

Abstract
Background
Third party-specific CTL, or veto CTL, are being assessed as a cellular therapeutic for the induction of T cell tolerance during transplantation. Conceptually, veto cell expressed antigens may induce B cell immune responses, and this may have deleterious consequences. Whether veto cells induce immunity, tolerance, or are ignored by B lymphocytes has, however, not been addressed.
Methods
CTL were retrovirally transduced with a model cell surface antigen to generate veto CTL. The impact of CTL-specific antigen expression on the activation and tolerization of antigen specific B cells was assessed in vitro and, using adoptive transfer models, in vivo.
Results
In vitro, CTL-expressed antigen induced an abortive proliferative response in specific B lymphocytes, whereby an initial proliferative burst was followed by cell death. In vivo, the administration of veto CTL also induced B cell tolerance. Specific immunoglobulin was not detected after subsequent immunization with a veto cell-expressed antigen. Modeling of this effect with antigen-specific B cell receptor (BCR) transgenic B lymphocytes demonstrated that antigen-specific B cells were eliminated by the veto CTL; cell division was accompanied by the exhaustion and depletion of responding cells. Veto-induced B cell tolerance could be wholly abrogated by treatment with the toll-like receptor ligand LPS, implying that this tolerance resulted from the absence of adequate supplemental signals during antigenic stimulation.
Conclusions
Veto CTL are effective promoters of B cell tolerance. Further assessment of their therapeutic potential in this regard is warranted.
Keywords: tolerance, antibody production, veto cell, cytotoxic T lymphocyte, B lymphocyte
Introduction
The induction of B cell tolerance is essential to successful solid organ and bone marrow transplantation. Host B cell responses to graft alloantigens or graft responses to host antigens are important components of graft-versus-host disease and graft rejection (1,2). Current pharmacotherapy for these conditions is often inadequate, and results in generalized immune suppression and its attendant complications. New therapies that antigen-specifically induce graft-host tolerance are therefore desired.
The observation that donor-specific transfusion (DST) prior to transplantation promotes graft acceptance demonstrated the potential for adoptive cellular therapy to induce graft-specific tolerance in the developed immune system (3-5). Dissection of the DST effect showed that infused allogeneic leukocytes effectively induce tolerance (6). CD8+ cytotoxic T lymphocyte (CTL) lines or clones were observed to mediate T cell tolerance (7,8). These cells, called veto cells, interact through their MHC with alloreactive CD8+ T-cells (precursor CTL, pCTL), inducing their death. The veto cells may be third party-specific and need not be stimulated through their TCR. Multiple cell types, including CD34+ hematopoietic progenitor cells and other myeloid cell types, are now recognized to possess veto activity (9). Nevertheless, interest in the application of CTL as veto cells has remained. In a study comparing relative efficacy of different veto populations, CTL proved the most potent (10). Veto CTL can eradicate both naïve and memory allo-specific T lymphocytes in vivo, and recent efforts have explored safe and effective approaches for their clinical application in conjunction with allogeneic transplantation (11,12).
B cell responses often depend on Th-cell assistance, and methods that specifically tolerize pathologic T cells may also limit undesirable B cell responses. Induction of pCTL tolerance, as may occur after the application of veto cells, unlike Th cell tolerance, would not be expected to impede B cell activity. Whether veto cells can independently induce B cell tolerance is unclear. Indeed, it may be hypothesized that antigens expressed by CD8+ veto cells will stimulate B lymphocytes. CTL, like activated CD4+ T lymphocytes, express CD40L, a critical B cell stimulatory molecule that could conceivably support specific B cell responses against alloantigens presented on veto CTL (13). Veto cells may therefore provoke antibody development while tolerizing pCTL.
To better define the impact of veto cells on B lymphocytes, we analyzed their ability to induce B cell tolerance or immunity in an antigen-specific system. CTL retrovirally transduced with a membrane-bound form of a model antigen, hen egg lysozyme (HEL), were transferred into mice, and the impact on the HEL-specific B cell response measured. Contrary to our expectations, this membrane bound antigen induced B cell tolerance rather than immunity. Unmanipulated mice receiving the veto cells produced little or no detectable antibody after subsequent immunization with HEL. This tolerance was blocked by the co-administration of the TLR ligand LPS, indicating that inadequate pro-inflammatory signaling in the context of Ag stimulation was responsible. Our results show that veto cells are effective inducers of antigen-specific B cell tolerance. They appear to act by initiating an abortive activation program in veto-cell reactive B cells.
Materials and Methods
Construct synthesis
cDNA for the leader, hinge and transmembrane (TM) domains of murine CD8 were amplified by PCR from splenic cDNA. HEL cDNA was constructed from overlapping oligonucleotides using PCR ligation. CD8 leader, HEL, and CD8 hinge/TM fragments were assembled by subcloning into the pBS-KS vector (Stratagene) using the indicated restriction sites (Fig. 1) and DNA sequence confirmed. The assembled construct was then subcloned into the MSCV-I-green flourescent protein (GFP) retroviral vector (14) at its EcoRI and XhoI cloning sites.
Figure 1. HEL expression on CTL.

(A) Diagram of the retroviral vector used to express HEL on CTL. Nucleotide sequence shows junctional segments between the chimeric gene's components. Amino acid sequence is shown above this and restriction sites used for cloning and segment identification below. Additional sequence can be found under the following GenBank accessions: G gallus lysozyme, NM_205281; CD8, XM_132621. (B) Retrovirus containing the MSCV-I-GFP vector or the HEL construct shown was used to transduce activated, purified CD8 T cells. Cells were surface stained with a HEL-specific antibody and flow cytometrically analyzed 5 days after transduction.
