

Abstract
Effective therapeutic options for Alzheimer’s disease (AD) are limited and much research is currently ongoing. The high attrition rate in drug development is a critical issue. Here, the quantitative pharmacology approach (QP-A) and model-based drug development (MBDD) provide a valuable opportunity to support early selection of the most promising compound and facilitate a fast, efficient, and rational drug development process. The aim of this analysis was to exemplify the QP-A by eventually predicting the clinical outcome of a proof-of-concept (PoC) trial of tesofensine in AD patients from two small phase IIa trials. Retrospective population pharmacokinetic/pharmacodynamic (PK/PD) modeling of tesofensine, its metabolite M1, and assessment scale-cognitive subscale data from two 4-week placebo-controlled studies in 62 mild AD patients was performed using non-linear mixed effects modeling. The final PK/PD model was used to predict data of a negative 14-week phase IIb PoC trial (430 AD patients). For the PK, one-compartment models for tesofensine and M1 with first-order absorption and elimination were sufficient. An extended Emax model including disease progression best described the PK/PD relationship using effect compartments. The placebo effect was also implemented in the final PK/PD model based on a published placebo model developed in a large AD cohort. Various internal evaluation techniques confirmed the reliability and predictive performance of the PK/PD model, which also successfully predicted the 14-week PoC data. For tesofensine, the dose concentration–effect relationship has successfully been described in mild AD patients demonstrating the supportive value of PK/PD models in QP-A/MBDD in early phases of clinical development for decision-making.
Key words: ADAS-COG, Alzheimer’s disease, PK/PD modeling, quantitative pharmacology, tesofensine
INTRODUCTION
The high attrition rate in drug development is a critical issue: only one in nine compounds will successfully pass the drug development process and will eventually obtain approval by regulatory agencies (1). Although several concepts have been proposed to streamline and improve the drug discovery and development process, e.g., the Critical Path Initiative led by the US Food and Drug Administration (FDA; 1–4), especially in the drug development process, traditional tools and concepts are still widespread. One major challenge is to identify the most promising compounds as early as possible before conducting more costly late-stage trials. One promising and supportive instrument as well as strategy might be the development, application, and implementation of reliable and robust mathematical models with predictive capabilities during drug development (3), also referred to as quantitative pharmacology approach (QP-A) and as model-based drug development (MBDD; 3,5–7). Today, quantitative pharmacology and MBDD have not been established throughout the pharmaceutical industry as standard approach and strategy, but they may represent a valuable opportunity to support an early selection of the most promising safe and/or efficacious compound and facilitate a fast, efficient, and rational drug development process (6,8,9). This concept seems to be particularly worthwhile in therapeutic areas with an emerging public health concern, e.g., in Alzheimer’s disease (AD): AD is a chronic progressive neurodegenerative disorder characterized by predominantly decline/loss of cognitive functions and activities of daily living, increased prevalence with age, and currently with only few (symptomatic) drug treatment options (10,11).
Tesofensine (NS2330), currently in clinical development for the treatment of obesity (12,13), is a central nervous system (CNS) active drug which was promoted as a potential candidate for the treatment of Alzheimer’s disease (14). A chance to reach the market for the treatment of AD of 15% was estimated (14) which is above the average of 8% for CNS active compounds (1). In vitro and in vivo investigations of tesofensine showed an inhibition of the presynaptic uptake of the neurotransmitters noradrenalin, dopamine, and serotonin. The cholinergic system was indirectly stimulated (14). Preclinical studies demonstrated the enhancement of the cognitive function, the short- and long-term memory, and the attention in animals. Tesofensine is mainly metabolized by CYP3A4 into M1 (15). Its trough concentrations in humans after oral administration of tesofensine at steady state were about one-third of the steady-state concentrations of tesofensine (16). In vivo investigation in mice resulted in a fivefold lower potency of the metabolite compared to the parent compound (17). Tesofensine and M1 showed long half-lives of >200 h in humans (16,18). First clinical results of two small 4-week phase IIa trials performed in mild AD patients were promising: a significant improvement in cognitive function was demonstrated (14). Subsequently, a 14-week phase IIb proof of concept (PoC) trial was performed in 430 patients with mild-to-moderate AD. Unfortunately, the trial did not meet the efficacy criteria to proceed with the phase III development program (19). Consequently, the question arose whether such tendency might have been foreseeable with data already available.
The aim of this analysis was to exemplify the application of the quantitative pharmacology approach in AD including prior knowledge, exposure–response relationship, and disease progression. In particular, a population pharmacokinetic/pharmacodynamic (PK/PD) model for tesofensine and M1 based on data obtained from two phase IIa studies was to be retrospectively developed to characterize the dose concentration–response relationship in mild AD patients. In this PK/PD model, disease progression (DP) of Alzheimer’s and the placebo effect should be integrated and the final model be evaluated for its performance. The final PK/PD model should then be used to predict the PK/PD of a 14-week phase IIb PoC trial with 430 mild-to-moderate AD patients. The predicted clinical outcome should be compared with the observed PK/PD measurements. Ultimately, the PK/PD model ought to be re-estimated with a combined dataset comprising the two phase IIa studies and the phase IIb PoC trial.
METHODS
Study Design
Two randomized, double-blind, placebo controlled, parallel study design phase IIa studies (S1, S2) were considered for model development. Patients participated in an inpatient phase (between 7 and 14 days) followed by an outpatient maintenance dose phase with weekly return visits on an ambulant basis through day 28 and follow-up visits at day 42. The studies were in accordance with the Declaration of Helsinki (1996 Version; 20), the ICH harmonized tripartite guideline for good clinical practice (21) and applicable regulatory requirements. Written informed consent was obtained from each patient and caregiver before study participation.
