

Abstract
Here we describe a generic, reverse transcriptase-loop-mediated isothermal amplification (RT-LAMP) assay, for the identification of Leishmania species from clinical samples. LAMP is an isothermal reaction recently developed as a point-of-care diagnostic tool. Primers were designed in the conserved region of the 18S ribosomal RNA (rRNA) gene; amplification was visualized by the pre-amplification addition of fluorescent detection reagent (FDR) and a simple UV lamp. By using a reverse-transcriptase step, the system detected infections between 10 and 100 parasites per mL. The assay was tested on a range of nucleic acid extracts from Leishmania species, visceral leishmaniasis (VL) patients from Sudan, and cutaneous leishmaniasis (CL) patients from Suriname. The sensitivity of RT-LAMP from the blood of VL patients was 83% (N = 30) compared with microscopy of bone-marrow and lymph-node aspirates; for CL patients the observed sensitivity was 98% (N = 43). The potential to use LAMP as a diagnostic tool for leishmaniasis is discussed.
Introduction
Leishmaniasis is caused by a single-celled, eukaryotic parasite transmitted by the bite of an infected sand fly. In humans, a range of clinical symptoms can be observed from self-healing skin lesions in cutaneous leishmaniasis (CL), lesions of the mucosal tracts in mucosal leishmaniasis (MCL) to the fatal form, visceral leishmaniasis (VL) also known as kala-azar.1 In addition, 5–60% (depending on geographical area) of treated VL patients develop the condition known as post-kala azar dermal leishmaniasis (PKDL), which becomes a reservoir for ongoing transmission of VL.2,3 This clinical spectrum of diseases is reflected in the number of Leishmania species that are able to infect humans; 20 species are known to cause disease, three can cause fatal VL, Leishmania donovani, Leishmania infantum, and Leishmania chagasi, the remainder can cause CL and/or MCL. Around 350 million people are at risk of infection and the annual incidence is estimated at 500,000 cases of VL and 1 to 1.5 million cases of CL.4
Accurate and fast diagnosis of Leishmania infection is absolutely essential to initiate correct and prompt treatment. Currently, the gold standard diagnostic is conventional microscopy of bone marrow, lymph-node or spleen aspirates for VL and a combination of microscopy and culturing of parasites from biopsy material for CL.5,6 Microscopy is widely used and has been used successfully in remote areas; however, this method is insensitive to low numbers of parasites, which is frequently observed in VL patients. In addition, aspirates for microscopy must be taken from the lymph nodes, bone marrow, or the spleen; spleenic aspirates are dangerous for the patient and the diagnostic sensitivity in other aspirates is lower than that of the spleen.7 For CL, microscopy of skin smears and culturing parasites are associated with low sensitivity.6,8
For VL a range of serological diagnostic techniques have been developed that are both sensitive and cost effective; the direct agglutination test (DAT) and the rK39 tests are extensively used in Asia and Africa9; rK39 is also the diagnostic of choice for kala-azar elimination from South-East Asia.10,11 However, antibodies detected by these tests can be found in the blood of infected patients for many months after the parasites have disappeared, making detection of active infection inaccurate; moreover, serological tests may not be used on immuno-compromised individuals. In addition, serological tests are not reliable for CL patients because of low or absent antibody titers in blood.12 Recently, molecular tests have become popular choices as diagnostics tools because of increased sensitivity and accuracy such as polymerase chain reaction (PCR)13 and quantification techniques such as real-time–PCR.14,15 While these techniques have increased sensitivity, can detect active infections, and can be used for CL patients; they also require sufficient technical skill, equipment, continual electricity, and are considerably more expensive than the serological tests.16 Therefore, many of these tests are not suitable for diagnosis in endemic regions, except in well-equipped laboratories. Isothermal amplification techniques such as nucleic acid sequence-based amplification (NASBA) for leishmaniasis have also been developed17; however, the post-amplification analysis with oligo-chromatography makes this technique time-consuming and contamination prone.
