was also inhibited by MMP-2 (66.0±7.4), suggesting a post-receptor effect on the downstream mechanisms of VSM contraction. To test the role of Ca2+ release from the sarcoplasmic reticulum, IVC was incubated in Ca2+-free (2 mM EGTA) Krebs with or without MMP-2. In Ca2+-free Krebs, caffeine did not cause contraction, suggesting limited role of the Ca2+-induced Ca2+-release mechanism, and Phe and AngII caused a small contraction (7.2±1.7 and 14.9±2.8) that was slightly increased by MMP-2 (10.4±3.0 and 33.8±10.0), suggesting little effect on IP3-induced Ca2+ release. To test the role of Ca2+ entry through membrane channels, after eliciting a transient Phe contraction in nominally 0 Ca2+ Krebs, increasing concentrations of CaCl2 (0.1, 0.3, 0.6, 1, 2.5 mM) were added and the [Ca2+]e-contraction relationship was constructed. The [Ca2+]e-contraction relation was reduced in MMP-2 treated IVC, suggesting inhibition of Ca2+ entry. In IVC treated with MMP-2, the Ca2+ channel blocker diltiazem (10-5M) did not cause any further inhibition of Phe contraction, suggesting that Ca2+ entry is already inhibited by MMP-2. To test whether MMP-2 actions involve generation of RGD and modulation of integrin receptors, experiments where repeated in IVC segments saturated with RGD (10-5 M), or pretreated with the αvβ3 integrin blocker cyclo-RGD. RGD-peptide caused only small relaxation of Phe contracted IVC (6.4±3.4%), and addition of MMP-2 to RGD-treated IVC caused further relaxation (69.7±3.0%). Pretreatement of IVC with cyclo-RGD did not significantly affect MMP-2 induced relaxation (55.0±5.0%).
Conclusions
In rat IVC, MMP-2 attenuates [Ca2+]e-dependent VSM contraction, without affecting Ca2+ release from intracellular Ca2+ stores. MMP-2 induced VSM relaxation may not involve RGD generation or activation of αvβ3 integrin receptor. MMP-2 induced inhibition of the Ca2+ entry mechanism of VSM contraction may play a role in the venous dilation associated with varicose vein formation.
Keywords: vascular smooth muscle, calcium, inferior vena cave, extracellular matrix, matrix metalloproteinase, varicose veins
INTRODUCTION
Varicose veins are a common venous disease with unclear mechanism. Varicose veins histology shows fragmentation of the elastic lamellae, loss of circular and longitudinal smooth muscle fibers, and damage of endothelium.1,2 The late stages of varicose veins are characterized by degeneration of extracellular matrix (ECM), endothelium, and venous smooth muscle (VSM) of the vein wall. In addition, alterations in collagen-to-elastin ratio are observed in varicose veins indicating an imbalance in the connective tissue matrix.3 There are several predisposing risk factors for varicose veins formation including female gender, pregnancy, obesity, aging and family history, suggesting a multifactorial etiology.4,5 However, the cellular mechanisms involved in early and recurrent varicose veins formation remain unclear.
In specimens of human varicose veins there is evidence of overexpression of matrix metalloproteinases (MMPs), and MMPs are found histologically in all layers of the vein wall.2,6,7 Also, thrombophlebitic varicose veins demonstrate a more pronounced expression and activity of MMPs than control veins.8 MMPs are highly conserved zinc-containing endopeptidases which are important in ECM tissue homeostasis, responsible for tissue turnover and remodeling, and MMPs proteolytic activity is balanced by the naturally occurring tissue inhibitors of MMPs (TIMPs).9,10 Other synthetic small molecule MMP inhibitors (hydroxamates, carboxylates, thiols) have been used to study MMP function in normal and diseased tissues. These inhibitors have variable affinity, specificity, and potency depending on the substitution groups affecting the zinc binding site or the catalytic domain of MMPs.11
Although the role of MMPs in varicose veins has largely been attributed to degradation of the ECM proteins, the effects of MMPs on VSM cell function particularly during the early stages of varicose veins formation are unclear. In a previous report we demonstrated that MMP-2 induced relatively rapid relaxation of phenylephrine contracted venous segments and suggested that it may involve VSM hyperpolarization and activation of K+ channels.12 The discovery of these novel rapid inhibitory effects of MMP-2 on venous function has made it necessary to further clarify their specificity. Also, the downstream mechanisms via which MMP-2 induced membrane hyperpolarization could lead to inhibition of VSM contraction are unclear. Furthermore, the cellular mechanisms via which MMP may affect VSM membrane channels are not clear.
Ca2+ is a major determinant of vascular smooth muscle contraction, and previous studies have shown that MMPs inhibit Ca2+ influx in the large conductance aortic tissue.13 Also, studies have suggested that integrins can detect injury-generated molecules derived from ECM proteins, and transfer the information to plasma membrane channels in vascular smooth muscle cells which then respond with either vasoconstriction or vasodilation.14 Peptides containing the amino acid sequence Arg-Gly-Asp (RGD) are important ligands that interact with the integrin receptors.15,16 These observations have led to the suggestion that the proteolytic activity of MMPs on ECM proteins could increase the generation of RGD peptides, which in turn affects VSM membrane integrins and channels activity. In the present Study, we further characterized the specificity of MMP-2 on VSM by testing its effects on vein architecture, and the reversibility of these effects using MMP inhibitors. Also, since membrane hyperpolarization is predicted to inhibit voltage-gated Ca2+ channels, we hypothesized that the venorelaxant effects of MMP-2 involve changes in the Ca2+ mobilization mechanisms of VSM contraction. Furthermore, we tested whether the effects of MMP on VSM function may involve an effect of RGD/integrins on membrane channels.