Tesofensine doses from 0.125–2 mg were orally administered every morning over 28 days: patients in dose groups S1.A and S1.B received 0.5 and 1.0 mg/day for 3 days, respectively, and 0.125 mg/day for 25 days. Patients in dose groups S1.C and S1.D received 1.5 and 2.0 mg/day for 3 days, respectively, and 0.25 mg/day for 25 days. Patients in dose groups S2.A and S2.B received 2.0 mg/day for 3 days and 0.5 and 1.0 mg/day for 25 days, respectively.
Observations
Plasma samples for the determination of tesofensine and M1 concentrations were taken for all dose groups at 0, 96, 168, 312, 648, 984, and 1,320 h. Additional samples were taken for S2.A at 174 and 654 h and for S2.B at 6, 102, 480, 486, 650, 652, 654, 655, 656, 658, 660, and 672 h. Tesofensine and M1 concentrations were determined by a fully validated HPLC-MS/MS (16). AD assessment scale-cognitive subscale (ADAS-Cog; 22) was assessed at screening, day 1, 28, and 42 by adequately trained staff at approximately the same time of the day, in the same test order, and by the same health care professional(s).
Data Analysis
The population PK/PD analyses, external prediction, quantitative predictive check (QPC), and visual predictive check (VPC) were performed using NONMEM, version VI and the ADVAN6 subroutine (23). The estimation method was the first-order conditional estimation method with interaction. Parameter estimates and standard errors of estimates expressed in percent are given. Interindividual variability (IIV) was modeled using exponential random effects models. Goodness-of-fit was analyzed using the objective function value (OBJF; 24) and various diagnostic methods, e.g. (conditional) weighted residuals (25). If models were classified as nested, one model was declared superior over the other model when the OBJF was reduced by 3.84 (p < 0.05, 1df; 23). Median profiles were simulated using Berkeley Madonna (Version 8.0.1). Figures were generated using SigmaPlot (Version 10.0); SAS (Version 9.1.3) was used for statistical analysis.
PK/PD model development was performed sequentially: first, DP and the placebo effect (26) were explored separately. For the latter, data from the placebo-treated patients were investigated using a published model (27) and other reasonable placebo models were explored if necessary. Subsequently, the PK models for tesofensine and M1 were developed. One- and two-compartment models were assessed for both compounds individually. Pharmacostatistical models were systematically evaluated to identify the best model. Subsequently, a combined PK model for parent drug and metabolite was built. Different formation processes and structural and statistical modifications were investigated.
The final combined PK model and the placebo effect model were then used for PK/PD model development with the PK and placebo-effect parameters initially fixed. Different types of PD models were investigated. Potency of M1 was assumed to be fivefold lower than tesofensine based on prior knowledge gained from the modeling of preclinical investigations (17). Afterwards, PK/PD parameters were simultaneously estimated. Ultimately, the final PK/PD model was re-estimated with a combined dataset comprising the two-phase IIa studies and the phase IIb PoC trial (see below). This re-estimation was performed after the external prediction (see below) was finished.
Model Evaluation
Bootstrap analysis (28,29) was performed using Perl speaks–NONMEM (30) for the final PK/PD model with 500 bootstrap datasets to obtain the bootstrap median values and standard errors.
VPC (31) was performed for the plasma concentration–time profiles to evaluate the adequacy of the developed PK/PD model for describing the data (central trend and variability). Simulation of 1,000 new datasets was carried out using the final model and fixed- and random-effects parameters. The concentration–time profiles were plotted for the fifth, 50th, and 95th percentile of the simulated data, and overlaid with the observed data.
QPC was performed for ADAS-Cog value at week 4. A total of 1,000 datasets was simulated using the final model and fixed- and random-effects parameter. For each dataset, the same number of patients, dosing history, number of observations, and sampling scheme as in the original data were used. Histogram of the simulated medians were constructed and overlaid with their fifth, 50th, and 95th percentile and the observed median.
Simulations for Prediction of PoC Trial
The final PK/PD model was used to predict the PK/PD of a phase IIb double-blind, randomized, dose-ranging, placebo-controlled, multicenter, safety, and efficacy study of three doses of tesofensine in mild to moderate AD patients. In brief, 430 patients were equally randomized to receive 0.25, 0.5, or 1.0 mg of tesofensine or placebo once daily for 14 weeks followed by a 6-week follow-up. The 14-week treatment period is in the same range as utilized in other PoC trials for new chemical entity compounds treating Alzheimer’s disease (32,33). A total of 222 active and 78 placebo patients completed the trial; 130 discontinued prematurely. Study groups were comparable for all recorded demographic variables. The majority of patients were females (63%) and the average age of patients was 75 years old. Most patients were Caucasians (>90%), the remaining patients were Black or Hispanic. No other anti-dementia medication was permitted. Blood sampling time points for the determination of tesofensine and M1 in plasma and further study details can be found elsewhere (16). ADAS-Cog was assessed at screening and weeks 0, 4, 9, 14, and 20. Mean baseline ADAS-Cog values were comparable in the four different dose groups ranging between 21.2 and 22.6.
Simulation of 1,000 new datasets was carried out for each of the active- and placebo-dose groups using the final model and the fixed- and random-effects parameter estimates. The concentration–time profiles were plotted for the median, fifth and 95th percentile of the simulated data, and overlaid with the observed data from the 300 completed patients. The observed individual ADAS-Cog values were corrected (i.e., subtracted) for the individual baseline value. The quality of the prediction was assessed by visual inspection of the simulated and overlaid observed data. In addition, ADAS-Cog values at week 14 were summarized by descriptive statistics and the statistical differences between the observed and predicted values were evaluated for each dose group using a chi-square test.
Additionally, the final model (including population parameter uncertainty) was used to simulate 500 replicates of the 14-week PoC study with the same study design using the Trial Simulator (Version 2.1.1). For simulation, the standard errors obtained from the variance–covariance matrix of NONMEM and provided in literature for the placebo model were used. The interdependence of the parameters was used for all unfixed parameters. An enrolment of 430 patients was assumed with a random dropout rate of 25%, a value pre-specified in the study protocol. ANOVA analyses (p < 0.05) were performed to evaluate the outcome (i.e., ADAS-Cog) at week 4 and 14. In case of a significant ANOVA results, a post-hoc analysis was performed to exactly evaluate the significant differences, i.e., whether the placebo group was significantly better or worse compared to the active dose groups.