Mice, cells, and antibodies
C57BL/6, MD-4 BCR transgenic (Tg) (C57BL/6-Tg(IghelMD4)4Ccg/J), B6.129S7-Rag1tm1Mom/J, and CD45.1-congenic B6.SJL-Ptprca Pep3b/BoyJ mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). MD-4 mice were maintained on a C57BL/6 background or bred with CD45.1-congenic mice. The MD-4 transgene was determined by staining peripheral blood with phycoerythrin (PE)-labeled anti-mouse IgMa (clone DS-1; Pharmingen, San Diego, CA, USA). Other antibodies used include PE rat anti-mouse Ig kappa light chain (Pharmingen); goat anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA, USA); goat anti-rat IgG (Jackson ImmunoResearch), 145-2C11 hamster anti-mouse CD3ε (Pharmingen) and 37.51 hamster anti-mouse CD28 (Pharmingen). All animal experimentation was performed in accordance with St. Jude Children's Research Hospital Institutional Animal Care and Use Committee requirements.
Flow cytometry
Analyses were performed on a FACScalibur (BD Biosciences) using Cellquest software (BD Biosciences) and sorting on a MoFlo cytometer (Dako Cytomation, Fort Collins, CO, USA).
Retroviral transduction and T cell culture
Retrovirus was produced as described (15). Briefly, 10 μg of receptor or empty vector construct and 10 μg of retrovirus helper DNA constructs were co-transfected into 293-T cells by calcium phosphate precipitation. At 16 h the cells were washed and cultured in Dulbecco's Modified Eagles Medium (MediaTech) / 10% fetal calf serum (FCS) for 48 h. Supernatant was collected twice daily and used to infect GP+E86 retroviral producer cells in the presence of 8 μg ml-1 polybrene. Transduced GP+E86 cells were flow cytometrically sorted for the presence of GFP, if needed, and expanded. To transduce T lymphocytes, freshly isolated LN cells from C57BL/6 mice were depleted of B cells by panning on goat anti-mouse IgG-coated plates and stimulated with soluble CD3- and CD28-specific antibodies (Abs). Alternatively whole splenocytes were stimulated with conA. rmIL-2 (2 ng ml-1; R&D Systems, Minneapolis, MN, USA) or 10 IU ml-1 rhIL-2 (NCI BRB Repository, Frederick, MD, USA) was added to the cultures. Medium was removed after 1 day and replaced with cleared supernatant from the GP+E86 retroviral producer cells with 8 μg ml-1 polybrene and rIL-2 and the cells were spun at 1800 r.p.m. for 90 min. in a Jouan CR422 tabletop centrifuge. A second cycle of spin infection was repeated on day 2. Transduced T cells were flow cytometrically sorted on day 4–5 for expression of GFP and CD8 and expanded by culturing in EHAA medium (Invitrogen)/10% FCS in the presence of rIL-2 for 5 days. The cells were restimulated every 7–10 days using 2 μg ml-1 conA, 2×106 ml-1 3000 rad-irradiated syngeneic splenocytes and rIL-2 for a maximum of two cycles. Transduced cells were washed and assayed as indicated. Assays were performed in the absence of exogenously added IL-2.
T cell proliferation analysis
The designated number of resting transduced CTL, 7-9 days after prior stimulation, were cultured with 2.5×105 of the indicated irradiated splenocyte population with or without 2 μg ml-1 conA. After 2 days, the cells were pulsed with 1 μCi 3H-thymidine for 16 h and harvested onto filtermats (Wallac-Perkin Elmer, Waltham, MA, USA). Proliferation was measured by liquid scintillation counting of incorporated 3H. All samples were analyzed in triplicate.
Cytokine analysis
Cultures were established as for proliferation analyses. 50 μl of culture supernatant was removed after 48 hours and IFN-γ concentration measured by ELISA (BD-Pharmingen).
In vitro cytotoxicity assay and quantitative flow cytometry
Transduced CTL (105), day 4–6 post stimulation, were incubated at a 1:1 ratio with MD-4 splenocytes for 6h. Splenocytes typically contained ∼50% B cell targets. At the end of the culture, 5000–10000 6 μm fluorescent TruCount beads (BD Biosciences, Duarte, CA, USA) were added. Samples were stained for the indicated target B cell population and then analyzed by quantitative flow cytometry. A pre-established TruCount bead event number was run to ensure that equivalent proportions of each sample were enumerated. Viable cell counts are plotted for individual samples assayed in triplicate.
In vitro B cell proliferation analysis
5,6-carboxyfluorescein (CFSE)-labeled MD-4 Tg splenocytes (2×105) were co-cultured with 105 transduced CTL. For labeling with CFSE (Molecular Probes, Carlsbad, CA, USA), splenocytes were harvested, washed, and incubated with 5 μM CFSE (Molecular Probes) for 7 min at 37 C. The cells were then washed three times with phosphate-buffered saline and cultured as indicated in the text. After 1, 2, or 3 days, cells were stained with B220 and IgMa and analyzed by quantitative flow cytometry as described above.
Adoptive transfer studies
HEL- or vector-modified CTL (∼107) and CFSE-labeled MD-4 splenocytes (∼107) were transferred into Rag1-/- or C57BL/6 mice via retro-orbital injection (through contralateral orbits). At the indicated time point, organs were collected and single cell suspensions made. For splenocytes, red blood cells were lysed. For liver cells, purification was performed by spinning over 37.5% Percoll (GE Healthcare) for 10 min at 1500 r.p.m. Cells were washed two times with Hank's buffered salt solution (HBSS), stained for the presence of Ag-specific B lymphocytes as described in the text, and analyzed by flow cytometry.
Immunization studies
Transduced CTL (107), 6 days after stimulation, were adoptively transferred at day -9, -6, and -3 into C57BL/6 mice. For some cohorts, 50 μg LPS (Sigma) in 100 μl saline was administered i.p. with each CTL treatment. Mice were phlebotomized and serum isolated on day 0. They were then immunized subcutaneously with 100 μg HEL (Sigma) in complete Freunds adjuvant (CFA). The mice were phlebotomized again at day 28 post-immunization. Sera were titered against purified HEL- or control vector-transduced CTL and anti-Ig kappa light chain was used as a secondary Ab.