A recent advance in diagnostics has been the development of loop-mediated isothermal reaction (LAMP).18 This nucleic acid amplification technique uses only one enzyme (Bst DNA polymerase) and is able to amplify large amounts of DNA within 30–60 minutes by the intricate design of primers and auto-strand displacement DNA synthesis.18 The reaction has several advantages; first, the reaction takes place between 60 and 65°C, and can therefore be completed without the use of a thermocycler. Second, the specificity of the reaction is high because of the design of six primers. Third, the product can be visualized directly using simple detection methods. Recently, the stability of LAMP reagents was studied and showed to be unaffected by ambient temperatures up to 37°C, potentially negating a cold-chain.19 Therefore, LAMP has emerged as a powerful tool for point-of-care diagnostics and has been implemented for other protozoan parasitic diseases such as the Plasmodium20,21 and the Trypanosoma,22,23 with high efficacy. Recently, several studies have included a reverse transcriptase step to specifically amplify RNA; this has mainly been used for amplification of RNA viruses such as human immunodeficiency virus (HIV)24 and avian flu viruses,25 moreover, by amplification of RNA it is possible to increase the sensitivity of the assay.
The objective of this study was to develop a quick, sensitive, and specific test that could be used with minimal equipment and no post-amplification handling. The aim was to design a pan-Leishmania assay that could be used for the diagnosis of both VL and CL and to test the assay on a range of clinical samples.
Material and Methods
Primer design.
Primers were designed using PrimerExplorer version 4.0 software (http://primerexplorer.jp/elamp4.0.0). The 18S ribosomal RNA (rRNA) gene was chosen for amplification because of the strong conservation across the Leishmania species for the design of a pan-Leishmania test. An alignment was made with the following species: Leishmania braziliensis braziliensis (GenBank accession nos.: M80292 & DQ182537), Leishmania mexicana amazonensis (M80293 & DQ182536), Leishmania tropica (M80294), Leishmania adleri (M80291), Leishmania donovani (M80295, GQ332356), Leishmania guyanensis (DQ182541), and Leishmania lainsoni (DQ182542). Eight novel primer sets were designed with the software; primer sequences were subject to a basic local alignment search tool (BLAST) analysis against human DNA representing a possible source of mammalian contamination and against other organisms included in the differential diagnosis of leishmaniasis, such as Schistosoma, Mycobacterium sp., Plasmodium, and Brucella sp.
Reference DNA.
Each novel primer set (N = 8) was tested with nucleic acid extract from L. donovani isolates (105 parasites per mL; strain MHOM/SD/68/IS), and a temperature gradient from 58 to 66°C. The primer set with the strongest amplification (Figure 1) was subsequently validated using reference DNA from 12 species of Leishmania, which are capable of infecting humans (L. donovani, L. infantum, L. chagasi, L. guyanensis, L. major, L. tropica, L. mexicana, L. braziliensis, L. amazoniensis, L. aethipoia, L. lainsoni, and L. peruviana) at concentration 105 parasites per mL. Three isolates of each species were tested to ensure all species and strains could be amplified by this pan-Leishmania test. Ten-fold dilutions of L. donovani (MHOM/SD/68/IS) and L. guyanensis (MHOM/BZ/75/M4147) were used to examine sensitivity; serial dilutions were made from cultures (concentration 107 parasite per mL, counted using a Bürker counting chamber, VWR International, USA) and DNA/RNA extracted with the Boom method26; a serial dilution in TE buffer was made from this stock solution. Samples of Plasmodium falciparum, Escherichia coli, Staphylococcus aureus, Mycobacterium tuberculosis, M. leprae, and other atypical Mycobacterium infections were tested to ensure differential diagnosis could be made with these primers. Trypanosoma brucei and Trypanosoma cruzi were also tested because of the close genetic relationship of Leishmania with the Trypanosoma.
Figure 1.
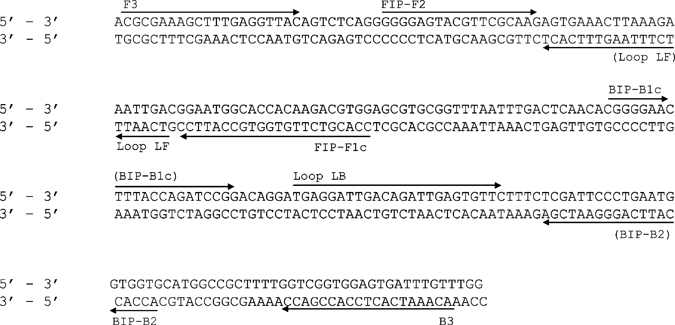
Schematic representation of the nucleic acid sequence of the 18S ribosomal RNA (rRNA) (accession no. GQ332356). Primers FIP and BIP are a combination of F2 & F1c and B2 and B1c, respectively (see Table 1 for full primer sequences).