RESULTS
Data Base
The dataset from the two phase IIa studies used for model development, consisted of 44 active and 18 placebo treated patients with mild AD reflected by a median ADAS-Cog (22) value at baseline of 8.5 points (range: 3.3–23.7). Patient characteristics of active and placebo group were comparable. The study population (60 Caucasians, two African–Americans) contained 44% females and had normal hepatic function. Median creatinine clearance, age, and weight was at 77 mL/min (range: 41–136), 70 years (range: 70–80), and 80 kg (range: 54–129), respectively.
The PK dataset included 357 tesofensine and 341 M1 plasma concentrations from the 44 active treated patients. Concentrations ranged between 0.267 and 30.5 ng/mL and 0.105 and 7.34 ng/mL for tesofensine and M1, respectively. ADAS-Cog measurements for PD modeling were available from 18 placebo and 44 active treated patients, contributing 176 active and 72 placebo measurements (range: 1.3–23.7 points).
Disease Progression and Placebo Effect Model
DP was incorporated a priori into the model. Due to the short treatment period of 4 weeks in the presented studies, DP could not be estimated. Alternatively, literature-based DP rate of 6 points per year (26) was implemented using a linear slope model where DP was added to the overall effect.
A published placebo model (27), developed in a large cohort of AD patients, was applied to describe the placebo effect of the 72 ADAS-Cog measurements from the 18 patients:
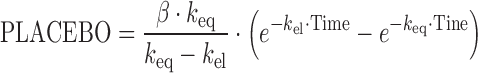 |
where keq is the rate constant defining the onset rate of the placebo effect, kel the rate constant defining the offset rate of the placebo effect, and β is a scaling parameter defining the size of the placebo effect. Due to the sparse data situation, parameters of the model could not be estimated. Subsequently, all structural and IIV parameters were fixed to literature values (34). Population and individual predictions were obtained by setting the number of evaluations to 0 (MAXEVAL = 0). Goodness-of-fit plots (Fig. 1a–b) indicated that the placebo model with literature values sufficiently described the data. QPC results for the ADAS-Cog value (week 4) suggested good predictivity (Fig. 2a); observed and predicted median values deviated less than 4%.
Fig. 1.

Goodness-of-fit plots for the final PK/PD model: population predictions (left panel) and individual predictions (right panel) versus ADAS-Cog measurements of placebo (a, b) and active treatment (g, h) and versus observed plasma concentrations of tesofensine (c, d), and M1 (e, f). Solid line indicates line of identity
Fig. 2.

Internal evaluation: quantitative predictive check for the ADAS-Cog at the end of treatment (4 weeks) of a placebo and b active treatment
Population PK Model
Plasma concentration–time profiles of tesofensine and M1 were best described by one-compartment models with first-order elimination processes for both compounds. Elimination of tesofensine from the central compartment contained a relative metabolic clearance (CLmet/F) which accounted for the formation of M1 from tesofensine and a relative non-metabolic clearance (CLnon-met/F) which accounted for any elimination pathways except the formation of M1 from tesofensine. Absorption of tesofensine was best modeled as a first-order absorption process (ka). Due to the sparse data situation in the absorption phase, ka was fixed to 0.69 h−1 based on results from a population PK analysis performed with phase I data (unpublished). Fixing of ka to 0.385 h−1, a value determined in a recent analysis (16), resulted in a significant increase of the OBJF. Simulations revealed (not shown) that the impact of a varying ka between 0.385 and 0.69 h−1on the overall plasma-concentration time profile is negligible. Based on prior knowledge gained in mice (17), the typical volume of distribution of M1 (V3/F) was set to 0.768-fold of the typical volume of distribution of tesofensine (V2/F) to overcome identifiably issues. In the final combined PK model IIV was included in non-metabolic clearance (ωCLnon-met/F), clearance of M1 (ωCLmet/F) and the central volumes of distribution of tesofensine (ωV2/F) and M1 (ωV3/F). A correlation between CLnon-met/F and V2/F was implemented in the model. Residual variability was modeled with a proportional residual error model for tesofensine and M1.
Population PK/PD Model
For the evaluation of the drug effect on the ADAS-Cog value, the placebo effect (PLACEBO) and disease progression (6 points/year) were incorporated a priori into the PK/PD model. The ADAS-Cog value in the PK/PD model was described according to the following equation: ADAS-Cog = Baseline + DRUG + PLACEBO + Disease Progression. A schematic illustration of the PK/PD model is shown in Fig. 3.
Fig. 3.

Schematic final PK/PD model
The drug effect of tesofensine and M1 (DRUG) on ADAS-Cog was best described by an extended Emax model accounting for the competitive interaction of both compounds. Effect compartments were necessary to establish the concentration–effect relationship.
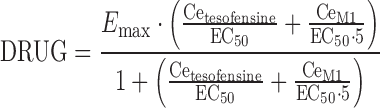 |
where Emax was the maximum effect attributable to the active treatment and EC50 was the concentration producing 50% of the maximum effect, Cetesofensine and CeM1 reflected the effect compartment concentration of tesofensine and M1, respectively. Due to the limited data keo was fixed to 0.0001 h−1, a value close to the estimate and additionally based on results from a sensitivity analysis: varying keo between 0.01–0.00001 h−1 resulted in an expected impact on the estimate of EC50. For keo values ≤0.0001 h−1, no difference was observed with respect to the OBJF, other parameter estimates, and goodness-of-fit assessment. For keo values >0.0001 h−1, a significant increase of the OBJF occurred. In addition, other parameter estimates and the goodness-of-fit were influenced. IIV was included in Emax (ωEmax). Residual variability was modeled with an additive error model.