Statistics
Representative data is shown for each experiment. Error bars indicate 1 standard deviation and p values indicate significance in 2-sided t-tests calculated with Excel spreadsheet software (Microsoft).
Results
Expression of a HEL neo-antigen on CTL
To express HEL on the surface of CTL, we synthesized a chimeric gene construct linking the mouse CD8α leader to coding sequence of HEL and the CD8 hinge and TM domains (Fig. 1a). This was incorporated into the murine stem cell virus-driven retroviral vector MSCV-I-GFP (14), which includes an internal ribosomal entry site-linked GFP. Retrovirus incorporating the chimeric construct or control vector was transduced into purified, TCR-stimulated CD8+ T cells, which were then flow cytometrically enriched for GFP-expressing cells. Surface expression of HEL was verified by staining with HEL-specific antibody (Fig. 1b). Therefore a modified CD8 sequence effectively anchors ectopically expressed HEL to the CTL surface.
The HEL-CD8 construct lacked a cytoplasmic signaling domain. As a result, we did not anticipate that engagement of a complementary ligand on cognate B cells, HEL-specific immunoglobulin, would stimulate effector functions in the modified CTL. Nevertheless, to assess this, we analyzed the impact of chimeric receptor-mediated B cell engagement on CTL stimulation. HEL-CTL and irradiated HEL-specific MD-4 BCR Tg B cells (16) were co-cultured and proliferation or IFN-γ production by the CTL determined (Fig. 2a, b). No impact of the Ag-specific B cells was detected by either measure, indicating that the HEL-specific B cells do not effectively stimulate the cognate Ag-modified CTL.
Figure 2. Cross stimulation by HEL-specific MD-4 B cells and HEL-modified CTL.

(A) 5×104 resting HEL- or vector-modified CTL transduced and flow cytometrically purified for the expression of CD8 and GFP were re-stimulated with the indicated number of irradiated MD-4 BCR Tg or control non-Tg C57BL/6 splenocytes 9 days after initial transduction. After 48 h, cultures were pulsed with 3H-thymidine and 3H incorporation determined 16 h later by scintillation counting. No proliferative response was observed. Viability of the modified CTL was confirmed by stimulation with control splenocytes in the presence of the mitogen conA. (B) 5×104 purified vector- of HEL-modified CTL, 9 days after transduction, were cultured alone, or co-cultured with 2.5×105 irradiated MD-4 Tg or non-Tg C57BL/6 splenocytes. 48 h later cell free supernatant was isolated and IFN-γ production measured by ELISA. Cells were additionally stimulated with non-Tg splenocytes in the presence of conA as a positive control. ND, none detected. (C) To determine if MD-4 B cell engagement of HEL on HEL-modified CTL induced B cell lysis, MD-4 splenocytes (∼50% B lymphocytes) were co-cultured in triplicate with HEL-CTL or vector-modified CTL for 6 h at a 1:1 ratio. B lymphocytes were then enumerated by staining with B220 and MD-4 allotype specific IgMa reactive antibodies and quantitative flow cytometry. A significant increase in survival (2-sided t-test, p<0.05) is apparent among B cells co-cultured with HEL-CTL in the experiment shown, but was inconsistently seen in additional analyses. (D, E) To determine if MD-4 B lymphocytes are stimulated to proliferate and expand in response to the HEL-CTL, CFSE-labeled MD-4 splenocytes were co-cultured with HEL- or vector-transduced CTL, or cultured untreated or in the presence of LPS for up to 3 days. Cells were analyzed at daily intervals by quantitative flow cytometry. Total number of viable cells detected are plotted (D). (E) Plots showing CFSE dilution with proliferation. Representative data is shown for each subfigure from 2 or more experimental repeats.
Ineffective stimulation of B cells by HEL-CTL
Allogeneic veto CTL can induce the apoptotic death of allospecific T lymphocytes. This killing is gradual and apparent over a period of days in co-culture assays, contrasting with the more rapid lysis of target cells, measurable over the course of hours, when CTL are stimulated through their TCR (17-19). To assess for CTL mediated killing of target B cells, the MD-4 B cells were first co-cultured with HEL- or control MSCV-transduced CTL for 6 h and MD-4 cell numbers determined by quantitative flow cytometry. The HEL-transduced CTL did not significantly diminish viable B cell numbers, indicating that they were incapable of targeted cytolysis of the MD-4 cells (Fig. 2c).
Ex-vivo cultured B cells die in vitro over several days in the absence of stimulation. To assess whether the HEL-modified CTL could stimulate the survival and expansion or alternatively accelerate the death of such cells, we labeled MD-4 B cells with CFSE and co-cultured them with HEL- or control vector-modified CTL. Cell numbers were assessed daily for 3 days by quantitative flow cytometry for B220+ cells and proliferation visualized by CFSE dilution. By day 3, virtually all of the B cells cultured in the absence of added CTL or with control vector-transduced CTL had died (Fig. 2d, e). None of these cells demonstrated evidence of proliferation during the time period. In contrast, B cells co-cultured with HEL-transduced CTL entered cell cycle. They expanded and underwent up to 2 division cycles by day 2. However, by day 3, despite continuing cell division, numbers of viable B cells had diminished. This loss of viable cell number was not evident when B cells were activated with a control mitogenic stimulus, LPS. Although proliferation induction was slower with LPS, cell expansion was better sustained and by 3 days of culture LPS-stimulated cells had overtaken the HEL-CTL cultured cells both in cell number and in proliferation cycles. Therefore, the HEL-modified CTL engage the MD-4 B cells and this promotes B cell proliferation. B cell expansion however is limited and the cells undergo an abortive activation program in which initial B cell expansion is followed by cell death.