MATERIALS AND METHODS
Solutions, Drugs and Chemicals
Normal Krebs solution contained: NaCl 120 mmol/L, KCl 5.9 mmol/L, NaHCO3 25 mmol/L, NaH2PO4 1.2 mmol/L, Dextrose 11.5 mmol/L (Fisher Scientific, Fair Lawn, NJ), CaCl2 2.5 mmol/L (BDH Laboratory Supplies Poole, England), MgCl2 1.2 mmol/L (Sigma, St. Louis, MO). Krebs solution was bubbled with 95% O2 and 5% CO2 for 30-45 min, at an adjusted pH 7.4. For nominally 0 Ca2+ Krebs, CaCl2 was omitted. For Ca2+-free Krebs, CaCl2 was omitted and 2mM EGTA (Sigma) was added. 96 mmol/L KCL was prepared as normal Krebs but with NaCl 5.9 mmol/L and KCl 120 mmol/L. Phenylephrine (Phe, Sigma), Angiotensin II (AngII, Sigma), MMP-2 (Human recombinant, Biomol, Plymouth Meeting, PA), diltiazem (Calbiochem, La Jolla, CA). TIMP-1 (Human recombinant, Calbiochem, La Jolla, CA), Ro 28-2653 (5-biphenyl-4-yl-5-[4-(-nitro-phenyl)-piperazin-1-yl]-pyrimidine-2,4,6-trione, Roche Diagnostics GmbH, Pharma Research Penzberg, Germany), and BB-94 (Batimastat, British Biotech, Ltd, Oxford, United Kingdom). Stock solutions of RGD (Arg-Gly-Asp) sequence, and Cyclo(Ala-Arg-Gly-Asp-3-Aminomethylbenzoyl), an RGD peptidomimetic antagonist of αvβ3 integrin,17 (Sigma) (10-2 M) were prepared in 0.1N HCl, and serial dilutions were prepared in deionized water. All other reagents were prepared in deionized water
We have previously performed dose-response curves with MMPs in rat aorta rings.13 We found that MMP-2 at 1 μg/ml caused maximal aortic relaxation. Our experiments on rat IVC have also shown that MMP-2 caused time-dependent relaxation that reached steady-state in 30 min. In order to maintain internal consistency of the data and to be able to compare the effects of MMP-2 in the presence of various inhibitors, we used the maximal MMP-2 concentration and measured its effect after 30 min treatment. The concentrations of the agonists KCl, Phe, and AngII and the Ca2+ channel blocker diltiazem are standard concentrations that are used in isometric contraction experiments. Maximal concentrations of TIMP-1,18,19 Ro 28-2653,20 and BB-94,21 were determined based on reported IC50. The RGD saturating concentration was determined by performing a dose-response curve. The concentration of Cyclo-RGD used was based on results from published reports.17,22,23
Animals and tissues
Male Sprague-Dawley rats (12 wk, 250-300g, Charles River lab, Wilmington, MA) were housed in the animal facility and maintained on ad libitum standard rat chow and tap water in 12 hr/12 hr light/dark cycle. Rats were euthanized by inhalation of CO2. The inferior vena cave (IVC) was rapidly excised, placed in oxygenated Krebs solution, and carefully dissected and cleaned of connective tissue under microscopic visualization. The IVC was portioned into 2 to 3 mm rings in preparation for isometric contraction experiments. All procedures followed the NIH guide for the Care of Laboratory Animal Welfare Act, and the guidelines of the Animal Care and Use Committee at Harvard Medical School.
Isometric contraction
IVC segments were suspended between two wire hooks, one hook was fixed and the other hook was connected to a Grass force displacement transducer (FT03, Astro-Med Inc., West Warwick, RI). Vein segments were stretched under 0.5 gm of resting tension and allowed to equilibrate for 30-45 min in a tissue bath filled with 50 ml Krebs solution continuously bubbled with 95% O2 5% CO2 at 37°C. The changes in isometric tension were recorded on a Grass polygraph (Model 7D, Astro-Med Inc.).
Control contraction was elicited in response to 96 mmol/L KCl. Once maximum contraction was reached and a plateau achieved (within 10-15 min) the tissue was washed 3 times in Krebs, 10 min each. The control contraction to 96 mmol/L KCl was repeated twice prior to further experimentation. IVC segments were stimulated with phenylephrine (Phe, 10-5 M) to elicit a contraction. When Phe contraction reached steady-state, MMP-2 (1 μg/ml) was added and the effects on contraction was observed for 30 min. To test the specificity of the effects of MMP-2, experiments were repeated in IVC segments treated with MMP inhibitors TIMP-1 (1 μg/ml), Ro 28-2653 (10-6 M), or BB-94 (10-6 M) for 15 min. Also, angiotensin II (AngII, 10-6 M) induced contraction was measured in IVC segments nontreated or pretreated with MMP-2. To test the effects of MMP-2 on the Ca2+ release mechanism, the effects of caffeine (25 mM), Phe (10-5 M) and AngII (10-6 M) induced contraction were measured in IVC segments incubated in Ca2+-free (2mM EGTA) Krebs and nontreated or pretreated with MMP-2. To evaluate the effects of MMP-2 on Ca2+ entry, IVC segments nontreated or pretreated with MMP-2 were incubated in Ca2+-free (2mM EGTA) Krebs for 5 min followed by nominally 0 Ca2+ Krebs for 5 min, stimulated with Phe (10-5 M), then increasing extracellular CaCl2 concentrations ([Ca2+]e) were added and the contractile response was measured after 5 min in each [Ca2+]e. To further test the effect of MMP-2 on Ca2+ entry, the effect of MMP-2 on Phe contraction was tested in IVC segments treated with the Ca2+ channel blocker diltiazem (10-5 M).
To test for possible role of RGD/integrin, the effect of MMP-2 on Phe contraction was tested in IVC segments treated with saturating concentrations of RGD. To determine the saturating concentrations of RGD, IVC segments were stimulated with either 96 mmol/L KCl or Phe (10-5 M), to produce a steady-state contraction, increasing RGD concentrations (10-9 to 10-4 M) were added and any relaxation response was observed. To test for the effectiveness of the RGD peptide used in this study, their effects on rat mesenteric artery rings were also tested. In other experiments, the effects of MMP-2 were examined in IVC segments treated with Cyclo(Ala-Arg-Gly-Asp-3-Aminomethylbenzoyl) (Cyclo-RGD) (10 μM), an RGD peptidomimetic antagonist of αvβ3 integrin, to determine if the relaxation effects of MMP-2 are reversed.
Histology
To test for changes in vein wall structure cryosections of IVC (6 μm thick) were placed on glass slides and prepared for staining with hematoxylin and eosin to assess for the integrity/thickness of the endothelium, smooth muscle layer, and adventitia in the vein wall. Stained sections were coded and labeled in a blinded fashion. Images were acquired on a Nikon microscope with digital camera mount and analyzed using Metamorph Image software.
Statistical Analysis
Data were analyzed and presented as means±SEM and compared using Student’s t-test for unpaired and paired data. Differences were considered statistically significant if P < 0.05.
RESULTS
Effect of MMP-2 on IVC Contraction in Ca2+-containing Krebs
In normal Krebs (2.5 mM Ca2+) IVC segments demonstrated significant contraction with Phe (10-5 M) that reached a peak of 133.2±17.5 mg/mg tissue in 3.9±0.4 min followed by a smaller steady-state contraction (73.4±11.6 mg/mg tissue) which was maintained for 30 min. Because of the variability in the time to peak and magnitude of the peak, we chose the steady-state Phe response for measuring and comparing the effects of MMP-2. Addition of MMP-2 (1 μg/ml) caused a significant and time dependent relaxation in Phe contraction. MMP-2 induced IVC relaxation was gradual with 50% of relaxation occurring at 16.0±0.9 min. MMP-2 caused a decrease in Phe contraction to 32.4±12.8 mg/mg tissue in 30 min or 39.5±10.5% of the control Phe contraction of 82.0±2.4% (p=0.03) (Fig 1A).
Fig. 1.