After finalizing the model, PK and PD parameters were unfixed and estimated simultaneously with good precision (Table I, relative standard errors (RSE) <50%). The population parameter estimates were similar to the median of the 500 bootstrap replicates with a relative bias of −4.7– +7.5%, and were within the 90% confidence intervals obtained from the bootstrap analysis (Table I). These results suggested unbiased parameter estimates of the model developed.
Table I.
Parameter Estimates of Final PK/PD Model
| Phase IIa studies dataset | Combined dataset | |||||
|---|---|---|---|---|---|---|
| NONMEM | Bootstrap analysesc | NONMEM | ||||
| Parameter | Value | RSE,% | Value | 5th–95th percentile | Value | RSE,% |
| Fixed Effects PK | ||||||
| ka [1/h] | 0.69 | FIX | 0.69 | FIX | 0.602 | 15.7 |
| V2/F [L] | 720 | 4.9 | 716 | 665–779 | 694 | 2.3 |
| CLnon-met/F [L/h] | 1.31 | 5.5 | 1.31 | 1.19–1.43 | 1.16 | 3.0 |
| CLmet/F [L/h] | 0.416 | 6.9 | 0.413 | 0.373–0.457 | 0.430 | 3.8 |
| V3/F [L]d | 553 | FIX | 550 | FIX | 533 | FIX |
| CLM1/F [L/h] | 1.17 | 8.2 | 1.15 | 1.03–1.31 | 1.01 | 3.9 |
| Fixed Effects PD | ||||||
| keo [1/h] | 0.0001 | FIX | 0.0001 | FIX | 0.0001 | FIX |
| EMAX | −1.46 | 29.3 | −1.57 | −2.57–−0.93 | −2.25 | 13.2 |
| EC50 [ng/mL] | 0.0139 | 49.6 | 0.0139 | 0.0064–0.123 | 0.407 | 26.3 |
| Fixed Effects Placebo | ||||||
| keq [1/h] | 0.00183 | FIX | 0.00183 | FIX | 0.00183 | FIX |
| kel [1/h] | 0.000473 | FIX | 0.000473 | FIX | 0.000473 | FIX |
| β | −1.42 | FIX | −1.42 | FIX | −1.42 | FIX |
| Random Effects | ||||||
| ωCLnon-met/F [CV%]a | 42.2 | 23.8 | 41.3 | 32.4–49.9 | 68.3 | 15.4 |
| ωCLM1/F [CV%]a | 21.3 | 23.9 | 20.3 | 15.7–24.8 | 26.8 | 11.4 |
| ωV2/F [CV%]a | 30.3 | 24.4 | 30.0 | 24.1–36.0 | 35.6 | 13.2 |
| ωV3/F [CV%]a | 42.5 | 39.3 | 41.2 | 27.4–52.1 | 40.1 | 16.0 |
| ωEMAX [CV%]a | 97.7 | 35.7 | 97.6 | 64.6–131 | 108 | 12.5 |
| ωβ [CV%]a | 128 | FIX | 128 | FIX | 128 | FIX |
| Corr CLnon–met/F_V2/F | 0.72 | 28.9 | 0.717 | 0.653–0.774 | 0.645 | 17.2 |
| prop errtesofensine [%]a | 14.9 | 23.5 | 14.4 | 11.7–17.6 | 24.3 | 9.1 |
| prop errM1 [%]a | 17.3 | 36.9 | 16.9 | 14.0–23.6 | 19.1 | 10.8 |
| add errADAS-Cog | ±2.3 | 19.5 | ±2.2 | 1.9–2.7 | ±4.0 | 11.3 |
RSE Relative standard error
aRSE is given on the variance scale
bRSE of the covariance estimate
cbased on 500 bootstrap runs
d0.768-fold of V2/F
Goodness-of-fit plots (Fig. 3c–h) revealed that the parent drug, metabolite, and ADAS-Cog data were well described by the model. The degree of ε-shrinkage was low (≤14%) on the tesofensine, the M1 concentrations, the active ADAS-Cog and the placebo ADAS-Cog observations indicating that the individual data were sufficiently informative on the parameters for estimating individual predictions (35).
Dose normalized VPC for tesofensine and M1 (Fig. 4) showed neither bias nor overestimation of variability of the PK part of the model. QPC results for the ADAS-Cog value (week 4) were satisfying (Fig. 2b); observed and predicted median values were similar showing less than 2% deviation.
Fig. 4.

Internal evaluation: visual predictive checks for a tesofensine and b M1. Observed concentrations (circles) and calculated fifth, 50th (median), and 95th percentiles (solid lines) from simulations are shown over time
Simulations
Simulations of the median PK profiles of tesofensine and M1 after a 14-week oral once-daily administration of 0.5 mg tesofensine are shown in Fig. 5a. Steady state of tesofensine and M1 effect compartment concentrations was not achieved at the end of treatment. Figure 5b provides an overview of the various single effects (disease progression, placebo effect without DP, and tesofensine + M1 drug effect without DP and placebo) and their combination (tesofensine + M1 drug effect plus placebo effect plus DP) after a 14-week administration of 0.5 mg tesofensine. The maximum effects of tesofensine and placebo were achieved after approximately ∼2–3 and ∼6 weeks, respectively. Median ADAS-Cog profiles over time after a 14-week treatment of 0.25, 0.5, and 1 mg tesofensine including DP and placebo effect are shown in Fig. 5c. Higher doses earlier achieved the maximum effect after ∼2 weeks; for the maximum effect and the effect-time course after ∼6–8 weeks, virtually no dose-dependent differences were observable.
Fig. 5.