Induction of B cell tolerance by HEL-CTL
These results opened the possibility that CTL veto cells could induce B cell tolerance in vivo. To test this we adoptively transferred HEL- or control vector-transduced CTL that were flow cytometrically sorted for vector-encoded GFP into syngeneic C57BL/6 mice. Three sequential transfers of modified CTL over a 9 day period were followed by immunization with HEL emulsified in CFA. Serum was isolated immediately prior to immunization (day 0) as well as 28 days later, and analyzed for HEL specific antibody by flow cytometric titration of serum against purified HEL-modified T cells. Analyses of serum collected on day 0 showed that the HEL-modified CTL were not in themselves immunogenic and did not induce detectable levels of antibody (Fig. 3a). By day 28 after immunization, serum from control-treated mice demonstrated high titer anti-HEL antibody (detectable to a 1:6400 dilution; Fig. 3b). However, mice receiving HEL-transduced CTL produced little or no HEL-specific antibody. In separate experiments, this unresponsiveness remained even after boosting the HEL-CTL treated and HEL-immunized mice with additional i.p. antigen (data not shown). Therefore, adoptively transferred HEL-modified CTL are able to functionally tolerize the HEL-specific B cell response.
Figure 3. HEL-CTL induce B cell tolerance in vivo.

HEL-CTL or control vector-transduced CTL (107) were adoptively transferred i.v. into C57BL/6 mice 9, 6, and 3 days prior to immunization with HEL/CFA. Serum was isolated prior to initial immunization with HEL (day 0) and on day 28 and antibody titers tested by staining T cells transduced with the HEL construct or with control vector with serum from the treated animals. (A) Serum collected from mice on day 0 prior to immunization. Plot shows mean fluorescence intensity (MFI) of staining against HEL-transduced cells at various dilutions or control MSCV vector-transduced cells at the highest titer only. (B) Serum collected from mice on day 28 after immunization. (C) Serum collected on day 0 from mice that also received 50 μg LPS i.p. with each infusion of the HEL- or vector-modified CTL. (D) Serum collected on day 28 from mice treated as in (C). Mean ± S.E.M. is shown for each value. 6-8 mice were studied per treatment group in this representative experiment.
For effective stimulation, naïve B cells require signals both through their Ag receptor as well as through accessory receptors (20,21). It was possible that the HEL-modified CTL, by providing BCR-signaling in the absence of necessary accessory signals prompted the death of the MD-4 B cells. Signaling through TLRs is particularly potent, and in some circumstances can even impart T cell independent expansion and differentiation (22). To test whether the presence of TLR ligand could disrupt tolerance induction, we treated mice with HEL- or vector-modified CTL prior to immunization as above, but LPS was administered i.p. with each pre-immunization cell transfer. Notably, transfer of the HEL-CTL in the presence of added LPS stimulation induced a detectable, though weak, HEL-specific antibody response even prior to initial immunization with HEL protein (Fig. 3c). This contrasts with the absence of such a response when LPS was omitted (Fig. 3a). After immunization, the titer and binding of HEL-specific antibody in the HEL-CTL- and control CTL-treated mice was similar (Fig. 3d). Therefore, LPS administered during veto cell treatment not only abrogates tolerance induction, but combined B-cell stimulation by LPS and HEL presented by the HEL-modified CTL leads to anti-HEL antibody production.
Elimination of HEL-specific B cells by HEL-CTL
To better understand the impact of HEL-modified CTL treatment on specific B cells, we co-transferred MD-4 Tg B cells and a single dose of HEL- or control vector-modified CTL into Rag-/- mice. B cells were enumerated 48 h after transfer. Substantial numbers of transferred B cells were detected in the lymphoid organs of mice receiving control vector-transduced CTL (Fig. 4). In contrast, few Tg B cells were detected after transfer of the HEL-modified CTL. Therefore, engagement with HEL-modified CTL leads to the depletion of Ag-specific B lymphocytes in immunocompromised mice.
Figure 4. Co-transfer of MD-4 B cells and modified CTL into Rag1-/- mice.

HEL- or control vector-transduced CTL (107) were adoptively transferred i.v. (retro-orbital) into B and T cell deficient Rag1-/- mice. MD-4 splenocytes (2×107) were transferred through the alternate retro-orbital plexus. On day 2, the mice were sacrificed and numbers of residual MD-4 B cells determined by staining with anti-B220 and MD-4 allotype-specific anti-IgMa antibodies. Percent B220+IgMa+ cells among splenocytes or mixed LN cells from control or HEL-CTL treated mice is shown. Representative data is shown from one of 3 mice per experimental group with essentially identical results.
To determine if a similar Ag-specific depletion of B lymphocytes could be observed in wild type mice, we repeated the experiment, though with adoptive transfer of cells into otherwise unmanipulated C57BL/6 mice (Fig. 5a, b). As for the Rag-/- mice, loss of B cells was apparent in both spleen and LN. This did not result from the migration of the B cells to alternative locations, as transferred B cells were also nearly absent in the bone marrow, liver, and lung. In some experiments, as in Figure 5, a virtually complete absence of the Ag-specific B cells was observed in all organs analyzed. In others, residual proliferating MD-4 B cells were observed in the spleen, though typically few if any cells were detected in the LN. The cells present were diminished in total number compared with control treated mice despite the cells having undergone multiple rounds of cell division (see for example Fig. 6a, b, left columns). Therefore, Ag present on the HEL-modified CTL can lead to MD-4 B cell stimulation and proliferation. However, this response is ineffective as the B cells fail to appreciably expand even when some cells persist and extensively divide.
Figure 5. Co-transfer of MD-4 B cells and modified CTL into wild type mice.
