

Abstract
A simple and fast nuclear magnetic resonance method for docking proteins using pseudo-contact shift (PCS) and 1HN/15N chemical shift perturbation is presented. PCS is induced by a paramagnetic lanthanide ion that is attached to a target protein using a lanthanide binding peptide tag anchored at two points. PCS provides long-range (~40 Å) distance and angular restraints between the lanthanide ion and the observed nuclei, while the 1HN/15N chemical shift perturbation data provide loose contact-surface information. The usefulness of this method was demonstrated through the structure determination of the p62 PB1-PB1 complex, which forms a front-to-back 20 kDa homo-oligomer. As p62 PB1 does not intrinsically bind metal ions, the lanthanide binding peptide tag was attached to one subunit of the dimer at two anchoring points. Each monomer was treated as a rigid body and was docked based on the backbone PCS and backbone chemical shift perturbation data. Unlike NOE-based structural determination, this method only requires resonance assignments of the backbone 1HN/15N signals and the PCS data obtained from several sets of two-dimensional 15N-heteronuclear single quantum coherence spectra, thus facilitating rapid structure determination of the protein–protein complex.
Electronic supplementary material
The online version of this article (doi:10.1007/s10858-010-9401-4) contains supplementary material, which is available to authorized users.
Keywords: Lanthanide-binding peptide tag, Two-point anchoring, Paramagnetic NMR, Pseudo-contact shift, Rigid-body docking, Autophagy
Introduction
The structure determination of a protein–protein complex is an important step in revealing the interaction mechanism; however, the application of X-ray crystallography or NMR spectroscopy to this end is not straightforward. In crystallography, complexes are often difficult to crystallize and the possibility of crystal artifacts must always be taken into consideration. In NMR spectroscopy, the protein structure is generally determined on the basis of the short distance restraints derived from nuclear Overhauser effects (NOEs), and it is often difficult to collect a sufficient number of distance restraints for precise structure determination.
Paramagnetic lanthanide ions induce several effects in observed nuclei, such as a pseudo-contact shift (PCS) and residual dipolar coupling (RDC) due to the anisotropy of the magnetic susceptibility tensor (Δχ-tensor). PCS provides long-range distance and angular information between the lanthanide ion and the observed nuclei situated up to ~40 Å apart from the lanthanide ion (Allegrozzi et al. 2000). Accordingly, the paramagnetic lanthanide ion can be used as a powerful probe for solution structure determination, especially for larger molecular weight proteins, multidomain proteins, and protein complexes. For metalloproteins, metal ions such as Ca2+ and Mg2+ can be replaced by the paramagnetic lanthanide ions, and paramagnetic lanthanide probes have been successfully applied to metalloproteins (Bertini et al. 2001, 2004, 2007; Barbieri et al. 2002; Pintacuda et al. 2006, 2007; Allegrozzi et al. 2000).
However, the application of paramagnetic lanthanide probes to non-metalloproteins requires a general method that attaches the lanthanide ions to the protein at a fixed position. Recently, the lanthanide binding peptide tag (LBT), which can be attached to the target protein by two anchoring points, a disulfide bridge and an N-terminal fusion, has been reported (Saio et al. 2009a). Several other lanthanide-binding tags have been also reported, including lanthanide-chelating reagents attached via disulfide bonds (Dvoretsky et al. 2002; Haberz et al. 2006; Pintacuda et al. 2004; Prudêncio et al. 2004; Ikegami et al. 2004; Leonov et al. 2005; Gaponenko et al. 2002, 2004; Vlasie et al. 2007; Keizers et al. 2007, 2008; Su et al. 2008b), and lanthanide-binding peptides attached through N- or C-terminal fusion (Gaponenko et al. 2000; Wöhnert et al. 2003; Martin et al. 2007; Ma and Opella 2000; Zhuang et al. 2008) or a disulfide bond (Su et al. 2006, 2008a). However, single-anchored tags tend to be mobile, while symmetrically designed, double-anchored synthetic chelators often suffer from peak doubling caused by enantiomeric conformers. Furthermore, most of the synthetic tags reported to date are not commercially available. Among these, the lanthanide-attaching method using a two-point anchored peptide tag has a number of advantages in terms of chiral purity, rigidity, and ready availability for protein NMR researchers (Saio et al. 2009a).
We applied this lanthanide tagging method, which introduces the lanthanide ion using two-point anchored peptide tag, to the structure determination of the protein–protein complex of p62 PB1. P62 is a multi-module adaptor protein that plays an important role in autophagy and the NF-κB signaling pathway. In autophagy, p62 interacts with ubiquitinated proteins via its UBA domain, and self-assembles through its PB1 domain to form large protein aggregates (Bjørkøy et al. 2005). The aggregates are then transported to the autophagosome through interaction with LC3 (Noda et al. 2008). The p62 PB1 domain forms a homo-oligomer in a front-to-back manner using its conserved interaction motifs, the OPCA motif and the conserved Lys motif (Saio et al. 2009b). In order to avoid the homo-oligomerization of the p62 PB1-PB1 complex, we introduced site-directed mutations into the interaction motifs and prepared two mutants that only limited 1:1 dimer formation. A monomer structure of the p62 PB1 mutant that abrogates homo-oligomerization has been already solved by NMR spectroscopy (Saio et al. 2009b), but the structure of p62 PB1-PB1 complex has not yet been solved. By attaching the lanthanide binding peptide tag to the one subunit of the dimer, we fixed the lanthanide ion on the protein and obtained inter-subunit structural information from the PCS. Here, we demonstrate a simple and fast method for the structure determination of protein–protein complexes in which the monomer structures are docked based on 1H/15N PCS and 1H/15N chemical shift perturbation data.