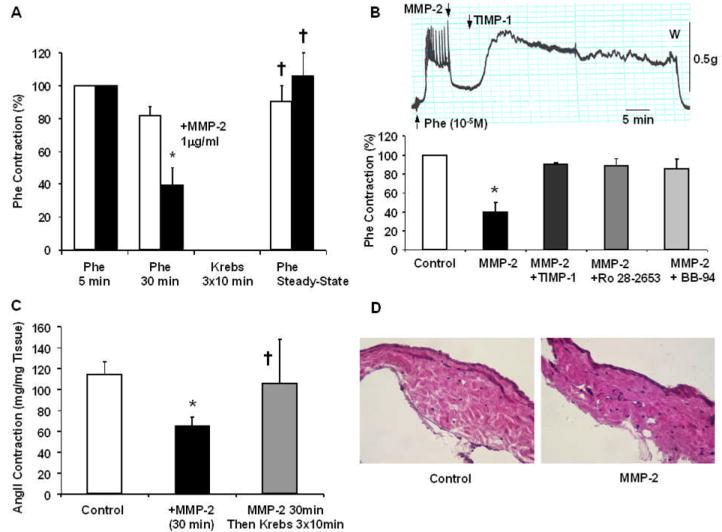
Reversibility and specificity of the effects of MMP-2. Rat IVC segments were treated with Phe (10-5 M), the steady-state contraction at 5 min and sustained contraction at 30 min were measured, the tissues were washed with Krebs, then a second control Phe contraction was elicited (A, Unfilled Bars). Other tissues were precontracted with Phe (10-5 M), treated with MMP-2 (1 μg/ml) for 30 min, washed in Krebs, then a second Phe contraction was elicited to demonstrate reversibility of the effects of MMP-2 (A, Solid Bars). To test the specificity of the effects of MMP-2, the experiments were repeated in IVC segments treated with MMP inhibitors TIMP-1 (1 μg/ml), Ro 28-2653 (10-6 M), and BB-94 (10-6 M) (B). In a representative tracing, when MMP-2 induced IVC relaxation reached steady-state TIMP-1 was added and reversal of the effects of MMP-2 was observed (B, upper panel). Aggregate data of Phe contraction in the absence and presence of MMP-2, and specific inhibitors of MMPs were also measured (B, lower panel). Other IVC segments were nontreated, pretreated with MMP-2 for 10 min, or pretreated with MMP-2 for 10 min followed by 3 washings in Krebs then stimulated with AngII (10-6M) and the peak contractile response was measured (C). Sections of IVC nontreated (control) and treated with MMP-2 were stained with hematoxylin and eosin to demonstrate the integrity of the vein wall and minimal evidence of structural degradation (D, Total Magnification 200). Data represent means±SEM of 4-24 measurements.
* Significantly different from control p<0.05
† Not significantly different from control
The effects of MMP-2 on Phe contraction were reversible. After 3 washes of MMP-2 treated IVC segments in Krebs, a significant Phe contraction could still be elicited, and the steady-state contraction was restored to levels greater than those observed in the presence of MMP-2, and not significantly different from tissues nontreated with MMP-2 (Fig. 1A).
In IVC segments treated with TIMP-1 the effect of MMP-2 on Phe contraction was blocked (Fig. 1B). Other MMP inhibitors specifically Ro 28-2653 and BB-94 prevented the effects of MMP-2, and the Phe contraction in IVC segments treated with MMP-2 plus the inhibitor was not significantly different from the control Phe contraction (p=0.19, p=0.22, p=0.28, for TIMP-1, Ro 28-2653, BB-94, respectively) (Fig. 1B). These finding support that the effects of MMP-2 on venous tissue relaxation are due to specific activity of MMP-2.
To evaluate whether the effects of MMP-2 are specific to Phe contraction and α-adrenergic receptors, the effects of MMP-2 on AngII contraction were tested. In control IVC segments AngII (10-6 M) produced a robust and transient contraction that reached a peak of 114.2±12.2 mg/mg tissue. In MMP-2 treated IVC there was a significant reduction in AngII contraction (66.0±7.4 mg/mg tissue, p<0.01). In IVC segments pretreated with MMP-2 followed by washing 3 times in Krebs, the tissues still responded to AngII and produced a contraction (106.0±41.8 mg/mg tissue) that was not significantly different from control AngII contraction in tissues non-treated with MMP-2 (p=0.86) (Fig. 1C).
In IVC tissue sections stained with H&E no apparent evidence of tissue degradation could be detected in control nontreated and MMP-2 treated tissues (Fig. 1D).
Effect of MMP-2 on IVC Contraction in Ca2+-Free Krebs
To determine the effects of MMP-2 on the venous intracellular Ca2+ release mechanisms, the effects of MMP-2 on caffeine (25mM), Phe (10-5M) and AngII (10-6M) contraction in Ca2+ -free (2mM EGTA) Krebs were examined. In Ca2+-free Krebs, caffeine (25 mM) did not elicit any detectable contraction, suggesting a limited role of the Ca2+-induced Ca2+-release mechanism in IVC contraction.
Phe (10-5M) caused a small but detectible contraction (7.2±1.7 mg/mg tissue) in Ca2+ -free Krebs, supporting a role of the IP3-induced Ca2+ release from intracellular Ca2+ stores. In IVC segments pretreated with MMP-2, Phe produced a measurable contraction (10.4±3.0 mg/mg) that was slightly greater but not significantly different from Phe contraction in control tissues nontreated with MMP-2 (p=0.39) (Fig. 2A).
Fig. 2.
Effect of MMP-2 on Phe- and AngII-induced IVC contraction in Ca2+ free Krebs. Rat IVC segments nontreated or treated with MMP-2 (1 μg/ml) were incubated in Ca2+-free (2 mM EGTA) for 5 min. The tissues were stimulated with Phe (10-5M) (A) or AngII (10-6M) (B) and the peak of the transient contraction was measured as an indicator of the intracellular Ca2+ release mechanism from the sarcoplasmic reticulum. Data represent means±SEM of 3-16 measurements.
Also, in Ca2+-free Krebs AngII (10-6M) caused a transient contraction that was slightly greater but not significantly different in MMP-2 treated (33.8±10.0 mg/mg tissue) and control nontreated tissues (14.9±2.8 mg/mg tissue, p=0.19) (Fig. 2B).