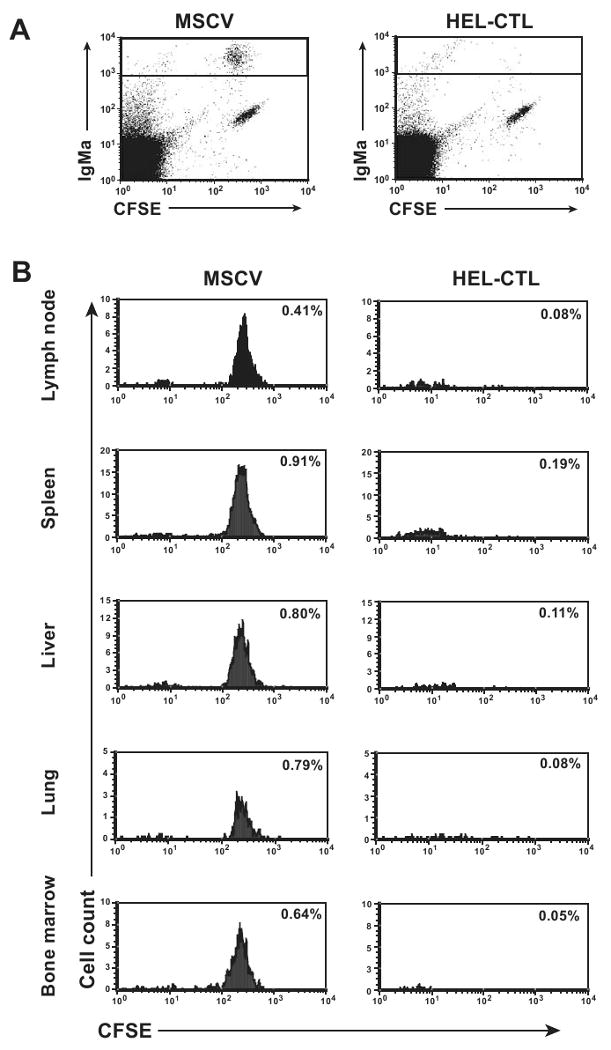
CFSE-labeled MD-4 splenocytes were adoptively transferred into wild-type C57BL/6 mice i.v. retro-orbitally and HEL- or control vector-modified CTL then transferred through the alternate retro-orbital plexus. 4 days later, the indicated organs were isolated and lymphocytes purified. Cells were stained for IgMa and analyzed by flow cytometry. (A) Representative dot plots of LN cells, gated for lymphocytes based on scatter properties, showing IgMa versus CFSE fluorescence. (B) Histogram plots of CFSE-staining among IgMa+ lymphocytes, gated based on the box shown in (A), is shown. Plots are normalized so that an equal number of scatter-gated lymphocytes were analyzed for the paired HEL-CTL and vector control plots to allow visual comparability. Percent of total lymphocytes that are IgMa+ MD-4 B cells is indicated. Representative plots are shown.
Figure 6. LPS promotes the survival of proliferating, HEL-CTL-treated MD-4 B cells.
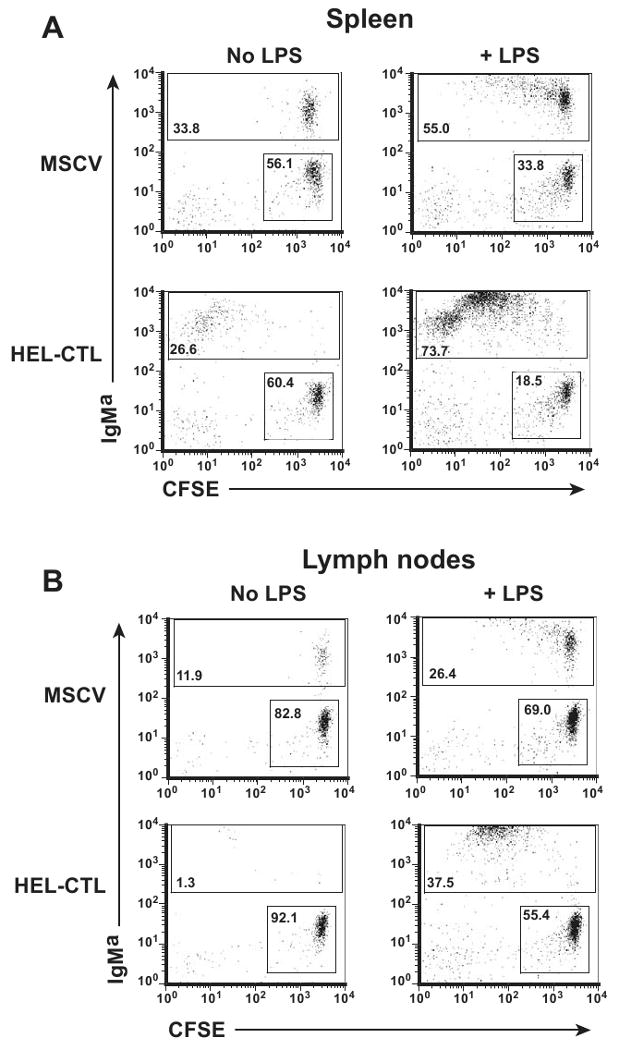
MD-4 mice were bred onto the CD45.1 background, and CFSE-labeled splenocytes transferred into congenic C57BL/6 (CD45.1-CD45.2+) mice. HEL-CTL or vector-control CTL were transferred as in figure 5. LPS or saline (No LPS) was administered i.p. on the day of transfer. Lymphocytes in the spleen and LN were isolated 6 d later, and gated on the adoptively transferred CD45.1+ cells. (A) IgMa and CFSE staining among splenocytes. Numbers of dots on the different plots were normalized to show approximately equivalent numbers of IgMa- CFSE+ cells, which comprise primarily transferred T cells. Percent of IgMa- CFSE+ and IgMa+ cells are indicated in the respective boxes. (B) Analyses were performed as in (A), but for LN cells. Representative plots are shown.