Methods
Plasmid construction
Wild-type p62 PB1 forms a homo-oligomer in front-to-back manner, thus making NMR analysis more difficult. On the basis of our previous study (Saio et al. 2009b), we therefore prepared two p62 PB1 mutants, hereafter referred to as DR and KE, that have mutations in the conserved interaction surfaces, the OPCA motif and the conserved Lys motif, respectively, and thus form a 1:1 dimer. For DR, site-directed mutations were introduced into the conserved acidic residues on the OPCA motif to form a D67A/D69R double mutation. To attach the lanthanide ion to DR, a lanthanide binding sequence comprised of 16 amino acids, CYVDTNNDGAYEGDEL (LBT) (Nitz et al. 2003, 2004; Su et al. 2006, 2008a), was attached to the N-terminus of DR, according to our previous report (Saio et al. 2009a), to which is hereafter referred as LBT-DR. LBT-DR was subcloned, together with a GST tag and a tobacco etch virus (TEV) protease cleavage site, into a pGSTV vector derived from the pET-21 plasmid (Novagen, USA). As a binding partner for LBT-DR, we prepared the KE mutant in which the two basic residues on the conserved basic surface, Lys7 and Arg94, were mutated to Glu and Ala, respectively. The KE mutant was subcloned, with a GST tag and HRV3C protease cleavage site, into a pGSPS vector derived from the pET-21 plasmid (Novagen).
P62 PB1 has two cysteine residues, Cys 26 and Cys42. We changed Cys42 on LBT-DR, and Cys26 and Cys42 on KE to serine in order to guarantee proper S–S formation between LBT and the Cys26 on DR.
Sample preparation
Proteins were expressed in E. coli strain BL21 (DE3) cells. For the unlabeled samples, cells were grown in Luria–Bertani media. For the uniformly 15N- or 13C/15N-labeled samples, cells were grown in M9 media containing 15NH4Cl (1 g/l), Celtone-N powder (0.2 g/l) (Cambridge Isotope Laboratories, USA) and unlabeled glucose (10 g/l), or 15NH4Cl (1 g/l), Celtone-CN powder (0.2 g/l) (Cambridge Isotope Laboratories, USA) and [U-13C] glucose (2 g/l), respectively. The uniformly 15N/2H-labeled sample was prepared by culturing cells in 100% 2H2O M9 medium using 15NH4Cl and [U-2H] glucose as the sole nitrogen and carbon sources. Cells were grown at 37°C to A600 of 0.8, and protein expression was induced by the application of Isopropyl β-D-1-thiogalactopyranoside to a final concentration of 0.5 mM for 16 h at 25°C. For the preparation of amino acid selectively 15N-labeled samples, the cells were grown at 37°C in 1 l of minimal media supplemented with 1 g 14NH4Cl and 200 mg of 19 unlabeled amino acids, respectively. Protein expression was induced at A600 of 0.8 by the addition of isopropyl β-D-1-thiogalactopyranoside to a final concentration of 0.5 mM and was cultured for 8 h at 25°C. Fifty mg of specific 15N-labeled amino acid was added to the medium 15 min before induction.
For the preparation of LBT-DR, the disrupted cells were centrifuged and the supernatant was applied to glutathione-Sepharose 4B resin (GE Healthcare, UK) for affinity purification. The GST tag was removed by incubation for 4 h at room temperature with TEV protease. The isolated protein was further purified by gel filtration chromatography on a Superdex 75 column (GE Healthcare). LBT-DR was also expressed in the inclusion body, and retrieved by high-pressure refolding (Schoner et al. 2005; Qoronfleh et al. 2007). Details of the refolding process will be published elsewhere. KE was prepared from the soluble fraction, according to the procedure described previously (Saio et al. 2009b).
After the gel filtration, LBT-DR was incubated with 1 mM 5, 5′-ditiobis(2-nitrobenzoic acid) (DTNB) for 2 h at room temperature, which linked the N-terminal Cys of LBT and the C26 on DR via an intramolecular disulfide bond (Saio et al. 2009a). The oxidized LBT-DR was then mixed with KE, followed by gel filtration chromatography on a Superdex 75 column.
NMR spectroscopy
For NMR measurements, the samples were prepared in 20 mM MES buffer (pH 6.5) with 50 mM NaCl. All NMR experiments were run on Inova 800, 600 or 500 MHz NMR spectrometers (Varian, USA) at 25°C. Spectra were processed using the NMRPipe program (Delaglio et al. 1995) and data analysis was performed with the help of the Olivia program developed in our laboratory (Yokochi et al. http://fermi.pharm.hokudai.ac.jp/olivia/). Intermolecular NOEs were obtained from a 3D 15N-edited NOESY experiment with a mixing time of 200 ms on 15N/2H-labeled LBT-DR complexed with unlabeled KE.
Tensor calculation
Δχ-tensors for LBT-DR were calculated from the PCS values, and the structure of DR was calculated using the Numbat program (Schmitz et al. 2008) by the equation
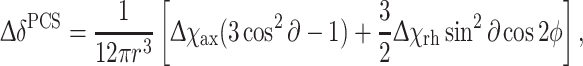 |
1 |
where ΔδPCS is the pseudo contact shift, r, ∂ and φ are polar coordinates of the nucleus with respect to the principal axes of the magnetic susceptibility tensor, and Δχax and Δχrh are the axial and rhombic components, respectively, of the anisotropic magnetic susceptibility tensor. Conformer 1 of the family of NMR structures of DR (PDB code: 2KKC) was used for the tensor fit.
Docking
PCS-based rigid body docking was carried out using the Xplor-NIH program (Schwieters et al. 2003, 2006), equipped with PARA restraints for Xplor-NIH (Banci et al. 2004). The coordinates of LBT-DR (including the metal) were held fixed, whereas KE was treated as a rigid body. As the starting structure of DR, conformer 1 of the family of NMR structures of DR (PDB code: 2KKC) was used, with the exception that Cys42 was replaced with Ser using the PyMOL program (http://www.pymol.org/). The structure of the KE mutant was built based on the structure of DR, with the six surface residues of DR (Lys7, Cys 26, Cys 42, Ala67, Arg69, and Arg94) changed to Glu, Ser, Ser, Asp, Asp, and Ala, respectively, in accordance with the amino acid sequence of KE.