Effect of MMP-2 on [Ca2+]e-Dependent Contraction
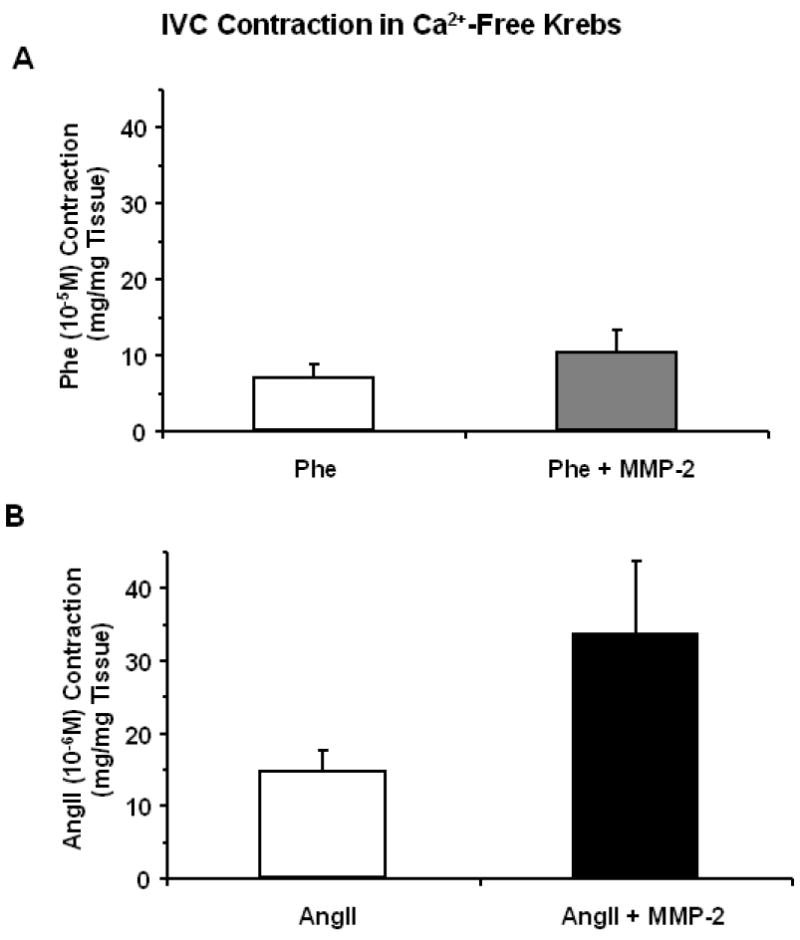
To determine if MMP-2 induced IVC relaxation involves modulation of extracellular Ca2+ entry through plasma membrane Ca2+ channels, the effects of MMP-2 on the [Ca2+]e-contraction relationship was examined. In IVC segments incubated in nominally 0 Ca2+ Krebs, the addition of increasing [Ca2+]e (0.1, 0.3, 0.6, 1.0, 2.5 mM) caused stepwise increases in contraction. In IVC segments pretreated with MMP-2 (1 μg/ml) for 10 min, the [Ca2+]e-contraction relation was significantly inhibited (Fig. 3).
Fig. 3.
Effect of MMP-2 on [Ca2+]e-contraction relationship. IVC segments incubated in 0 Ca2+ Krebs were either nontreated (A) or treated with MMP-2 (1 μg/ml) for 10 min (B). After eliciting a transient Phe contraction in 0 Ca2+ Krebs, increasing concentrations of CaCl2 were added, the Ca2+-dependent contraction was measured and the [Ca2+]e-contraction relationship was constructed (C). Data represent means±SEM of 4 measurements.
* Statistically significant p<0.05
Effect of MMP-2 in the Presence of Ca2+ Channel Blockers
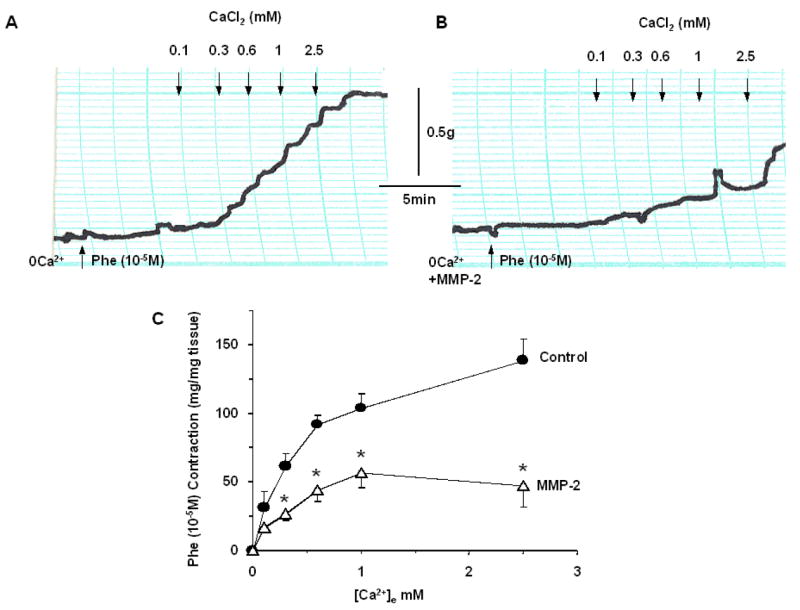
In IVC segments precontracted with Phe (10-5M), the Ca2+ channel blocker diltiazem (10-6 M) alone caused significant reduction of Phe contraction to 55.5±6.2%, that was slightly less than the relaxation induced by MMP-2 alone (36.1±7.3%, p=0.056). In IVC segments contracted with Phe then treated with MMP-2, addition of diltiazem did not cause any further relaxation (38.0±8.8%, p=0.4), indicating that Ca2+ entry is already inhibited by MMP-2. Interestingly, in IVC first treated with diltiazem followed by the addition of MMP-2 there was a substantial relaxation of Phe contraction to 6.7±1.3% (p<0.001) (Fig. 4).
Fig. 4.
Effects of MMP-2 during Ca2+ channel blockade. IVC segments were contracted with Phe (10-5M), then treated with MMP-2 (1 μg/ml) for 10 min followed by the Ca2+ channel blocker diltiazem (10-6M) (A), or treated first with diltiazem followed by MMP-2 (B). Bar graphs (C) represent means±SEM of 3 measurements. Note that in IVC treated with MMP-2, diltiazem did not cause any further reduction in Phe contraction (A,C). In tissues treated with diltiazem the addition of MMP-2 caused further inhibition of Phe contraction (B,C).
* Statistically significant p<0.05
** Significantly different from tissue treated with MMP-2 or diltiazem alone p<0.01
Effect of MMP-2 in the Presence of RGD (Arg-Gly-Asp) Peptides
To test whether the effects of MMP-2 on membrane channels involve generation of RGD and possible effects on membrane integrins, we tested the effects of MMP-2 in IVC segments treated with RGD peptides. In IVC segments contracted with 96 mM KCl, increasing concentrations of RGD did not inhibit contraction, and at 10-5 M RGD concentration only 3.6±1.8% relaxation could be detected (Fig. 5). Similarly, in IVC segments contracted with Phe, increasing RGD concentrations up to 10-5 M caused minimal tissue relaxation (6.4±3.4%). In parallel experiments on mesenteric artery segments, RGD caused concentration-dependent relaxation (Fig. 5C). To ensure that we did not overlook any vasodilatory effects of RGD in venous tissue, we increased RGD concentration to 10-4M, which still caused minimal tissue relaxation (6.3±2.4%, p=ns). Because there was no difference in venous relaxation between the RGD concentrations 10-5M and 10-4M, we tested the effects of MMP-2 on Phe contraction in IVC segments saturated with RGD (10-5M). In RGD saturated IVC, MMP-2 caused significant relaxation of Phe contraction to (69.7±3.0%, p=0.0001 compared to RGD alone) (Fig. 5A,D). Also, addition of Cyclo(Ala-Arg-Gly-Asp-3-Aminomethylbenzoyl) (10 μM), an RGD peptidomimetic antagonist of αvβ3 integrin receptor, did not reverse MMP-2 induced relaxation (55.0±5.0%, p=0.1 compared with MMP-2 relaxation in the absence of cyclo-RGD) (Fig. 5B,D).
Fig. 5.