We next tested whether TLR signaling could overcome the effect of the Ag-modified CTL on Ag-specific B cell depletion. We administered the TLR4 ligand LPS or saline i.p. on the day of transfer of MD-4 B lymphocytes and HEL- or control vector-modified CTL. In the absence of LPS substantial numbers of MD-4 B cells persisted after co-transfer with vector-transduced CTL. After co-transfer with HEL-CTL, despite extensive proliferation of residual MD-4 B cells, total B cell numbers were diminished (Fig. 6a, b left columns). In contrast, when LPS was co-administered, a modest proliferative response was observed by a subset of B cells in the control CTL treated mice (Fig. 6a, b right columns). More significantly, strong proliferation and expansion of the MD-4 B cells was seen both in the spleen and the LN of mice treated with HEL-CTL and LPS. Therefore TLR signaling converts an abortive activation program triggered by the HEL-CTL into an effective one characterized by the prominent expansion of Ag-specific B cells. Implicitly, LPS provides important survival signals for the HEL-CTL stimulated B cells.
Discussion
Allogeneic CTL can induce tolerance among naïve and even sensitized cognate allospecific CD8+ T cells (7,11), and these veto cells have shown potential in the clinical induction of allograft tolerance (12). Although other cell types may also serve as veto cells, CTL express high levels of death ligands documented to be important in veto-mediated tolerance, and are particularly potent (17,18). Whether and how veto CTL tolerize the B cell compartment has not been adequately assessed, and is a subject of importance considering the role of antibody responses in both graft-versus-host disease and graft rejection (23-25). Conceivably, third-party-specific allogeneic veto cells may be used to tolerize allo-MHC- or minor alloantigen-specific B cells. Alternatively, antigen-specific B cells may be targeted by antigen-modified veto cells.
We demonstrate that CTL expressing a model B cell antigen, HEL, are effective inducers of B cell tolerance. In vitro, these veto cells do not directly lyse Ag-specific B cells in short-term cytolysis assays, implying that active killing of the naive B cells is not an important mechanism of their action. Rather, the veto cells stimulate B cell proliferation. However, this proliferation is short lived. Initial B cell expansion is not sustained and a loss of viable Ag-specific B cells is detected within 3 days of stimulation. Therefore, the veto cells activate the B cells, but this activation is ineffective and fails to induce sustained growth and survival.
These results bear similarities with veto-induced tolerance of T lymphocytes, in which pCTL that encounter veto cells die off over several days of co-culture (17). However, the mechanism of action of veto cells in B cell tolerance remains to be determined and may differ from tolerance in T cells, which is fas dependent. Indeed, whereas ligation of TLR or CD40 on resting B cells increases susceptibility of B cells to fas-mediated killing, BCR ligation generates resistance to fas-mediated death (26-28). The B cells targeted by veto cells will be stimulated through their BCR by veto cell-expressed antigen, and this should induce protection against fas-derived death signaling. They may also receive additional signals, for instance through CD40 by veto cell-expressed CD40L. However, BCR ligation has been observed to be dominant to other signals, and combined BCR and CD40 signaling also leads to resistance to fas-induced apoptosis (29).
Alternatives to fas mediated cell death for the veto-induced B cell tolerance may also be considered. BCR signaling in anergic B cells is incomplete and unable to induce fas resistance (30). If suboptimal stimulation of B cells and chronic antigen exposure due to prolonged engagement with veto CTL induces anergy, subsequent encounters with CD40L+ FasL+ veto cells may be adequate to further upregulate fas and induce fas mediated apoptosis. It must be noted however, that B cells stimulated by the veto CTL did proliferate well early after stimulation, even if this was not associated with later population expansion (Fig. 2d, e). As anergic cells are non-proliferative, if anergy induction is a primary mechanism of veto cell activity, it is only developing after an initial proliferative burst. Further evaluation of signaling in B cells exposed to veto CTL will be important to better define the roles of anergy, fas – fasL, and other candidate effector molecules in tolerance induction.
Our in vivo data are consistent with the in vitro findings. Treatment with modified CTL is able to induce Ag-specific B cell tolerance in wild type mice. Modeling of this tolerance through the combined adoptive transfer of Ag-modified-CTL and cognate Ag-specific B cells indicates that the B cells are eliminated. Nevertheless, some B cells proliferate, though as with the in vitro co-cultures total cell numbers do not increase despite multiple rounds of cell division. Anergy development subsequent to an initial proliferative burst may here too explain the B cell loss, as anergy is associated with a markedly diminished in vivo lifespan (31). Alternatively, inadequate expansion may be associated with cellular exhaustion due to the unavailability of pro-survival signals. Indeed, LPS, a TLR4 ligand, can convert veto-induced B cell tolerance into an effective immune response.
TLR signals are a potent route to B cell co-stimulation. Recent studies have demonstrated that TLR signaling can act independently of Th cell mediated co-stimulation, and in the presence of DCs can support B cell activation, antibody production, and even affinity maturation (22). TLR signaling induces both pro- and anti-apoptotic pathways, though in normal B lymphocytes anti-apoptotic effects dominate (32,33). It remains possible that the LPS does not act by enhancing B cell survival but rather directly inhibits veto activity instead. However, this would seem unlikely considering the well established role of TLR ligands in B cell activation. Further, studies of T cell suppression by veto CTL failed to identify any direct effect of LPS signaling on veto function (34).
The ultimate utility of veto cells in immune tolerance induction remains to be determined and is the subject of active investigation (12). They may be potentially applied in different ways. Donor-derived veto cells may be capable of tolerizing alloantigen-specific B cells, such as those specific for major histocompatibility complex molecules, when administered prior to solid organ or hematopoietic cell transplantation. CTL genetically modified to express specific antigens, such as the HEL-CTL here, may also be capable of tolerizing B cells specific for pre-defined protein antigens. Examples may include coagulation factors, enzymes, and other proteins introduced through biologic, gene, or cellular therapies into deficient individuals. These introduced proteins can include neo-antigens that may otherwise induce neutralizing antibody responses (35).