At the start of the docking calculation, the relative orientation and position of KE were randomized to generate 100 starting structures that were located within 100 Å from the DR mutant. The coordinates of DR and the metal, on the other hand, were fixed, with the position of the metal determined by tensor-fits from PCSs observed for LBT-DR. Next, the rigid body docking calculation was performed based on the PCS and contact-surface restraints. During the calculation, the coordinates of DR and the metal were fixed, whereas those of KE were freely rotated and translated. For the PCS restraints, a pseudo atom representing the tensor axis was introduced. The atom representing the origin of the axis was restrained within 0.3 Å of the metal, while the coordinates of the tensor were freely rotated around the origin. The target function was calculated based on three terms: a square-well quadratic term for ambiguous distance restraints (E NOE; Clore and Schwieters 2003), the least square energy penalty for PCS restrains (E PCS; Banci et al. 2004), and a quartic van der Waals repulsion term (E repel). Ambiguous distance restraints were set with upper-limit of 5 Å. During the minimization process, the force constant for E NOE and E PCS were held constant at 0.01 kcal mol−1 Å−2 and 0.8 kcal mol−1 ppm−2, respectively. The force constraint for E repel was geometrically increased over 14 cycles from 0.004 to 1 kcal mol−1 Å−4. The van der Waals radius scale factor was decreased from 1.0 to 0.78. The Xplor-NIH script for the docking calculation is provided as Supporting information.
Results and discussion
Construct design for LBT attachment
A lanthanide binding sequence, CYVDTNNDGAYEGDEL (LBT) (Nitz et al. 2003, 2004; Su et al. 2006, 2008a), was introduced to DR according to the procedure reported previously (Saio et al. 2009a). In the DR structure (PDB code: 2KKC), the distance between the Cα atoms of Ser3 and Cys26 is around 5 Å, which is roughly consistent with the Cα distance between the N- and C-terminal residues of LBT (PDB code: 1TJB, Nitz et al. 2004), thus we fused the LBT sequence to the N-terminus of Ser3 with a three-residue linker sequence (His-Met-Gly). To assess the effects of the LBT attachment, we compared the backbone chemical shifts of the 15N-labeled LBT-DR/unlabelled KE containing 1 eq Lu3+ and 15N-labeled DR/unlabelled KE complexes (Fig. 1). Those residues with chemical shifts significantly affected by the introduction of LBT were found to be located around the anchoring points, while other residues showed only negligible chemical shift changes, indicating that the DR structure was maintained in LBT-DR.
Fig. 1.

Chemical shift differences in the backbone amide groups of DR between 15N-labeled LBT-DR/unlabeled KE containing 1 eq Lu3+ and 15N-labeled DR/unlabeled KE. Δ (ppm) was defined as ((Δ1HN)2 + (Δ15N/5)2)1/2 and plotted against the residue number of DR. The residues for which the signals disappeared upon the introduction of LBT are represented as gray bars. The residues for which the signals shifted (Δ ppm > 0.1) or disappeared are mapped onto the structure of p62 PB1
Resonance assignment and PCS measurement
For the backbone amide resonance assignment of the LBT-DR/KE complex, a standard set of triple resonance NMR spectra was measured using the 13C15N-labeled LBT-DR/unlabelled KE and unlabelled LBT-DR/13C15N-labeled KE complexes, both of which contain 1 eq diamagnetic lanthanide Lu3+. Resonance assignment was accomplished with reference to those of free-state DR (Saio et al. 2009b). The assignment rate of the backbone amide signals of LBT-DR and KE was 91 and 98%, respectively.
1H-15N HSQC spectra of the 15N-labeled LBT-DR/unlabelled KE and unlabelled LBT-DR/15N-labeled KE complexes were recorded in the presence of 1 equivalent of lanthanide ions (Lu3+, Tb3+, Dy3+, Er3+ and Tm3+), where Lu3+ was used as a diamagnetic reference (Fig. 2a, b). Since the 1H and 15N of each amide group are spatially close, the PCS has similar ppm values in both 1H and 15N dimensions (Saio et al. 2009a). Thus, by overlaying the spectra recorded with different lanthanide ions, the signals were found to be located in a straight line. Based on this, the 1H-15N HSQC cross-peaks of the paramagnetic samples could be readily assigned using the assignment of the diamagnetic samples. Most PCS-induced resonances for KE were assigned based on the above procedure. On the other hand, the PCS-based assignment of LBT-DR was not so straightforward due to the complexity of the PCS-induced spectra resulting from the spatial proximity of LBT-DR to the lanthanide ion (Fig. 2a). For reliable PCS assignments for the LBT-DR mutant, amino acid selective 15N-labeled LBT-DR samples were prepared by the selective 15N labeling of Arg, Phe, or Leu. The 1H-15N HSQC spectra of these three samples were recorded in the presence of 1 equivalent of lanthanide ions (Lu3+, Tb3+, Dy3+, Er3+ or Tm3+: Fig. 2c and supporting information Figure S1). In case of the 15N-Arg-labeled LBT-DR/unlabeled KE complex, all resonances of the arginine residues in the LBT-DR mutant were assigned (Fig. 2c). The reliable assignments obtained using the amino acid selective labeling were used for further assignments of the uniformly 15N-labeled LBT-DR mutant samples. Finally, a total of 205 PCS-shifted signals for LBT-DR were assigned (38, 49, 58, and 60 signals from Dy3+-, Tb3+-, Er3+-, and Tm3+-containing samples, respectively), whereas a total of 316 PCS-shifted signals were assigned for KE (75, 79, 82, and 80 signals from Dy3+-, Tb3+-, Er3+-, and Tm3+-containing sample, respectively: supporting information Table S1).
Fig. 2.

1H-15N HSQC spectra of 15N-labeled LBT-DR/unlabeled KE (a), 15N-labeled KE/unlabeled LBT-DR (b), and 15N-Arg-labeled LBT-DR/unlabeled KE (c) in complex with Lu3+ (gray), Tb3+ (orange), Dy3+ (red), Er3+ (green), and Tm3+ (blue). Spectra were obtained using 800 (a and b) and 600 MHz (c) NMR spectrometers at 25°C
Determination of the metal position
For the PCS-based docking calculation, the position of the lanthanide ion relative to the coordinates of DR is required. As the structure of p62 PB1 containing LBT and the metal has not been determined, the metal position was determined based on the backbone amide proton PCS of 15N-labeled LBT-DR/unlabeled KE in complex with Dy3+, Tb3+, Er3+ and Tm3+, using the numbat program (Schmitz et al. 2008); Based on the PCS values from the four lanthanide ions, Δχ-tensors for each lanthanide were simultaneously fitted as well as a conserved metal position due to their isomorphous nature. Subsequently, the Δχ-tensor parameters were further refined for each lanthanide ion (Fig. 3a–c; Table 1), where the position of the metal was allowed to vary within a range of ±0.3 Å. The magnitude of the refined tensor was almost the same as that before refinement: within 10% difference (data not shown). The Δχ-tensor parameters were well defined with the principal axes for the four lanthanides oriented in similar directions and the correlations between the experimental and back-calculated PCS values were good (Fig. 3a, b, and supporting information Figure S2 and S3). It should also be noted that the magnitude of the tensor was comparable to that reported previously (for LBT attached to GB1; Saio et al. 2009a). Thus, we concluded that the position of the lanthanide ion as well as the Δχ-tensor parameters were accurately determined. The Δχ-tensor parameters were used as an input parameter for docking calculations.