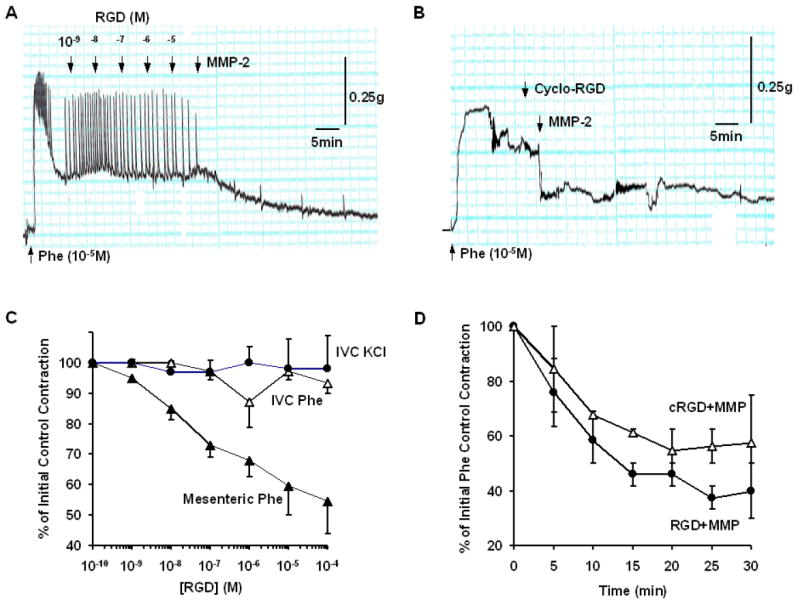
Role of RGD/integrins in MMP-2 induced IVC relaxation. IVC segments were contracted with Phe (10-5M) (A,C) or 96 mmol/L KCl (C). Increasing concentrations of RGD peptide were added and their effects on contraction was measured. For comparison, the effects of RGD on Phe-induced contraction in rat mesenteric artery were also measured (C). In other experiments, IVC segments were saturated with 10-5 M RGD or pretreated with the αvβ3 integrin receptor antagonist cyclo-RGD (10-5M), MMP-2 (1 μg/ml) was added and its effect on contraction was measured over a 30 min period (A,B) and presented as % of initial control contraction (D). Data represent the average of 2-4 measurements.
DISCUSSION
The main findings of the present study are: 1) MMP-2 induced relaxation of rat IVC contraction is specific and reversible; 2) MMP-2 induced venous relaxation does not appear to involve degradation of the vein architecture, or the VSM contractile proteins or specific receptors; 3) MMP-2 inhibits [Ca2+]e dependent VSM contraction, but not intracellular Ca2+ release mechanisms; and 4) MMP-2 induced VSM relaxation may not involve generation of RGD or modulation of membrane αvβ3 integrin receptor.
We have previously shown that MMP-2 and -9 cause relaxation of rat aorta that reaches a maximum in 1 hr.13 Because MMPs may have different distribution and function in arteries as compared to veins, we have examined the effects of MMPs on rat IVC. We tested the effects of MMP-2 as a prototype, but with the understanding that different MMPs may have different expression, location, activity and functions in different venous preparations. Consistent with our previous reports,12,13 we observed that MMP-2 caused significant relaxation of Phe contraction in IVC. In the this study, the specificity of these novel effects of MMPs on venous tissue function and the cellular and molecular targets involved were systematically examined.
MMP-induced IVC relaxation is specific because it was reversed by TIMP-1, an endogenous inhibitor of many MMPs including MMP-2,24 and was also blocked by other MMP inhibitors with variable specificity and affinity such as Ro 28-2653 and BB-94. The prevention of MMP-2 induced venous relaxation by three chemically unrelated MMP inhibitors supports the contention that the venodilatory effects are related to specific MMP activity.
Because MMPs induce proteolysis and ECM turnover,9,25,26 one would predict that MMP-induced venous relaxation is due to destruction of vein architecture, membrane receptors or contractile proteins. This is unlikely because: 1) The effects of MMP-2 were reversible, and in IVC segments treated with MMP then washed in Krebs, a significant Phe and AngII contraction could still be elicited, 2) Histological analysis of MMP treated IVC revealed intact vein architecture, 3) MMP-2 did not inhibit Phe or AngII contraction in Ca2+-free Krebs; instead, a slight increase in contraction was observed, and 4) In normal Krebs, MMP-induced inhibition of IVC contraction to Phe, an α-adrenergic receptor agonist, and AngII, an angiotensin type 1 receptor agonist, indicate that the effects of MMP are not related to changes in a particular receptor, but may involve a common pathway downstream from the membrane receptor.
In vascular smooth muscle, agonist-receptor interaction is coupled to increased release of inositol-1,3,5-trisphosphate (IP3) and activation of plasma membrane Ca2+ channels. IP3 induces Ca2+ release from the intracellular Ca2+ stores. Parallel activation of Ca2+ channels promotes Ca2+ influx from the extracellular space.27 To test whether MMP-2 induced VSM relaxation involves inhibition of Ca2+ release the effects of MMP were examined in Ca2+-free solution. In Ca2+-free Krebs, Phe and AngII produced transient contraction, suggesting that they activate IP3-induced Ca2+ release. AngII-induced contraction was significantly greater than that induced by Phe, and may reflect greater distribution of angiotensin type-1 receptors than α-adrenergic receptors or their downstream signaling mechanisms in rat IVC. The observation that MMP-2 did not inhibit Phe or AngII contraction in Ca2+-free solution suggests that it does not block the IP3-sensitive Ca2+ release.28-30 In effect, MMP-2 caused a slight increase in Phe and AngII contraction in Ca2+-free Krebs. MMP-2 may facilitate the turnover of phosphatidyl-4,5,bisphosphate and thereby increase the generation of IP3 during receptor activation. This is supported by reports that platelet activation is associated with MMP-2 release, and that MMP-2, added before stimulation with subthreshold doses of different agonists, potentiates platelet activation, IP3 formation and activation of PI3-K pathway.31
In contrast, when Phe contraction was measured at increasing [Ca2+]e, MMP-2 caused significant venous relaxation, suggesting inhibition of extracellular Ca2+ influx through Ca2+ channels. Our findings in rat IVC are consistent with the previous observation that MMPs inhibit Ca2+ influx in rat aorta.13 However, the present study evaluated the effects of MMPs on veins which are structurally and functionally different form arteries. Also, while the effects of MMP-2 on the aorta may have implications in abdominal aortic aneurysm, the present observations on venous tissue may have implications in varicose veins. Ca2+ enters vascular smooth muscle cells via voltage-gated, ligand-gated and store-operated Ca2+ channels. There are also several types of voltage-gated Ca2+ channel (CaV1.1-1.4, 2.1-2.3, 3.1-3.3) broadly classified into L-, T- and N-type channels.32,33 Ca2+ antagonists such as diltiazem, verapamil and nifedipine block L-type Ca2+ channels.34 In this study, diltiazem inhibited Phe-induced contraction, suggesting the presence of functional voltage-gated Ca2+ channels in rat IVC. Also, diltiazem did not cause any further relaxation in MMP-2 treated IVC, indicating that the L-type channels are already inhibited by MMP-2. Importantly, MMP-2 caused further relaxation in diltiazem treated IVC (Fig. 4), suggesting that during blockade of L-type Ca2+ channels, MMP-2 may inhibit other subclasses of voltage-gated channels (T- or N-type) or perhaps ligand-gated, store-operated Ca2+ channels or non-specific cation channels.27,35