Simulated pharmacokinetic and pharmacodynamic profiles using the final PK/PD model: a simulated median plasma and effect compartment concentration–time profiles after once-daily oral dosing of 0.5 mg tesofensine; b simulated median ADAS-Cog over time after a 14 week once daily oral administration of placebo, 0.25, 0.5, and 1.0 mg tesofensine and for disease progression; c simulated median ADAS-cog (including disease progression) over time after a 4- and 14-week administration of placebo and 0.5 mg tesofensine and for disease progression, respectively
The final model was used to predict the data from a 14-week PoC study in mild-to-moderate AD patients. From the 222 active treated patients who finished the trial, seven patients without measurements on week 14 were excluded. Overall data from 215 patients, contributing 1,546 tesofensine, 1,380 M1 concentrations, and 814 ADAS-Cog measurements, were available. Results are shown in Figs. 6 and 7a–c: overall, tesofensine and M1 concentrations as well as ADAS-Cog measurements were very well predicted and showed no bias or overestimation of variability.
Fig. 6.

External PK predictions for tesofensine (left panels) and M1 (right panels) for the 0.25 mg (a, b), 0.5 mg (c, d), and 1.0 mg (e, f) dose group. Observed concentrations (circles) and calculated fifth, 50th (median), and 95th percentiles (solid lines) from simulations are shown over time
Fig. 7.

External PD predictions of the baseline-subtracted ADAS-Cog measurements: observed concentrations (circles) and calculated median, fifth and 95th percentiles (solid lines) from simulations are shown over time for the 0.25 (a), 0.5 (b), 1.0 mg (c), and placebo (d) dose group. Box-whisker plot (e) (box 25th, 50th, and 75th percentile, whiskers tenth and 90th percentile, open circles fifth and 95 percentile) of observed (OBS) and simulated (SIM) ADAS-Cog values at week 14 of the 0.25, 0.5, 1.0 mg, and placebo dose group
From 78 placebo-treated patients who finished the trial, five patients without measurements on week 14 were excluded. Overall data from 73 patients, contributing 291 ADAS-Cog measurements, was available. Results are shown in Fig. 7d: ADAS-Cog measurements were satisfactorily predicted. Bias in model prediction was not observed as the calculated median represented the trend of the observed data with a slight tendency of underprediction at the late time points. The majority of observed data were within the 90%-prediction interval.
Figure 7e and Table II compare the predicted and observed (baseline subtracted) ADAS-Cog values at the end of treatment (week 14) of all treatment groups. Observed outcomes for the 0.25, 0.5, and 1 mg dose group at week 14 were very well predicted. No significant differences between the distributions of the observed and predicted values (median and variability) were apparent. Only the placebo group was slightly underpredicted. Statistical observation (chi-square test, p < 0.05) revealed no statistical difference between the simulated and observed results of each tesofensine dose group (0.25, 0.5, and 1 mg). The simulated results of the placebo group were statistically significantly different from the observed values.
Table II.
Statistical Evaluation of the ADAS-Cog Value Distribution at Week 14 of the Observed and Simulated Phase IIb Data
| Placebo | 0.25 mg | 0.5 mg | 1 mg | |||||
|---|---|---|---|---|---|---|---|---|
| OBS | SIM | OBS | SIM | OBS | SIM | OBS | SIM | |
| N | 71 | 1,000 | 83 | 1,000 | 66 | 1,000 | 56 | 1,000 |
| P1 | −22.33 | −14.23 | −12.67 | −14.93 | −15.00 | −17.29 | −13.67 | −16.06 |
| P5 | −11.67 | −4.99 | −8.67 | −8.80 | −11.33 | −9.16 | −10.67 | −9.78 |
| P10 | −8.67 | −3.30 | −7.33 | −6.55 | −10.00 | −6.53 | −8.33 | −6.71 |
| P25 | −5.33 | −1.34 | −4.67 | −3.72 | −4.33 | −3.71 | −4.33 | −3.75 |
| Median | −1.67 | 0.58 | −1.67 | −1.47 | −1.17 | −1.30 | −2.00 | −1.39 |
| Mean | −1.94 | 0.08 | −1.55 | −1.99 | −1.70 | −2.04 | −0.98 | −1.99 |
| P75 | 1.33 | 2.37 | 1.33 | 0.57 | 2.00 | 0.59 | 1.67 | 0.61 |
| P95 | 6.67 | 4.68 | 6.00 | 3.23 | 6.00 | 3.31 | 11.33 | 3.35 |
| P99 | 13.67 | 6.13 | 13.00 | 5.64 | 7.67 | 5.06 | 17.67 | 5.47 |
OBS observed; SIM simulated
Additionally, a power analysis was performed. The assumed dropout rate of 25% was similar to the observed dropout rate of ∼30% (130 of 430 enrolled patients discontinued prematurely). In 82.8% and 85.2% of the 500 simulated PoC trial replicates, one or more active groups were significantly better compared to placebo at week 4 and 14, respectively (p < 0.05). In the remaining 7.2% and 4.8% of the studies, no significant difference was observed at week 4 and 14, respectively.
Re-estimation of Final PK/PD Model with Combined Dataset
The dataset utilized for model development was combined with the dataset of the phase IIb PoC study. The combined dataset consisted of 90 placebo and 361 tesofensine treated patients contributing 2,309 and 2,040 plasma concentrations of tesofensine and M1, respectively. In total, 1,508 ADAS-Cog (339 placebo, 1,169 tesofensine) measurements were available. The final model developed with the small phase IIa dataset could successfully be re-estimated with the combined dataset. In addition, it was investigated whether the full or at least parts of the placebo model could be estimated with the larger dataset. Finally, none of these parameters could successfully be estimated. Consequently, these parameters remained fixed to the literature values used before. The parameter estimates of the re-estimated model are provided in Table I. Except for EC50 with a higher and more precisely estimated value (higher precision also for Emax), overall parameter estimates were in the same range as the estimates from the phase IIa datasets. This was further confirmed by simulations of 0.5 mg tesofensine administration over 14 weeks with the final estimates of the phase IIa dataset and the combined dataset. The PK profiles were very similar (Fig. 8a) as well as the maximum effect in ADAS-Cog after tesofensine treatment despite a slight different in the initial shape of the ADAS-cog-time profile most probably due the different EC50 values (Fig. 8b).