Fig. 3.

Input parameter determination for the docking calculation. a and b Comparison of experimental and back-calculated PCSs of backbone amide protons observed for 15N-labeled LBT-DR/unlabeled KE in complex with Tb3+ (a) and Tm3+ (b). The tensors were calculated using the monomer structure of DR. The ideal correlations are indicated. c View of the experimentally determined isosurfaces corresponding to a PCS of ±4.6 and ±1.2 ppm, respectively. Positive and negative PCS values are indicated by blue and red, respectively. d Chemical shift perturbation of the backbone amide groups of KE upon complex formation with LBT-DR at a ration of 1:1. Δ (ppm) was defined as ((Δ1HN)2 + (Δ15N/5)2)1/2 and plotted against the residue number of KE. The residues with Δ (ppm) > 0.5, except for Asp90, are shown in red. Asp90, which showed a chemical shift change larger than 0.5 ppm but was not used as a contact surface restraint as it did not satisfy the inclusion criteria, is shown in cyan. e The mapping of the results with large chemical shift changes on the structure of p62 PB1. Residues are colored according to the color codes used in (d)
Table 1.
Δχ-tensor parameters for lanthanide ions in complex with LBT-DR/KE, determined on the basis of the monomer structure of DR and the PCS values obtained from LBT-DR signals
| Tb3+ | Tm3+ | Dy3+ | Er3+ | |
|---|---|---|---|---|
| Δχaax | 40.8 ± 1.1 | −27.2 ± 1.3 | 28.6 ± 1.5 | −10.4 ± 0.3 |
| Δχarh | 20.7 ± 0.9 | −18.9 ± 1.0 | 21.7 ± 1.0 | −9.1 ± 0.2 |
| αb | 119 | 114 | 124 | 114 |
| βb | 106 | 107 | 108 | 107 |
| γb | 15 | 2 | 37 | 14 |
aΔχax and Δχrh values are in 10−32 [m3] and error estimates were obtained by Monte-Carlo protocol using the 100 partial PCS data sets in which 30% of the input data were randomly deleted. During the Monte–Carlo analysis, the metal position was allowed to vary within a range of ±0.3 Å
bEuler angle rotations in ZXZ convention (degrees)
The contact-surface restraints
The DR and KE mutants were docked based on the PCS and contact-surface restraints. The contact surface restraints were generated from the 1HN/15N backbone chemical shift differences between the free and bound states of KE (Fig. 3d). The chemical shift of backbone 1HN/15N is sensitive to the chemical environment of the two nuclei, which is very useful for the identification of the interaction surface on proteins. Unlike NOE-based analysis, the chemical shift perturbation of the backbone signals can easily be obtained without time-consuming side-chain assignment. Combined with the backbone PCS restraints, backbone chemical shift perturbation mapping ensures fast and reliable structure determination of protein–protein complexes. However, chemical shift perturbations can result either from a direct ligand interaction or from a conformational rearrangement around the observed nuclei, and it is possible that the signals of the residue on the opposite side of the interaction surface induce sizable perturbations, as a consequence of a change in the local structure. Thus, we selected interfacial residues according to the three criteria proposed by Clore and Schwieters (2003): (A) significant chemical shift perturbation is observed upon complex formation, (B) at least one or two atoms of the residue are exposed on the surface of the protein, and (C) the selected residue is involved in a cluster of residues on a contiguous, single binding surface. On binding with LBT-DR, several 1H-15N HSQC signals for KE indicated significant chemical shift perturbations (Fig. 3d, e). Asp67, Glu68, Asp69, Asp71, Val73, Phe75, Ser76, Ser77, and Asp90 all indicated large chemical shift perturbations. The eight residues other than Asp90 fulfilled the above-mentioned criteria, whereas Asp90, which indicated a sizable chemical shift difference and whose atoms are exposed on the surface of the protein, failed to comply with criterion C in that Asp90 is located on the opposite side of the continuous cluster comprised of the other eight residues. Thus, we concluded that Asp67, Glu68, Asp69, Asp71, Val73, Phe75, Ser76, and Ser77 are involved in the binding surface, and we converted the chemical shift perturbation information into the contact-surface restraints. It should be noted that the chemical shift perturbation mapping of LBT-DR, on binding with KE, indicated well-defined contact-surface area (Supporting information Figure S4).
The contact-surface restraints were set up as distance restraints between the atoms of the selected residues of KE and all atoms of DR using the r −6 averaging option (Ubbink et al. 1998; Clore and Schwieters 2003; Díaz-Moreno et al. 2005). For the r −6 averaging option, the distance between selected sets of atoms is averaged according to the equation
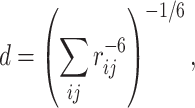 |
2 |
where r ij represents the distance between the atom i in the selected residue of KE and atom j in all residues in DR. Averaging the minus 6th power of the distance emphasizes the smaller distance values, thus a restraint is satisfied when at least one pair of the atoms locate close to each other.
Rigid-body docking
The docking calculation was carried out using the Xplor-NIH program (Schwieters et al. 2003, 2006) with a rigid body minimization protocol (Clore 2000; Tang and Clore 2006). For the calculation, a total of 459 backbone 1H and 15N PCS restraints derived from Tb3+ and Tm3+ as well as contact-surface restraints based on the chemical shift perturbation were used. The contact-surface restraints were added only to restrict the binding surface of KE. Details of the calculation are described in the “Method” section. A total of 100 structures were calculated, each of which started from the randomly arranged KE coordinates around DR. During the calculation, DR and the metal were held in a fixed position, while KE was freely rotated and translated as a rigid body. In the present calculation, we excluded Asp90 from the contact-surface restraints as it failed to satisfy all inclusion criteria (see above). However, test calculations showed that the inclusion of Asp90 in the contact-surface restraints had no effects on the results (data not shown). This may be due to the ambiguity of the distance restraints defined using the r −6 averaging option.