The question remains as of how MMPs inhibit Ca2+ entry through membrane channels. MMP may directly or indirectly interact and alters the Ca2+ channel activity. In a recent report we suggested that MMP-induced venous relaxation may involve membrane hyperpolarization and activation of large conductance K+ channels (BKCa).12 Since membrane hyperpolarization is expected to inhibit voltage-gated Ca2+ channels, then the inhibitory effects of MMP on Ca2+ channels may be secondary to its activation of K+ channels. Whether Ca2+ channels are affected first or secondary to changes in K+ channel activity, it remains to be clarified how MMP may modify membrane channel activity in VSM. Soluble RGD peptide bind to smooth muscle αvβ3 integrin receptors cause K+ channel activation leading to smooth muscle hyperpolarization and relaxation.36 Also, studies in smooth muscle cells isolated from rat cremaster arterioles have demonstrated a relationship between αvβ3 integrin receptors and the RGD stimulated dilation and lowering of [Ca2+]i.22 In addition, αvβ3 and α5β1 integrins are differentially linked through intracellular signaling pathways to L-type Ca2+ channel, and thereby alter Ca2+ influx in vascular smooth muscle.37 These studies, together with our previous report that MMP-2 induced IVC relaxation may involve membrane hyperpolarization and activation of BKCa,12 and the present observation that MMP-2 inhibits [Ca2+]e dependent IVC contraction, have led us to hypothesize that MMP-2 may function via RGD/αvβ3 integrin receptor. If MMP-2 functions via an RGD/αvβ3 integrin receptor relaxation pathway, then RGD peptides should cause relaxation of VSM, MMP-2 should have no effect in VSM saturated with RGD, and MMP-2 induced relaxation should be prevented by blockers of αvβ3 integrin receptors. Consistent with other reports evaluating the effect of RGD on various arteries,23,38-41 our preliminary experiments on rat mesenteric artery suggest that RGD peptides cause concentration-dependent relaxation (Fig. 5), confirming that the concentrations of the RGD used are effective. However, maximal concentrations of RGD (10 to 100 μM) caused only a small relaxation in rat IVC. Also, in IVC segments pretreated with saturating concentrations of RGD MMP-2 caused significant venorelaxation. Furthermore, MMP-2 induced IVC relaxation was not reversed by αvβ3 integrin antagonist, supporting that the inhibitory effects of MMP-2 on [Ca2+]e-dependent VSM contraction do not involve αvβ3 integrin receptor. Some shortcomings are that we only used RGD (Arg-Gly-Asp) tripeptide to induce venous relaxation, and several RGD-containing peptidomimetics (GPenGRGDSPCA, GRGDNP, RGDRGD, GRGDSP) could have variable effects ranging from vasodilation to constriction depending on the vessel type and RGD motif used.23,36,38,42-44 Also, we only examined the role of αvβ3 integrin, while other integrins may be expressed in rat IVC, and should be further characterized.41,44,45 Furthermore, MMP may function via a mechanism(s) not involving RGD/integrins. For example, MMPs may interact with cell surface proteins such as ICAM-1 or protease-activated receptors (PARs) and activate signaling pathways that could change smooth muscle Ca2+ channels activity.46-48 This is supported by reports that proteases such as thrombin activate PARS and cause vascular smooth muscle relaxation by inhibiting Ca2+ influx.49
The present ex vivo study examined the acute effects of one member of a large MMP family on rat IVC. Whether MMP-2 exerts long-term inhibition of VSM should be examined in chronic and in vivo studies. Also, whether other MMPs exert similar effects on other types of veins from rat and human remain to be examined. Furthermore, while acute exposure of veins to MMPs may mainly affect VSM function while causing limited histological and structural changes, chronic exposure of vein wall to MMPs may lead to additional degradation of the ECM, structural changes and extensive venodilation. Although the pathogenesis of varicose veins is unclear, studies in animals and human suggest a role of MMPs in varicose vein formation. While animal models of venous hypertension have demonstrated increased tissue levels of MMP-2 and -9 as well as valve remodeling,50,51 a mechanistic explanation of MMPs causing VSM dilation is lacking. Also, in human saphenous vein specimens and venous segments with thrombophlebitis from patients with chronic venous insufficiency, there is overexpression of multiple MMPs in all layers of the vein wall.2,6,8 What is not known is the implication of these MMPs and whether their presence is an epiphenomenon, or antecedent and directly contributory to venous dilation, valve dysfunction, tortuosity and varicose vein formation. This study demonstrates relatively rapid MMP-2 induced dilation of venous segments, which are specific and reversible and not specific to a particular vasoconstrictor receptor/agonist. The mechanism of MMP-2 induced venous relaxation appears to involve inhibition of Ca2+ influx through membrane channels, but may not be linked to the αvβ3 integrin receptor. Future studies defining the mechanism(s) linking MMPs and membrane channel activity will be helpful in understanding the pathogenesis of varicose veins and designing new treatment options for de novo and recurrent varicosities.
Acknowledgments
This work was supported by grants from the National Heart, Lung and Blood Institute (HL-65998, HL-70659).