Fig. 8.

Simulated pharmacokinetic (a) and pharmacodynamic (b) profiles using the final PK/PD model with parameter estimates of the phase IIa dataset and the combined phase IIa and IIb dataset once-daily oral dosing of 0.5 mg tesofensine for 14 weeks
DISCUSSION
For AD patients, the US FDA has approved five drugs of only moderate and temporary benefit that are associated with a high non-responder rate. A number of drug candidates are currently in clinical development (10). Scientific methods of pharmacometrics bridging different disciplines such as biology, pharmacology of drug (and placebo), and disease as well as prior knowledge from other nonclinical and clinical trials were employed to exemplify the quantitative pharmacology approach to improve decision making in drug development. A population PK/PD model for tesofensine was successfully developed and further utilized for simulations to probe its features.
The PK/PD model was successful in describing the plasma concentration-time-effect profiles of tesofensine and its metabolite M1 under consideration of disease progression and the placebo effect. The structural PK model developed with the phase IIa dataset revealed two one-compartment models for tesofensine and M1. The data was very well described and, according to the principle of parsimony, the simple one-compartment model was assessed as sufficient. In general, the PK model presented was almost identical, regarding structure and parameter estimates, as in a recently published model (16). Additionally, the re-estimation with the combined phase IIa and IIb dataset revealed very similar PK characteristics.
The ADAS-Cog measurements were successfully described by an extended Emax model which has already been applied in a pharmacological PK/PD study of tesofensine and M1 (22). The maximum effect achievable with the phase IIa dataset was estimated as a ∼1.5 point ADAS-Cog value reduction from the patients’ baseline under consideration of the placebo effect and disease progression with a high IIV of 98% CV. The efficacy observed of tesofensine in mild AD patients was comparable with the efficacy observed in other compounds approved for AD (36). Simulations revealed that the maximum effect was achieved after ∼2–4 weeks (considering DP and placebo effect) and depending on the dose strength. A comparable fast onset was also observed in other approved AD compounds (37–40). After ∼6–8 weeks, no difference was observable between the active dose groups. This finding is in agreement with the results of the PoC study where no statistically significant difference between the three active doses was observed.
The presented analysis utilized two assumptions based on investigations in mice (17). First, the volume of distribution of the metabolite M1 was fixed to the 0.768-fold of the volume of distribution of tesofensine. As the developed PK model allowed the successful description and prediction of the pharmacokinetics of M1 in the PoC trial, this assumption seems justified. In addition, in mice, the potency of M1 was found to be fivefold lower than the potency of tesofensine for the dopamine reuptake transporter. This ratio was also incorporated for this data analysis in humans. So far, it has not been confirmed whether this ratio investigated in a different pharmacodynamic setting can be translated into humans. However, as this is the only in vivo investigation available so far and the overall contribution of M1 to the total effect might be low, the assumption seems acceptable.
Data analysis showed that effect compartments were necessary to link the PD effect with the plasma concentration–time profiles. Due to the sparse data situation, the rate constant keo was fixed to a small value resulting in a long effect half-life. Simulations showed that concentrations of the EC50 value in the effect compartments concentrations were quickly achieved resulting in the fast onset of the effect. After the end of treatment, a slow decrease of the effect compartment concentrations was observable resulting in a slow disappearance of the ADAS-Cog improvement which was comparable to, e.g., galantamine (38). Nevertheless, the follow-up visit (week 20) of the external PoC prediction was well predicted. With more frequent ADAS-Cog sampling, a more precise keo value might have been estimated resulting in a more precise disappearance of the effect. Considering the potential long-term treatment of AD patients, this aspect might be of less importance for tesofensine.
In the PK/PD model, all components influencing the ADAS-Cog profile were considered: additionally to the drug effect, the disease progression and the placebo effect were investigated. For AD, DP is well known, showing an increase of the ADAS-Cog value of ∼6 points/year (26). As the phase IIa studies were performed only for 4 weeks, DP could not be determined by the model. However, a reasonable DP literature value of 6 points/year increase was implemented in the models to reflect pathophysiological reality. By the successful prediction of the 14-week trial, the implementation in the models seems to be justified. A published placebo model (34) could not be applied for parameter estimation probably due to the sparse data situation. Fixing the parameters to literature values and applying them to the 4-week study data showed satisfactory results. As the published model showed adequate predictability for the 4-week data and was developed based on a large AD cohort, this model was utilized for model development.
The final PK/PD model, based on data of 4 weeks, demonstrated its unbiased descriptive and predictive performance by the successful internal bootstrap, QPC, and VPC evaluations and the adequate prediction of the PK of tesofensine and M1 and the ADAS-Cog values of the 14-week PoC study. Especially the ADAS-Cog values at the end of tesofensine treatment were very well predicted. The median values of the observed and simulated ADAS-Cog values for each of the three dose groups (0.25, 0.5, 1 mg) showed low bias and also statistical evaluation revealed no difference between observed and simulated values. The median placebo effect at the end of treatment compared to baseline was slightly underpredicted (simulated: +0.58) compared to the rather high effect observed (observed: −1.67). Additionally, the statistical evaluation revealed a significant difference between simulation and observation. Nevertheless, it should be noticed that the external prediction of the 4- and 14-week data was satisfying.
The power analysis of 500 simulated PoC trials revealed a comparably high probability of 82.8% and 85.2% for tesofensine to show superiority over placebo after 4 and 14 weeks of treatment, respectively. Despite the slightly different time course (Fig. 5) of the drug effect and the placebo effect, the probability was comparable. As the placebo effect washes out over time, a higher probability of success might be expected. This was confirmed by a trial simulation of tesofensine over 52 weeks. In such a scenario, again a higher probability of 96% was predicted for tesofensine to show superiority over placebo.