The PCS-isosurface observed with a paramagnetic lanthanide ion is symmetric, thus a PCS data set derived from one lanthanide ion causes four degenerate solutions obtained by rotation around the x, y, and z axes of the principal axis of the Δχ-tensor. In principle, the degeneracy can be overcome by adding a second PCS data set from another lanthanide ion, as the direction of the principal axis of the Δχ-tensor of a second lanthanide ion would be different from that of the first (Pintacuda et al. 2006). In our test calculations using only PCS restraints, the combined use of multiple PCS data sets couldn’t overcome the degeneracy (data not shown), presumably due to the minute difference in the orientation of the principle axes of the Δχ-tensors (Table 1). However, only one of the four degenerate solutions satisfied the contact surface restraints, thus a combination of the contact surface and PCS restraints allows the identification of a proper solution from among the four degenerate solutions.
An overlay of the 10 lowest energy structures and a ribbon model of the lowest energy structure of the DR/KE complex are shown in Fig. 4a and b. These structures have an average backbone rmsd of 0.31 Å. The atomic coordinates and structural restraints for the DR/KE complex have been deposited in the Protein Data Bank, www.pdb.org (PDB code: 2KTR). The conserved acidic residues comprising the OPCA motif form two acidic patches, A1 (Asp67, Glu68, Asp69 and Asp71) and A2 (Glu80), and the basic residues around the conserved Lys motif similarly form B1 (Lys7, Arg22 and Arg94) and B2′ (Arg21) patches (Fig. 4c). As shown in the open-book representation of the complex structure (Fig. 4c), A1 and A2 of KE interacts with B1 and B2′ of DR, respectively. In addition to these electrostatic interactions, a hydrophobic interaction is also formed between Val73 of KE and Ile20 of DR, at the center of the interaction surface. This interaction mechanism of the p62 PB1-PB1 complex supports the previous notion that both electrostatic and hydrophobic interactions are involved in the PB1-PB1 complex formation (Ogura et al. 2009; Hirano et al. 2005; Wilson et al. 2003). It should be noted that the positions of A1, A2, and Val73 of KE are complimentary to those of B1, B2′, and Ile20 of DR, respectively.
Fig. 4.

The docking structure of the DR/KE complex. a Stereo view of the ensemble of the 10 lowest energy structures of the DR/KE complex. The structures of DR are superimposed. The metal position is represented as a yellow sphere. b Ribbon representation of the lowest energy structure. c Electrostatic surface potentials of the interaction surface of the DR/KE complex are shown in an open-book style. The conserved acidic and basic regions are circled in magenta and cyan, respectively. Positive and negative surface potentials are drawn in blue and red, respectively. The structures were drawn using the PyMOL program with APBS tools (http://www.pymol.org/)
Validation of the calculated structure
The docking structure of the DR/KE complex was independently validated using intermolecular NOEs observed in a sample prepared with 15N/2H-labeled LBT-DR and unlabeled KE. Representative examples of the intermolecular NOEs are listed in Table 2 and mapped on the PCS-derived structure of DR/KE (Fig. 5a), and they can be seen to agree with the DR/KE structure described above. Several NOEs were detected involving the Val73 of KE, which is located at the center of the interaction surface (Fig. 4c). Most of the intermolecular NOEs were observed between the atoms located at a distance of 4–6 Å, whereas the distances between Arg22 Hε and Met83 CεH3, Phe23 HN and Met83 CεH3, and Ser24 HN and Met83 CεH3 were around 10 Å. This is explained by the side chain direction of the Met83. In conformer 1 of the family of NMR structures of DR, used for the docking calculation, the methyl group of Met83 points in the opposite direction to the interaction surface, though it is closer to the interaction surface in another conformer.
Table 2.
Representative examples of intermolecular NOEs between LBT-DR and KE
| Proton group in LBT-DR | Proton group on KE |
|---|---|
| Ala8 HN | Val73 Cγ1H3 |
| Ala8 HN | Val73 Cγ2H3 |
| Arg21 HN | Val73 Cγ1H3 |
| Arg21 Hε | Ala74 CβH2 |
| Arg22 HN | Val73 Cγ1H3 |
| Arg22 Hε | Asp67 CβH2 |
| Arg22 Hε | Glu68 HN |
| Arg22 Hε | Met83 CεH3 |
| Phe23 HN | Met83 CεH3 |
| Ser24 HN | Met83 CεH3 |
Fig. 5.

Validation of the docking structure. a Intermolecular NOEs observed for 15N/2H-labeled LBT-DR complexed with unlabeled KE. The atoms between which NOEs were observed are displayed in a stick model and connected by dotted lines. The structure is shown in the direction rotated by 30°, along the longitudinal axis, from that in Fig. 4a and b. b and c Comparison of experimental and back-calculated PCSs of backbone amide protons observed for LBT-DR/KE in complex with Tb3+ (b) and Tm3+ (c). The tensors were calculated using the whole lowest energy structure of the LBT-DR/KE complex
We calculated the Δχ-tensor parameters based on the docking structure and the PCS values obtained both from LBT-DR and KE (Table 3 and supporting information Figure S5). The Δχ-tensor parameters were well defined and, furthermore, comparable to those determined for LBT-DR (Table 1 and supporting information Figure S2). It should be noted that correlations between the experimental and back-calculated PCS values were good, which also supports the compatibility of the docking structure (Fig. 5b, c, and supporting information Figure S6). The magnitudes of Δχ-tensors calculated for the KE part alone were similar to those for LBT-DR and the LBT-DR/KE complex, which suggests stable complex formation between LBT-DR and KE (Supporting information Table S2). We also observed RDCs for LBT-DR/KE containing Tm3+. The magnitudes of the RDC values observed for KE were up to 14 Hz using 800 MHz NMR at 25°C, which were nearly identical to those observed for LBT-DR. This also supports the stability of the LBT-DR/KE complex.