List of Abbreviations
- AngII
angiotensin II
- [Ca2+]e
extracellular Ca2+ concentration
- EGTA
ethylene glycol-bis(2-aminoethyl ether)-N,N,N’,N’-tetraacetic acid
- IVC
inferior vena cava, MMP, matrix metalloproteinase
- Phe
phenylephrine
- RGD
Arg-Gly-Asp
- Cyclo-RGD
Cyclo(Ala-Arg-Gly-Asp-3-Aminomethylbenzoyl)
- TIMP
tissue inhibitor of MMP
- VSM
venous smooth muscle
References
- 1.Goldman MP, Fronek A. Anatomy and pathophysiology of varicose veins. J Dermatol Surg Oncol. 1989;15:138–45. doi: 10.1111/j.1524-4725.1989.tb03020.x. [DOI] [PubMed] [Google Scholar]
- 2.Woosside KJ, Hu M, Burke A, Murakami M, Pounds LL, Killewich LA, Daller JA, Hunter GC. Morphologic characteristics of varicose veins: possible role of metalloproteinases. J Vasc Surg. 2003;38:162–9. doi: 10.1016/s0741-5214(03)00134-4. [DOI] [PubMed] [Google Scholar]
- 3.Gandhi RH, Irizarry E, Nackman GB, Halpern VJ, Mulcare RJ, Tilson MD. Analysis of the connective tissue matrix and proteolytic activity of primary varicose veins. J Vasc Surg. 1993;18:814–20. [PubMed] [Google Scholar]
- 4.Carpentier PH, Maricq HR, Biro C, Pancot-Makinen CO, Franco A. Prevalence, risk factors, and clinical patterns of chronic venous disorders of lower limbs: a population-based study in France. J Vasc Surg. 2004;40:650–9. doi: 10.1016/j.jvs.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 5.Ruckley CV, Evans CJ, Allan PL, Lee AJ, Fowkes FG. Chronic venous insufficiency: clinical and duplex correlations. The Edinburgh Vein Study of venous disorders in the general population. J Vasc Surg. 2002;36:520–5. doi: 10.1067/mva.2002.126547. [DOI] [PubMed] [Google Scholar]
- 6.Gillespie DL, Patel A, Fileta B, Chang A, Barnes S, Flagg A, Kidwell M, Villavicencio JL, Rich NM. Varicose veins possess greater quantities of MMP-1 than normal veins and demonstrate regional variation in MMP-1 and MMP-13. J Surg Res. 2002;106:233–8. doi: 10.1006/jsre.2002.6455. [DOI] [PubMed] [Google Scholar]
- 7.Pascarella L, Penn A, Schmid-Schonbein GW. Venous hypertension and the inflammatory cascade: major manifestations and trigger mechanisms. Angiology. 2005;56(Suppl 1):S3–10. doi: 10.1177/00033197050560i102. [DOI] [PubMed] [Google Scholar]
- 8.Kowalewski R, Sobolewski K, Wolanska M, Gacko M. Matrix metalloproteinases in the vein wall. Int Angiol. 2004;23:164–9. [PubMed] [Google Scholar]
- 9.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92:827–39. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 10.Raffetto JD, Khalil RA. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol. 2008;75:346–59. doi: 10.1016/j.bcp.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raffetto JD, Khalil RA. Matrix metalloproteinases in venous tissue remodeling and varicose vein formation. Curr Vasc Pharmacol. 2008 doi: 10.2174/157016108784911957. in press. [DOI] [PubMed] [Google Scholar]
- 12.Raffetto JD, Ross RL, Khalil RA. Matrix metalloproteinase-2 induced venous dilation via hyperpolarization and activation of K+ channels: Relevance to varicose vein formation. J Vasc Surg. 2007;45:373–80. doi: 10.1016/j.jvs.2006.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chew DKW, Conte MS, Khalil RA. Matrix metalloproteinase-specific inhibition of Ca2+ entry mechanisms of vascular contraction. J Vasc Surg. 2004;40:1001–10. doi: 10.1016/j.jvs.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 14.Martinez-Lemus LA, Wu X, Wilson E, Hill MA, Davis GE, Davis MJ, Meininger GA. Integrins as unique receptors for vascular control. J Vasc Res. 2003;40:211–33. doi: 10.1159/000071886. [DOI] [PubMed] [Google Scholar]
- 15.Davis MJ, Wu X, Nurkiewicz TR, Kawasaki J, Davis GE, Hill MA, Meininger GA. Integrins and mechanotransduction of the vascular myogenic response. Am J Physiol Heart Circ Physiol. 2001;280:H1427–33. doi: 10.1152/ajpheart.2001.280.4.H1427. [DOI] [PubMed] [Google Scholar]
- 16.Takada Y, Ye X, Simon S. The integrins. Genome Biol. 2007;8:215. doi: 10.1186/gb-2007-8-5-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kerr JS, Wexler RS, Mousa SA, Robinson CS, Wexler EJ, Mohamed S, Voss ME, Devenny JJ, Czerniak PM, Gudzelak A, Jr, Slee AM. Novel small molecule alpha v integrin antagonists: comparative anti-cancer efficacy with known angiogenesis inhibitors. Anticancer Res. 1999;19:959–68. [PubMed] [Google Scholar]
- 18.Nagase H, Woessner JF., Jr Matrix metalloproteinases. J Biol Chem. 1999;274:21491–4. doi: 10.1074/jbc.274.31.21491. [DOI] [PubMed] [Google Scholar]
- 19.Ries C, Pitsch T, Mentele R, Zahler S, Egea V, Nagase H, Jochum M. Identification of a novel 82 kDa proMMP-9 species associated with the surface of leukaemic cells: (auto-)catalytic activation and resistance to inhibition by TIMP-1. Biochem J. 2007;405:547–58. doi: 10.1042/BJ20070191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maquoi E, Sounni NE, Devy L, Olivier F, Frankenne F, Krell HW, Grams F, Foidart JM, Noël A. Anti-invasive, antitumoral, and antiangiogenic efficacy of a pyrimidine-2,4,6-trione derivative, an orally active and selective matrix metalloproteinases inhibitor. Clin Cancer Res. 2004;10:4038–47. doi: 10.1158/1078-0432.CCR-04-0125. [DOI] [PubMed] [Google Scholar]
- 21.Kruse MN, Becker C, Lottaz D, Köhler D, Yiallouros I, Krell HW, Sterchi EE, Stöcker W. Human meprin alpha and beta homo-oligomers: cleavage of basement membrane proteins and sensitivity to metalloprotease inhibitors. Biochem J. 2004;378:383–9. doi: 10.1042/BJ20031163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.D’Angelo G, Mogford JE, Davis GE, Davis MJ, Meininger GA. Integrin-mediated reduction in vascular smooth muscle [Ca2+]i induced by RGD-containing peptide. Am J Physiol. 1997;272:H2065–70. doi: 10.1152/ajpheart.1997.272.4.H2065. [DOI] [PubMed] [Google Scholar]
- 23.Hein TW, Platts SH, Waitkus-Edwards KR, Kuo L, Mousa SA, Meininger GA. Integrin-binding peptides containing RGD produce coronary arteriolar dilation via cyclooxygenase activation. Am J Physiol Heart Circ Physiol. 2001;281:H2378–84. doi: 10.1152/ajpheart.2001.281.6.H2378. [DOI] [PubMed] [Google Scholar]
- 24.Meng Q, Malinovskii V, Huang W, Hu Y, Chung L, Nagase H, et al. Residue 2 of TIMP-1 is a major determinant of affinity and specificity from matrix metalloproteinases but effects of substitutions do not correlate with those of the corresponding P1’ residue of substrate. J Biol Chem. 1999;274:10184–9. doi: 10.1074/jbc.274.15.10184. [DOI] [PubMed] [Google Scholar]