The presented PD models are primarily descriptive and developed based on a smaller number of observations collected over 4 weeks in a few patients and finally used to predict larger and longer clinical trials. It would be favorable having more data per patient available and using AD-specific mechanistic models for analyses. Unfortunately, such models are not available yet and a better data base is difficult to achieve due to ethical and practical considerations. Despite the limitations, analyses with PK/PD models from small datasets can be extremely valuable, e.g. for future trial design (41) and/or, under consideration of other factors, for early decision making. In addition, the re-estimation with the large combined dataset showed that the parameter estimates from the 4-week dataset were comparable and indicated the value of the analyses of early clinical data.
Notwithstanding the good predictivity of the model, it should be noted that the relatively small sample size coupled with borrowed parameters for the disease progression and placebo model might smooth over important subpopulations in understanding response of disease to treatment. This should be considered if early decisions are made, as there might be subpopulations that benefit substantially from the treatment but due to the small sample size, they might only be identified after the completion of a larger study.
CONCLUSIONS
In summary, the analysis illustrated the bridging of knowledge of underlying processes in the patient (‘system’) and generation of compound-specific knowledge. Additionally, it is the first description of the dose-concentration-effect relationship of tesofensine and its metabolite M1 in mild AD patients and the results of a large PoC trial. The analysis demonstrated the value of the availability of PK/PD models in early phases of clinical development to complementarily support the further drug development process. As an example of QP-A/MBDD with successful prediction of clinical outcome by the development of PK/PD models (including PD effect, DP and placebo effect) from early clinical phase data, such analyses might be able to contribute in combination with other factors (e.g. medical need, uncertainty in model or model parameters, expected subpopulations) to an early decision-making process. Finally, the presented analysis also identifies the need for a major repository of disease progression data and placebo response data at an individual level and/or the availability of respective models with Bayesian priors for Alzheimer’s disease and across therapeutic areas to support parameter identifiability especially in early drug development.
Acknowledgments
The authors wish to thank Ole Østerberg for his very helpful contribution.
Conflict of Interest
This study was financially supported by Boehringer Ingelheim Pharma GmbH & Co KG, Biberach an der Riss, Germany. CK received research fund from Boehringer Ingelheim Pharma. TL, AS, DT, and HGS are current employees of Boehringer Ingelheim Pharma GmbH & Co KG.
References
- 1.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711–5. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 2.Tufts Center for the Study of Drug Development. Impact Report: Fastest drug developers consistently best peers on key performance metrics, Tufts University, Boston, 2006.
- 3.US Food and Drug Administration. Innovation or Stagnation: Challenge and opportunity on the critical path to new medical products, Rockville, 2004.
- 4.Frantz S. Pipeline problems are increasing the urge to merge. Nat Rev. 2006;5:977–9. doi: 10.1038/nrd2206. [DOI] [PubMed] [Google Scholar]
- 5.Bhattaram VA, Booth BP, Ramchandani RP, et al. Impact of pharmacometrics on drug approval and labeling decisions: a survey of 42 new drug applications. AAPS J. 2005;7:E503–12. doi: 10.1208/aapsj070351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lalonde RL, Kowalski KG, Hutmacher MM, et al. Model-based drug development. Clin Pharmacol Ther. 2007;82:21–32. doi: 10.1038/sj.clpt.6100235. [DOI] [PubMed] [Google Scholar]
- 7.Zhang L, Sinha V, Forgue ST, Callies S, Ni L, Peck R, Allerheiligen SR. Model-based drug development: the road to quantitative pharmacology. J Pharmacokinet Pharmacodyn. 2006;33:369–93. doi: 10.1007/s10928-006-9010-8. [DOI] [PubMed] [Google Scholar]
- 8.Stanski D. Model-based drug development: a critical path opportunity. http://wwwfdagov/oc/initiatives/criticalpath/stanski/stanskihtml. Last accessed 10 Aug 2009.
- 9.Zhang L, Pfister M, Meibohm B. Concepts and challenges in quantitative pharmacology and model-based drug development. AAPS J. 2008;10:552–9. doi: 10.1208/s12248-008-9062-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Association A. Alzheimer's disease facts and figures. Alzheimer's Dementia. 2009;5:234–70. doi: 10.1016/j.jalz.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 11.Burns A, Iliffe S. Alzheimer's disease. Br Med J. 2009;338:467–71. doi: 10.1136/bmj.b158. [DOI] [PubMed] [Google Scholar]
- 12.Astrup A, Madsbad S, Breum L, Jensen TJ, Kroustrup JP, Larsen TM. Effect of tesofensine on bodyweight loss, body composition, and quality of life in obese patients: a randomised, double-blind, placebo-controlled trial. Lancet. 2008;373:1906–13. doi: 10.1016/S0140-6736(08)61525-1. [DOI] [PubMed] [Google Scholar]
- 13.Astrup A, Meier DH, Mikkelsen BO, Villumsen JS, Larsen TM. Weight loss produced by tesofensine in patients with Parkinson's or Alzheimer's disease. Obesity. 2008;16:1363–9. doi: 10.1038/oby.2008.56. [DOI] [PubMed] [Google Scholar]
- 14.Thatte U. NS-2330 NeuroSearch. Curr Opin Investig Drugs. 2001;2:1592–4. [PubMed] [Google Scholar]
- 15.Lehr T, Staab A, Trommeshauser D, Schaefer HG, Kloft C. Semi-mechanistic population pharmacokinetic drug-drug interaction modelling of a long half-life substrate and itraconazole. Clin Pharmacokinet, 2010;49:53–66. [DOI] [PubMed]
- 16.Lehr T, Staab A, Tillmann C, Trommeshauser T, Raschig A, Schaefer HG, Kloft C. Population pharmacokinetic modelling of NS2330 (tesofensine) and its major metabolite in patients with Alzheimer's disease. Br J Clin Pharmacol. 2007;64:36–48. doi: 10.1111/j.1365-2125.2007.02855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lehr T, Staab A, Tillmann C, Nielsen EO, Trommeshauser D, Schaefer HG, Kloft C. Contribution of the active metabolite M1 to the pharmacological activity of tesofensine in vivo: a pharmacokinetic-pharmacodynamic modelling approach. Br J Pharmacol. 2008;153:164–74. doi: 10.1038/sj.bjp.0707539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lehr T, Staab A, Tillmann C, Trommeshauser D, Schaefer HG, Kloft C. A quantitative enterohepatic circulation model: development and evaluation with tesofensine and meloxicam. Clin Pharmacokinet 2009;48:529–542. [DOI] [PubMed]
- 19.Press release: NeuroSearch Continues the Development of Tesofensine (NS2330) and Boehringer Ingelheim Returns All Rights to NeuroSearch, 2009.