Table 3.
Δχ-tensor parameters for lanthanide ions in complex with LBT-DR/KE, determined on the basis of the docking structure of DR/KE and the PCS values obtained both from LBT-DR and KE signals
| Tb3+ | Tm3+ | Dy3+ | Er3+ | |
|---|---|---|---|---|
| Δχaax | 36.4 ± 1.5 | −23.4 ± 1.3 | 21.9 ± 0.8 | −10.0 ± 0.4 |
| Δχarh | 23.8 ± 0.7 | −20.1 ± 0.5 | 20.4 ± 0.7 | −9.9 ± 0.3 |
| αb | 117 | 108 | 120 | 119 |
| βb | 103 | 105 | 104 | 103 |
| γb | 7 | 178 | 28 | 11 |
aΔχax and Δχrh values are in 10−32 [m3] and error estimates were obtained by Monte–Carlo protocol using the 100 partial PCS data sets in which 30% of the input data were randomly deleted. During the Monte–Carlo analysis, the metal position was allowed to vary within a range of ±0.3 Å
bEuler angle rotations in ZXZ convention (degrees)
Conclusion
A paramagnetic lanthanide ion provides valuable information for NMR protein structural analysis as PCS contains both long-range distance and angular information, which cannot be replaced by other probes, such as spin labels, NOEs, or paramagnetic metal ions (e.g., Cu2+, Mn2+, or Gd3+), that only yield distance dependent information. Bertini et al. (2009) demonstrated accurate solution structure determinations of multi-domain metalloproteins utilizing paramagnetic lanthanide probes. Despite the advantages associated with the use of lanthanide probes, the application of this approach has been limited to certain metal-binding proteins. To apply this method to non metal-binding proteins, a wide variety of lanthanide ion anchoring tags have been developed, including lanthanide binding peptide tags and synthetic chelating reagents (Su and Otting 2009). However, lanthanide tagging has not yet been applied to protein structural analysis apart from a limited number of studies (Gaponenko et al. 2002, 2004; Zhuang et al. 2008; Xu et al. 2009). Recently, Feng et al. (2007) reported the structural analysis on the homo-oligomeric domain, Par-3 NTD, and utilized the lanthanide tagging method to obtain structural information for the complex. However, the high mobility of the lanthanide ion prevented the quantitative analysis of PCS. In general, the flexibility of the lanthanide binding tag prevents the wider application of lanthanide probes.
Recently, we reported a two-point anchoring method for a lanthanide binding peptide tag that fulfills both the need for ready availability for protein NMR researchers and higher rigidity (Saio et al. 2009a). Using this lanthanide tagging method, we here determined the protein–protein complex structure of the p62 PB1 homo-dimer, based on the distance and angular restraints from backbone 1H/15N PCSs and the contact-surface restraints derived from backbone chemical shift perturbations. These two kinds of restraints can easily be obtained by the measurement of 1H-15N HSQC spectra, as long as the backbone assignment of the target is available. On the other hand, NOE-based methods require experiments, far less sensitive than 1H-15N HSQC spectra, for side chain assignment and NOE collection, which is time-consuming and often difficult, especially for larger molecular weight targets. Using the two-point anchored peptide tag, which is readily available and holds a lanthanide ion in a fixed position, the application of the paramagnetic lanthanide probe will become more widely used.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Allegrozzi M, Bertini I, Janik MBL, Lee YM, Liu G, Luchinat C. Lanthanide-induced pseudocontact shifts for solution structure refinements of macromolecules in shells up to 40 Å from the metal ion. J Am Chem Soc. 2000;122:4154–4161. doi: 10.1021/ja993691b. [DOI] [Google Scholar]
- Banci L, Bertini I, Cavallaro G, Giachetti A, Luchinat C, Parigi G. Paramagnetism-based restraints for Xplor-NIH. J Biomol NMR. 2004;28:249–261. doi: 10.1023/B:JNMR.0000013703.30623.f7. [DOI] [PubMed] [Google Scholar]
- Barbieri R, Bertini I, Cavallaro G, Lee YM, Luchinat C, Rosato A. Paramagnetically induced residual dipolar couplings for solution structure determination of lanthanide binding proteins. J Am Chem Soc. 2002;124:5581–5587. doi: 10.1021/ja025528d. [DOI] [PubMed] [Google Scholar]
- Bertini I, Janik MB, Liu G, Luchinat C, Rosato A. Solution structure calculations through self-orientation in a magnetic field of a cerium(III) substituted calcium-binding protein. J Magn Reson. 2001;148:23–30. doi: 10.1006/jmre.2000.2218. [DOI] [PubMed] [Google Scholar]
- Bertini I, Del Bianco C, Gelis I, Katsaros N, Luchinat C, Parigi G, Peana M, Provenzani A, Zoroddu MA. Experimentally exploring the conformational space sampled by domain reorientation in calmodulin. Proc Natl Acad Sci USA. 2004;101:6841–6846. doi: 10.1073/pnas.0308641101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini I, Gupta YK, Luchinat C, Parigi G, Peana M, Sgheri L, Yuan J. Paramagnetism-based NMR restraints provide maximum allowed probabilities for the different conformations of partially independent protein domains. J Am Chem Soc. 2007;129:12786–12794. doi: 10.1021/ja0726613. [DOI] [PubMed] [Google Scholar]
- Bertini I, Kursula P, Luchinat C, Parigi G, Vahokoski J, Wilmanns M, Yuan J. Accurate solution structures of proteins from X-ray data and a minimal set of NMR data: calmodulin-peptide complexes as examples. J Am Chem Soc. 2009;131:5134–5144. doi: 10.1021/ja8080764. [DOI] [PubMed] [Google Scholar]