- 25.Liu YE, Wang M, Greene J, Su J, Ullrich S, Li H, Sheng S, Alexander P, Sang QA, Shi YE. Preparation and characterization of recombinant tissue inhibitor of metalloproteinase 4 (TIMP-4) J Biol Chem. 1997;272:20479–83. doi: 10.1074/jbc.272.33.20479. [DOI] [PubMed] [Google Scholar]
- 26.Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. 2002;90(3):251–62. [PubMed] [Google Scholar]
- 27.Khalil RA, van Breemen C. Sustained contraction of vascular smooth muscle: calcium influx or C-kinase activation? J Pharmacol Exp Ther. 1988;244:537–42. [PubMed] [Google Scholar]
- 28.Pattni K, Banting G. Ins(1,4,5)P3 metabolism and the family of IP3-3Kinases. Cell Signal. 2004;16:643–54. doi: 10.1016/j.cellsig.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 29.Szlufcik K, Missiaen L, Parys JB, Callewaert G, De Smedt H. Uncoupled IP3 receptor can function as a Ca2+-leak channel: cell biological and pathological consequences. Biol Cell. 2006;98:1–14. doi: 10.1042/BC20050031. [DOI] [PubMed] [Google Scholar]
- 30.Mikoshiba K. IP3 receptor/Ca2+ channel: from discovery to new signaling concepts. J Neurochem. 2007;102:1426–46. doi: 10.1111/j.1471-4159.2007.04825.x. [DOI] [PubMed] [Google Scholar]
- 31.Falcinelli E, Guglielmini G, Torti M, Gresele P. Intraplatelet signaling mechanisms of the priming effect of matrix metalloproteinase-2 on platelet aggregation. J Thromb Haemost. 2005;3:2526–35. doi: 10.1111/j.1538-7836.2005.01614.x. [DOI] [PubMed] [Google Scholar]
- 32.Triggle DJ. Calcium channel antagonists: clinical uses--past, present and future. Biochem Pharmacol. 2007;74:1–9. doi: 10.1016/j.bcp.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 33.Romero M, Sánchez I, Pujol MD. New advances in the field of calcium channel antagonists: cardiovascular effects and structure-activity relationships. Curr Med Chem Cardiovasc Hematol Agents. 2003;1:113–41. doi: 10.2174/1568016033477487. [DOI] [PubMed] [Google Scholar]
- 34.Triggle DJ. L-type calcium channels. Curr Pharm Des. 2006;12:443–57. doi: 10.2174/138161206775474503. [DOI] [PubMed] [Google Scholar]
- 35.Khalil RA, van Breemen C. Intracellular free calcium concentration/force relationship in rabbit inferior vena cava activated by norepinephrine and high K+ Pflugers Arch. 1990;416:727–34. doi: 10.1007/BF00370622. [DOI] [PubMed] [Google Scholar]
- 36.Mogford JE, Davis GE, Platts SH, Meininger GA. Vascular smooth muscle alpha v beta 3 integrin mediates arteriolar vasodilation in response to RGD peptides. Circ Res. 1996;79:821–6. doi: 10.1161/01.res.79.4.821. [DOI] [PubMed] [Google Scholar]
- 37.Wu X, Mogford JE, Platts SH, Davis GE, Meininger GA, Davis MJ. Modulation of calcium current in arteriolar smooth muscle by alphav beta3 and alpha5 beta1 integrin ligands. J Cell Biol. 1998;143:241–52. doi: 10.1083/jcb.143.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lipke DW, Soltis EE, Fiscus RR, Yang L, Newman PS, Aziz SM. RGD-containing peptides induce endothelium-dependent and independent vasorelaxations of rat aortic rings. Regul Pept. 1996;63:23–9. doi: 10.1016/0167-0115(96)00020-1. [DOI] [PubMed] [Google Scholar]
- 39.Srivatsa SS, Fitzpatrick LA, Tsao PW, Reilly TM, Holmes DR, Jr, Schwartz RS, Mousa SA. Selective alpha v beta 3 integrin blockade potently limits neointimal hyperplasia and lumen stenosis following deep coronary arterial stent injury: evidence for the functional importance of integrin alpha v beta 3 and osteopontin expression during neointima formation. Cardiovasc Res. 1997;36:408–28. doi: 10.1016/s0008-6363(97)00184-3. [DOI] [PubMed] [Google Scholar]
- 40.Platts SH, Mogford JE, Davis MJ, Meininger GA. Role of K+ channels in arteriolar vasodilation mediated by integrin interaction with RGD-containing peptide. Am J Physiol. 1998;275(4 Pt 2):H1449–54. doi: 10.1152/ajpheart.1998.275.4.H1449. [DOI] [PubMed] [Google Scholar]
- 41.Bakker EN, Balt JC, Pfaffendorf M, Spaan JA, VanBavel E. Vasomotor effects of arg-gly-asp (RGD) peptides are limited and not related to endothelium-derived hyperpolarizing factor-mediated relaxation in rat mesenteric arteries. Clin Exp Pharmacol Physiol. 2001;28:873–6. doi: 10.1046/j.1440-1681.2001.03537.x. [DOI] [PubMed] [Google Scholar]
- 42.Goligorsky MS, DiBona GF. Pathogenetic role of Arg-Gly-Asp-recognizing integrins in acute renal failure. off. Proc Natl Acad Sci U S A. 1993;90:5700–4. doi: 10.1073/pnas.90.12.5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yip KP, Marsh DJ. An Arg-Gly-Asp peptide stimulates constriction in rat afferent arteriole. Am J Physiol. 1997;273(5 Pt 2):F768–76. doi: 10.1152/ajprenal.1997.273.5.F768. [DOI] [PubMed] [Google Scholar]
- 44.Chan WL, Holstein-Rathlou NH, Yip KP. Integrin mobilizes intracellular Ca(2+) in renal vascular smooth muscle cells. Am J Physiol Cell Physiol. 2001;280:C593–603. doi: 10.1152/ajpcell.2001.280.3.C593. [DOI] [PubMed] [Google Scholar]
- 45.Martinez-Lemus LA, Crow T, Davis MJ, Meininger GA. alphavbeta3- and alpha5beta1-integrin blockade inhibits myogenic constriction of skeletal muscle resistance arterioles. Am J Physiol Heart Circ Physiol. 2005;289:H322–9. doi: 10.1152/ajpheart.00923.2003. [DOI] [PubMed] [Google Scholar]
- 46.Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Proteinase-activated receptors. Pharmacol Rev. 2001;53:245–82. [PubMed] [Google Scholar]
- 47.Fiore E, Fusco C, Romero P, Stamenkovic I. Matrix metalloproteinase 9 (MMP-9/gelatinase B) proteolytically cleaves ICAM-1 and participates in tumor cell resistance to natural killer cell-mediated cytotoxicity. Oncogene. 2002;21:5213–23. doi: 10.1038/sj.onc.1205684. [DOI] [PubMed] [Google Scholar]
- 48.Marutsuka K, Hatakeyama K, Sato Y, Yamashita A, Sumiyoshi A, Asada Y. Protease-activated receptor 2 (PAR2) mediates vascular smooth muscle cell migration induced by tissue factor/factor VII complex. Thromb Res. 2002;107:271–6. doi: 10.1016/s0049-3848(02)00345-6. [DOI] [PubMed] [Google Scholar]
- 49.Hamilton JR, Nguyen PB, Cocks TM. Atypical protease-activated receptor mediates endothelium-dependent relaxation of human coronary arteries. Circ Res. 1998;82:1306–11. doi: 10.1161/01.res.82.12.1306. [DOI] [PubMed] [Google Scholar]
- 50.Takase S, Pascarella L, Bergan JJ, Schmid-Schonbein GW. Hypertension-induced venous valve remodeling. J Vasc Surg. 2004;39:1329–34. doi: 10.1016/j.jvs.2004.02.044. [DOI] [PubMed] [Google Scholar]
- 51.Pascarella L, Schmid-Schonbein GW, Bergan JJ. An animal model of venous hypertension: the role of inflammation in venous valve failure. J Vasc Surg. 2005;41:303–11. doi: 10.1016/j.jvs.2004.10.038. [DOI] [PubMed] [Google Scholar]