- 20.Recommendations guiding physicians in biomedical research involving human subjects. J Am Med Assoc. 1997;277:925–6. [PubMed]
- 21.Note for Guidance on Good Clinical Practice(CPMP/ICH/135/95) Step 5, Consolidated Guideline 1.5.96 including post Step 4 errata, 1996.
- 22.Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer's disease. Am J Psychiatry. 1984;141:1356–64. doi: 10.1176/ajp.141.11.1356. [DOI] [PubMed] [Google Scholar]
- 23.Beal SL, Sheiner LB, Group NP. NONMEM users guides. San Francisco: University of California; 1998. [Google Scholar]
- 24.Akaike H. A new look at the statistical model identification. IEEE Trans Automat Contr. 1974;19:716–23. doi: 10.1109/TAC.1974.1100705. [DOI] [Google Scholar]
- 25.Jonsson EN, Karlsson MO. Xpose—an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed. 1999;58:51–64. doi: 10.1016/S0169-2607(98)00067-4. [DOI] [PubMed] [Google Scholar]
- 26.Chan PLS, Holford NHG. Drug treatment effects on disease progression. Annu Rev Pharmacol Toxicol. 2001;41:625–59. doi: 10.1146/annurev.pharmtox.41.1.625. [DOI] [PubMed] [Google Scholar]
- 27.Holford NHG, Peace KE. Results and validation of a population pharmacodynamic model for cognitive effects in Alzheimer patients treated with tacrine. Proc Natl Acad Sci USA. 1992;89:11471–5. doi: 10.1073/pnas.89.23.11471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ette EI. Stability and performance of a population pharmacokinetic model. J Clin Pharmacol. 1997;37:486–95. doi: 10.1002/j.1552-4604.1997.tb04326.x. [DOI] [PubMed] [Google Scholar]
- 29.Parke J, Holford NHG, Charles BG. A procedure for generating bootstrap samples for the validation of nonlinear mixed-effects population models. Comput Methods Programs Biomed. 1999;59:19–29. doi: 10.1016/S0169-2607(98)00098-4. [DOI] [PubMed] [Google Scholar]
- 30.Lindbom L, Ribbing J, Jonsson EN. Perl-speaks-NONMEM (PsN)—a Perl module for NONMEM-related programming. Comput Methods Programs Biomed. 2004;75:85–94. doi: 10.1016/j.cmpb.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 31.Post TM, Freijer JI, Ploeger BA, Danhof M. Extensions to the visual predictive check to facilitate model performance evaluation. J Pharmacokinet Pharmacodyn. 2008;35:185–202. doi: 10.1007/s10928-007-9081-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilkinson D, Murray J. Galantamine: a randomized, double-blind, dose comparison in patients with Alzheimer's disease. Int J Geriatr Psychiatry. 2001;16:852–7. doi: 10.1002/gps.409. [DOI] [PubMed] [Google Scholar]
- 33.Forette F, Anand R, Gharabawi G. A phase II study in patients with Alzheimer's disease to assess the preliminary efficacy and maximum tolerated dose of rivastigmine (Exelon) Eur J Neurol. 1999;6:423–9. doi: 10.1046/j.1468-1331.1999.640423.x. [DOI] [PubMed] [Google Scholar]
- 34.Holford NHG, Peace KE. Methodologic aspects of a population pharmacodynamic model for cognitive effects in Alzheimer patients treated with tacrine. Proc Natl Acad Sci USA. 1992;89:11466–70. doi: 10.1073/pnas.89.23.11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karlsson MO, Savic RM. Diagnosing model diagnostics. Clin Pharmacol Ther. 2007;82:17–20. doi: 10.1038/sj.clpt.6100241. [DOI] [PubMed] [Google Scholar]
- 36.Jann MW, Shirley KL, Small GW. Clinical pharmacokinetics and pharmacodynamics of cholinesterase inhibitors. Clin Pharmacokinet. 2002;41:719–39. doi: 10.2165/00003088-200241100-00003. [DOI] [PubMed] [Google Scholar]
- 37.Burns A, Rossor M, Hecker J, Gauthier S, Petit H, M ller HJ, Rogers SL, Friedhoff LT. The effects of donepezil in Alzheimer's disease—results from a multinational trial. Dement Geriatr Cogn Disord. 1999;10:237–44. doi: 10.1159/000017126. [DOI] [PubMed] [Google Scholar]
- 38.Corey-Bloom J. Galantamine: a review of its use in alzheimer's disease and vascular dementia. Int J Clin Pract. 2003;57:219–23. [PubMed] [Google Scholar]
- 39.Holford NHG, Peace K. The effect of tacrine and lecithin in Alzheimer's disease a population pharmacodynamic analysis of five clinical trials. Eur J Clin Pharmacol. 1994;47:17–23. doi: 10.1007/BF00193473. [DOI] [PubMed] [Google Scholar]
- 40.Reisberg B, Ferris S, Doody R, St ffler A, bius HJM, Schmitt F. Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med. 2003;348:1333–41. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- 41.Lockwood P, Ewy W, Hermann D, Holford N. Application of clinical trial simulation to compare proof-of-concept study designs for drugs with a slow onset of effect; an example in Alzheimer's disease. Pharm Res. 2006;23:2050–9. doi: 10.1007/s11095-006-9048-8. [DOI] [PubMed] [Google Scholar]