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore GM. Accurate and rapid docking of protein-protein complexes on the basis of intermolecular nuclear overhauser enhancement data and dipolar couplings by rigid body minimization. Proc Natl Acad Sci USA. 2000;97:9021–9025. doi: 10.1073/pnas.97.16.9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore GM, Schwieters CD. Docking of protein-protein complexes on the basis of highly ambiguous intermolecular distance restraints derived from 1H/15 N chemical shift mapping and backbone 15 N–1H residual dipolar couplings using conjoined rigid body/torsion angle dynamics. J Am Chem Soc. 2003;125:2902–2912. doi: 10.1021/ja028893d. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister G, Zhu W, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Díaz-Moreno I, Díaz-Quintana A, De la Rosa MA, Ubbink M. Structure of the complex between plastocyanin and cytochrome f from the cyanobacterium Nostoc sp. PCC 7119 as determined by paramagnetic NMR. The balance between electrostatic and hydrophobic interactions within the transient complex determines the relative orientation of the two proteins. J Biol Chem. 2005;280:18908–18915. doi: 10.1074/jbc.M413298200. [DOI] [PubMed] [Google Scholar]
- Dvoretsky A, Gaponenko V, Rosevear PR. Derivation of structural restraints using a thiol-reactive chelator. FEBS Lett. 2002;528:189–192. doi: 10.1016/S0014-5793(02)03297-0. [DOI] [PubMed] [Google Scholar]
- Feng W, Wu H, Chan LN, Zhang M. The Par-3 NTD adopts a PB1-like structure required for Par-3 oligomerization and membrane localization. EMBO J. 2007;26:2786–2796. doi: 10.1038/sj.emboj.7601702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaponenko V, Dvoretsky A, Walsby C, Hoffman BM, Rosevear PR. Calculation of z-coordinates and orientational restraints using a metal binding tag. Biochemistry. 2000;39:15217–15224. doi: 10.1021/bi001381w. [DOI] [PubMed] [Google Scholar]
- Gaponenko V, Altieri AS, Li J, Byrd RA. Breaking symmetry in the structure determination of (large) symmetric protein dimers. J Biomol NMR. 2002;24:143–148. doi: 10.1023/A:1020948529076. [DOI] [PubMed] [Google Scholar]
- Gaponenko V, Sarma SP, Altieri AS, Horita DA, Li J, Byrd RA. Improving the accuracy of NMR structures of large proteins using pseudocontact shifts as long-range restraints. J Biomol NMR. 2004;28:205–212. doi: 10.1023/B:JNMR.0000013706.09264.36. [DOI] [PubMed] [Google Scholar]
- Haberz P, Rodriguez-Castañeda F, Junker J, Becker S, Leonov A, Griesinger C. Two new chiral EDTA-based metal chelates for weak alignment of proteins in solution. Org Lett. 2006;8:1275–1278. doi: 10.1021/ol053049o. [DOI] [PubMed] [Google Scholar]
- Hirano Y, Yoshinaga S, Takeya R, Suzuki NN, Horiuchi M, Kohjima M, Sumimoto H, Inagaki F. Structure of a cell polarity regulator, a complex between atypical PKC and Par6 PB1 domains. J Biol Chem. 2005;280:9653–9661. doi: 10.1074/jbc.M409823200. [DOI] [PubMed] [Google Scholar]
- Ikegami T, Verdier L, Sakhaii P, Grimme S, Pescatore B, Saxena K, Fiebig KM, Griesinger C. Novel techniques for weak alignment of proteins in solution using chemical tags coordinating lanthanide ions. J Biomol NMR. 2004;29:339–349. doi: 10.1023/B:JNMR.0000032611.72827.de. [DOI] [PubMed] [Google Scholar]
- Keizers PH, Desreux JF, Overhand M, Ubbink M. Increased paramagnetic effect of a lanthanide protein probe by two-point attachment. J Am Chem Soc. 2007;129:9292–9293. doi: 10.1021/ja0725201. [DOI] [PubMed] [Google Scholar]
- Keizers PH, Saragliadis A, Hiruma Y, Overhand M, Ubbink M. Design, synthesis, and evaluation of a lanthanide chelating protein probe: CLaNP-5 yields predictable paramagnetic effects independent of environment. J Am Chem Soc. 2008;130:14802–14812. doi: 10.1021/ja8054832. [DOI] [PubMed] [Google Scholar]
- Leonov A, Voigt B, Rodriguez-Castañeda F, Sakhaii P, Griesinger C. Convenient synthesis of multifunctional EDTA-based chiral metal chelates substituted with an S-mesylcysteine. Chem Eur J. 2005;11:3342–3348. doi: 10.1002/chem.200400907. [DOI] [PubMed] [Google Scholar]
- Ma C, Opella SJ. Lanthanide ions bind specifically to an added “EF-hand” and orient a membrane protein in micelles for solution NMR spectroscopy. J Magn Reson. 2000;146:381–384. doi: 10.1006/jmre.2000.2172. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Hähnke MJ, Nitz M, Wöhnert J, Silvaggi NR, Allen KN, Schwalbe H, Imperiali B. Double-lanthanide-binding tags: design, photophysical properties, and NMR applications. J Am Chem Soc. 2007;129:7106–7113. doi: 10.1021/ja070480v. [DOI] [PubMed] [Google Scholar]
- Nitz M, Franz KJ, Maglathlin RL, Imperiali B. A powerful combinatorial screen to identify high-affinity terbium(III)-binding peptides. Chembiochem. 2003;4:272–276. doi: 10.1002/cbic.200390047. [DOI] [PubMed] [Google Scholar]
- Nitz M, Sherawat M, Franz KJ, Peisach E, Allen KN, Imperiali B. Structural origin of the high affinity of a chemically evolved lanthanide-binding peptide. Angew Chem Int Ed Engl. 2004;12:3682–3685. doi: 10.1002/anie.200460028. [DOI] [PubMed] [Google Scholar]
- Noda NN, Kumeta H, Nakatogawa H, Satoo K, Adachi W, Ishii J, Fujioka Y, Ohsumi Y, Inagaki F. Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells. 2008;13:1211–1218. doi: 10.1111/j.1365-2443.2008.01238.x. [DOI] [PubMed] [Google Scholar]