The ability of LPS to override veto-cell induced B cell tolerance is significant. Inflammation or infection may be associated with the presence of exogenous or endogenous TLR ligands able to convert veto tolerance into immunity. The use of veto cells in the context of inflammatory conditions may therefore have deleterious effects. Further, in regards to clinical application, it is important to consider that veto cells are unlikely to tolerize long lived plasma cells, which downregulate surface BCR and should thus cease to be targets of the veto cells. Therefore, in contrast to the established effectiveness of veto cells against T cells (11), their effectiveness against B cell responses in the primed immune system may be limited. In summary, our data support the utility of veto CTL as a cellular therapeutic for B cell tolerance, suggest potential mechanisms for this tolerance, but also illustrate potential limitations in their application.
Acknowledgments
The authors thank Richard Cross, Jennifer Smith, Yuxia He and Grieg Lenon for assistance with flow cytometry and cell sorting.
This work was supported by the National Institutes of Health Grant R01 AI056153 (to T.L.G.) and by the American Lebanese Syrian Associated Charities (ALSAC)/St. Jude Children's Research Hospital (to P.N. and T.L.G.).
Abbreviations
- CTL
Cytotoxic T lymphocyte
- HEL
hen egg lysozyme
- DST
donor specific transfusion
- TLR
toll like receptor
- LPS
lipopolysaccharide
- pCTL
precursor CTL
- rm
recombinant mouse
- rh
recombinant human
- BCR
B cell receptor
- GFP
green fluorescent protein
- Ab
antibody
- FCS
fetal calf serum
- CFA
complete Freunds adjuvant
- CFSE
5,6-carboxyfluorescein
- Tg
transgenic
Footnotes
P. Nguyen designed and performed research, analyzed data, and assisted in writing the paper. T.L. Geiger, designed research, analyzed data, and wrote the paper.
The authors have no commercial interests relating to this work.
Reference List
- 1.Soiffer R. Immune modulation and chronic graft-versus-host disease. Bone Marrow Transplant. 2008;42 1:S66–S69. doi: 10.1038/bmt.2008.119. [DOI] [PubMed] [Google Scholar]
- 2.Zarkhin V, Li L, Sarwal M. “To B or not to B?” B-cells and graft rejection. Transplantation. 2008;85:1705–1714. doi: 10.1097/TP.0b013e318177793e. [DOI] [PubMed] [Google Scholar]
- 3.Brennan DC, Mohanakumar T, Flye MW. Donor-specific transfusion and donor bone marrow infusion in renal transplantation tolerance: a review of efficacy and mechanisms. Am J Kidney Dis. 1995;26:701–715. doi: 10.1016/0272-6386(95)90432-8. [DOI] [PubMed] [Google Scholar]
- 4.Fabre JW, Morris PJ. The effect of donor strain blood pretreatment on renal allograft rejection in rats. Transplantation. 1972;14:608–617. doi: 10.1097/00007890-197211000-00013. [DOI] [PubMed] [Google Scholar]
- 5.Armstrong HE, Bolton EM, McMillan I, Spencer SC, Bradley JA. Prolonged survival of actively enhanced rat renal allografts despite accelerated cellular infiltration and rapid induction of both class I and class II MHC antigens. J Exp Med. 1987;165:891–907. doi: 10.1084/jem.165.3.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sato S, Iwata H, Kitagawa S, et al. The lymphoid cell populations required for induction of tolerance of different subsets of alloantigen-reactive T cells. Transplantation. 1991;52:862–867. doi: 10.1097/00007890-199111000-00021. [DOI] [PubMed] [Google Scholar]
- 7.Fink PJ, Shimonkevtz RP, Bevan MJ. Veto Cells. Ann Rev Immunol. 1988;6:115–37. doi: 10.1146/annurev.iy.06.040188.000555. [DOI] [PubMed] [Google Scholar]
- 8.Rammensee HG, Bevan MJ. Mutual tolerization of histoincompatible lymphocytes. Eur J Immunol. 1987;17:893–95. doi: 10.1002/eji.1830170625. [DOI] [PubMed] [Google Scholar]
- 9.Reisner Y, Gur H, Reich-Zeliger S, Martelli MF, Bachar-Lustig E. Hematopoietic stem cell transplantation across major genetic barriers: tolerance induction by megadose CD34 cells and other veto cells. Ann N Y Acad Sci. 2003;996:72–9. doi: 10.1111/j.1749-6632.2003.tb03235.x. [DOI] [PubMed] [Google Scholar]
- 10.Reich-Zeliger S, Bachar-Lustig E, Gan J, Reisner Y. Tolerance induction by veto CTLs in the TCR transgenic 2C mouse model. I. Relative reactivity of different veto cells. J Immunol. 2004;173:6654–6659. doi: 10.4049/jimmunol.173.11.6654. [DOI] [PubMed] [Google Scholar]
- 11.Reich-Zeliger S, Bachar-Lustig E, Bar-Ilan A, Reisner Y. Tolerance induction in presensitized bone marrow recipients by veto CTLs: effective deletion of host anti-donor memory effector cells. J Immunol. 2007;179:6389–6394. doi: 10.4049/jimmunol.179.10.6389. [DOI] [PubMed] [Google Scholar]
- 12.Ophir E, Reisner Y. Induction of tolerance in organ recipients by hematopoietic stem cell transplantation. Int Immunopharmacol. 2009;9:694–700. doi: 10.1016/j.intimp.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 13.Lefrancois L, Olson S, Masopust D. A critical role for CD40-CD40 ligand interactions in amplification of the mucosal CD8 T cell response. J Exp Med. 1999;190:1275–1284. doi: 10.1084/jem.190.9.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Persons DA, Allay JA, Allay ER, et al. Retroviral-mediated transfer of the green fluorescent protein gene into murine hematopoietic cells facilitates scoring and selection of transduced progenitors in vitro and identification of genetically modified cells in vivo. Blood. 1997;90:1777–1786. [PubMed] [Google Scholar]