- Ogura K, Tandai T, Yoshinaga S, Kobashigawa Y, Kumeta H, Ito T, Sumimoto H, Inagaki F. NMR structure of the heterodimer of Bem1 and Cdc24 PB1 domains from Saccharomyces cerevisiae. J Biochem. 2009;146:317–325. doi: 10.1093/jb/mvp075. [DOI] [PubMed] [Google Scholar]
- Pintacuda G, Moshref A, Leonchiks A, Sharipo A, Otting G. Site-specific labelling with a metal chelator for protein-structure refinement. J Biomol NMR. 2004;29:351–361. doi: 10.1023/B:JNMR.0000032610.17058.fe. [DOI] [PubMed] [Google Scholar]
- Pintacuda G, Park AY, Keniry MA, Dixon NE, Otting G. Lanthanide labeling offers fast NMR approach to 3D structure determinations of protein–protein complexes. J Am Chem Soc. 2006;128:3696–3702. doi: 10.1021/ja057008z. [DOI] [PubMed] [Google Scholar]
- Pintacuda G, John M, Su XC, Otting G. NMR structure of protein-ligand complexes by lanthanide labeling. Acc Chem Res. 2007;40:206–212. doi: 10.1021/ar050087z. [DOI] [PubMed] [Google Scholar]
- Prudêncio M, Rohovec J, Peters JA, Tocheva E, Boulanger MJ, Murphy ME, Hupkes HJ, Kosters W, Impagliazzo A, Ubbink M. A caged lanthanide complex as a paramagnetic shift agent for protein NMR. Chemistry. 2004;10:3252–3260. doi: 10.1002/chem.200306019. [DOI] [PubMed] [Google Scholar]
- Qoronfleh MW, Hesterberg LK, Seefeldt MB. Confronting high-throughput protein refolding using high pressure and solution screens. Protein Expr Purif. 2007;55:209–224. doi: 10.1016/j.pep.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Saio T, Ogura K, Yokochi M, Kobashigawa Y, Inagaki F. Two-point anchoring of a lanthanide-binding peptide to a target protein enhances the paramagnetic anisotropic effect. J Biomol NMR. 2009;44:157–166. doi: 10.1007/s10858-009-9325-z. [DOI] [PubMed] [Google Scholar]
- Saio T, Yokochi M, Inagaki F (2009b) The NMR structure of the p62 PB1 domain, a key protein in autophagy and NF-κB signaling pathway. J Biomol NMR (in press) [DOI] [PubMed]
- Schmitz C, Stanton-Cook MJ, Su XC, Otting G, Huber T. Numbat: an interactive software tool for fitting Deltachi-tensors to molecular coordinates using pseudocontact shifts. J Biomol NMR. 2008;41:179–189. doi: 10.1007/s10858-008-9249-z. [DOI] [PubMed] [Google Scholar]
- Schoner BE, Bramlett KS, Guo H, Burris TP. Reconstitution of functional nuclear receptor proteins using high pressure refolding. Mol Genet Metab. 2005;85:318–322. doi: 10.1016/j.ymgme.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/S1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- Schwieters CD, Kuszewski JJ, Clore GM. Using Xplor–NIH for NMR molecular structure determination. Progr Nucl Magn Resn Spectr. 2006;48:47–62. doi: 10.1016/j.pnmrs.2005.10.001. [DOI] [Google Scholar]
- Su XC, Otting G (2009) Paramagnetic labelling of proteins and oligonucleotides for NMR. J Biomol NMR. (ahead of print) [DOI] [PubMed]
- Su XC, Huber T, Dixon NE, Otting G. Site-specific labelling of proteins with a rigid lanthanide-binding tag. ChemBioChem. 2006;7:1599–1604. doi: 10.1002/cbic.200600142. [DOI] [PubMed] [Google Scholar]
- Su XC, McAndrew K, Huber T, Otting G. Lanthanide-binding peptides for NMR measurements of residual dipolar couplings and paramagnetic effects from multiple angles. J Am Chem Soc. 2008;130:1681–1687. doi: 10.1021/ja076564l. [DOI] [PubMed] [Google Scholar]
- Su XC, Man B, Beeren S, Liang H, Simonsen S, Schmitz C, Huber T, Messerle BA, Otting G. A dipicolinic acid tag for rigid lanthanide tagging of proteins and paramagnetic NMR spectroscopy. J Am Chem Soc. 2008;130:10486–10487. doi: 10.1021/ja803741f. [DOI] [PubMed] [Google Scholar]
- Tang C, Clore GM. A simple and reliable approach to docking protein–protein complexes from very sparse NOE-derived intermolecular distance restraints. J Biomol NMR. 2006;36:37–44. doi: 10.1007/s10858-006-9065-2. [DOI] [PubMed] [Google Scholar]
- Ubbink M, Ejdebäck M, Karlsson BG, Bendall DS. The structure of the complex of plastocyanin and cytochrome f, determined by paramagnetic NMR and restrained rigid-body molecular dynamics. Structure. 1998;6:323–335. doi: 10.1016/S0969-2126(98)00035-5. [DOI] [PubMed] [Google Scholar]
- Vlasie MD, Comuzzi C, van den Nieuwendijk AM, Prudêncio M, Overhand M, Ubbink M. Long-range-distance NMR effects in a protein labeled with a lanthanide-DOTA chelate. Chem Eur J. 2007;13:1715–1723. doi: 10.1002/chem.200600916. [DOI] [PubMed] [Google Scholar]
- Wilson MI, Gill DJ, Perisic O, Quinn MT, Williams RL. PB1 domain-mediated heterodimerization in NADPH oxidase and signaling complexes of atypical protein kinase C with Par6 and p62. Mol Cell. 2003;12:39–50. doi: 10.1016/S1097-2765(03)00246-6. [DOI] [PubMed] [Google Scholar]
- Wöhnert J, Franz KJ, Nitz M, Imperiali B, Schwalbe H. Protein alignment by a coexpressed lanthanide-binding tag for the measurement of residual dipolar couplings. J Am Chem Soc. 2003;125:13338–13339. doi: 10.1021/ja036022d. [DOI] [PubMed] [Google Scholar]
- Xu X, Keizers PH, Reinle W, Hannemann F, Bernhardt R, Ubbink M. Intermolecular dynamics studied by paramagnetic tagging. J Biomol NMR. 2009;43:247–254. doi: 10.1007/s10858-009-9308-0. [DOI] [PubMed] [Google Scholar]
- Zhuang T, Lee HS, Imperiali B, Prestegard JH. Structure determination of a galectin-3-carbohydrate complex using paramagnetism-based NMR constraints. Protein Sci. 2008;17:1220–1231. doi: 10.1110/ps.034561.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.