- 15.Alli R, Nguyen P, Geiger TL. Retrogenic modeling of experimental allergic encephalomyelitis associates T cell frequency but not TCR functional affinity with pathogenicity. J Immunol. 2008;181:136–145. doi: 10.4049/jimmunol.181.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goodnow CC, Crosbie J, Adelstein S, et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 1988;334:676–682. doi: 10.1038/334676a0. [DOI] [PubMed] [Google Scholar]
- 17.Reich-Zeliger S, Gan J, Bachar-Lustig E, Reisner Y. Tolerance induction by veto CTLs in the TCR transgenic 2C mouse model. II. Deletion of effector cells by Fas-Fas ligand apoptosis. J Immunol. 2004;173:6660–6666. doi: 10.4049/jimmunol.173.11.6660. [DOI] [PubMed] [Google Scholar]
- 18.Reich-Zeliger S, Zhao Y, Krauthgamer R, Bachar-Lustig E, Reisner Y. Anti-third party CD8+ CTLs as potent veto cells: coexpression of CD8 and FasL is a prerequisite. Immunity. 2000;13:507–515. doi: 10.1016/s1074-7613(00)00050-9. [DOI] [PubMed] [Google Scholar]
- 19.George JF, Thomas JM. The molecular mechanisms of veto mediated regulation of alloresponsiveness. J Mol Med. 1999;77:519–526. doi: 10.1007/s001099900027. [DOI] [PubMed] [Google Scholar]
- 20.Richards S, Watanabe C, Santos L, Craxton A, Clark EA. Regulation of B-cell entry into the cell cycle. Immunol Rev. 2008;224:183–200. doi: 10.1111/j.1600-065X.2008.00652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hodgkin PD, Basten A. B cell activation, tolerance and antigen-presenting function. Curr Opin Immunol. 1995;7:121–129. doi: 10.1016/0952-7915(95)80037-9. [DOI] [PubMed] [Google Scholar]
- 22.Herlands RA, Christensen SR, Sweet RA, Hershberg U, Shlomchik MJ. T cell-independent and toll-like receptor-dependent antigen-driven activation of autoreactive B cells. Immunity. 2008;29:249–260. doi: 10.1016/j.immuni.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanfilippo F, Baldwin WM. Antibody and complement in graft rejection. Transplant Proc. 1997;29:179–180. doi: 10.1016/s0041-1345(96)00645-8. [DOI] [PubMed] [Google Scholar]
- 24.Zaja F, Bacigalupo A, Patriarca F, et al. Treatment of refractory chronic GVHD with rituximab: a GITMO study. Bone Marrow Transplant. 2007;40:273–277. doi: 10.1038/sj.bmt.1705725. [DOI] [PubMed] [Google Scholar]
- 25.Patriarca F, Skert C, Sperotto A, et al. The development of autoantibodies after allogeneic stem cell transplantation is related with chronic graft-vs-host disease and immune recovery. Exp Hematol. 2006;34:389–396. doi: 10.1016/j.exphem.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 26.Rothstein TL, Wang JK, Panka DJ, et al. Protection against Fas-dependent Th1-mediated apoptosis by antigen receptor engagement in B cells. Nature. 1995;374:163–165. doi: 10.1038/374163a0. [DOI] [PubMed] [Google Scholar]
- 27.Schattner EJ, Elkon KB, Yoo DH, et al. CD40 ligation induces Apo-1/Fas expression on human B lymphocytes and facilitates apoptosis through the Apo-1/Fas pathway. J Exp Med. 1995;182:1557–1565. doi: 10.1084/jem.182.5.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mizuno T, Zhong X, Rothstein TL. Fas-induced apoptosis in B cells. Apoptosis. 2003;8:451–460. doi: 10.1023/a:1025534223168. [DOI] [PubMed] [Google Scholar]
- 29.Lagresle C, Mondiere P, Bella C, Krammer PH, Defrance T. Concurrent engagement of CD40 and the antigen receptor protects naive and memory human B cells from APO-1/Fas-mediated apoptosis. J Exp Med. 1996;183:1377–1388. doi: 10.1084/jem.183.4.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rathmell JC, Townsend SE, Xu JC, Flavell RA, Goodnow CC. Expansion or elimination of B cells in vivo: dual roles for CD40- and Fas (CD95)-ligands modulated by the B cell antigen receptor. Cell. 1996;87:319–329. doi: 10.1016/s0092-8674(00)81349-5. [DOI] [PubMed] [Google Scholar]
- 31.Fulcher DA, Basten A. Reduced life span of anergic self-reactive B cells in a double-transgenic model. J Exp Med. 1994;179:125–134. doi: 10.1084/jem.179.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Banerjee A, Grumont R, Gugasyan R, White C, Strasser A, Gerondakis S. NF-kappaB1 and c-Rel cooperate to promote the survival of TLR4-activated B cells by neutralizing Bim via distinct mechanisms. Blood. 2008;112:5063–5073. doi: 10.1182/blood-2007-10-120832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richards S, Watanabe C, Santos L, Craxton A, Clark EA. Regulation of B-cell entry into the cell cycle. Immunol Rev. 2008;224:183–200. doi: 10.1111/j.1600-065X.2008.00652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zimring JC, Chadwick TE, Hillyer CD, Roback JD. Effect of mediators of innate immunity and inflammation on CD8+ veto cells. Transplantation. 2004;78:1597–1600. doi: 10.1097/01.tp.0000144322.48212.14. [DOI] [PubMed] [Google Scholar]
- 35.Reding MT. Immunological aspects of inhibitor development. Haemophilia. 2006;12 6:30–5. doi: 10.1111/j.1365-2516.2006.01363.x. discussion 35-6. [DOI] [PubMed] [Google Scholar]