Clinical samples.
Visceral leishmaniasis patients.
Blood samples (N = 30) from patients with VL caused by L. donovani from Barbar El-Fugara village, Gedarif State, Eastern Sudan were tested by RT-LAMP. Patients were confirmed as parasitologically positive by microscopic examination of bone marrow or lymph-node aspirates. Two hundred microliters (200 μL) EDTA blood was mixed with 1.2 mL L6 lysis-buffer (50 mM Tris HCl, 5 M GuSCN, 20 mM EDTA, 0.1% Triton- X-100); samples were transported at room temperature to the University of Khartoum in Sudan where DNA/RNA was extracted with the Boom method.26 Blood (N = 50) was collected from apparently healthy children, without clinical symptoms or suspicion of past and present VL or any other current infection/disease, also from Gedarif state, Sudan. Ethical clearance was obtained from the Medical Faculty of the University of Khartoum and the National Ethical Committee at the Federal Ministry of Health, Sudan. DNA/RNA extractions were transported to Netherlands on dry ice.
Cutaneous leishmaniasis patients.
Skin biopsies from parasitologically confirmed CL patients were collected from 17 patients in Suriname before treatment and during follow-up; follow-up biopsies were taken weekly, ranging from 1 to 4 weeks (total samples N = 43). Patients were diagnosed by direct microscopy of skin smears and treated with pentamidine over a 3- to 4-week period depending on lesion size and the physician's judgment, as is common practice in Suriname. A single skin biopsy (2 mm in diameter) was collected under local anesthesia (xylocaine) with a sterile disposable skin biopsy puncher from the active edge of the lesion according to World Health Organization (WHO) recommendations (WHO, 1990). Skin biopsies from lesions of CL patients were mixed with 950 μL L6 lysis buffer and stored at −70°C until further processing. The DNA/RNA was extracted with the Boom method26 at the department of Dermatology, Academic Hospital Paramaribo, Suriname. Work performed in Suriname was reviewed and approved by the Medical Ethical Committee of the Academic Medical Center, Amsterdam, The Netherlands (MEC 03/228). The DNA/RNA extractions were transported to The Netherlands on dry ice.
Quantitative reverse-transcriptase PCR (qRT-PCR).
All samples were analyzed with quantitative reverse-transcriptase PCR (qRT-PCR)14 (Table 1). This method is able to amplify all Leishmania species and quantifies parasitemia in patient samples. The 2.5 µL RNA/DNA extraction material was added to 22.5 µL amplification mix (I-script, Bio-Rad, USA) and amplified in the IQ5-cycler (Bio-Rad) using the following program: 10 minutes at 50°C, 5 minutes at 95°C followed by 45 cycles of 30 sec at 95°C and 45 sec at 60°C. In each experiment, 10-fold serial dilutions of Leishmania parasite extracts (MHOM/SD/68/IS) were included for quantification.
Table 1.
Primers used for the reverse transcriptase-loop-mediated isothermal amplification (RT-LAMP) assay, sequencing of the LAMP product;* are qRT-PCR primers taken from Van der Meide (2008)14
| Primer name | Purpose | Sequence 5' to 3' |
|---|---|---|
| FIP | LAMP assay | CCA CGT CTT GTG GTG CCA TTC C-GG GGA GTA CGT TCG CAA G |
| BIP | LAMP assay | CGG GGA ACT TTA CCA GAT CCG G-AC CAC CAT TCA GGG AAT CGA |
| F3 | LAMP assay | CGC GAA AGC TTT GAG GTT AC |
| B3 | LAMP assay | ACA AAT CAC TCC ACC GAC C |
| LF | LAMP assay | GTC AAT TTC TTT AAG TTT CAC T |
| LB | LAMP assay | TGA GGA TTG ACA GAT GGA GTG TTC |
| F2 | Sequencing LAMP product | GGG GAG TAC GTT CGC AAG |
| B2 | Sequencing LAMP product | ACCACCATTCAGGGAATCGA |
| qRT-PCR forward primer* | qRT-PCR | CCA AAG TGT GGA GAT CGA AG |
| qRT-PCR reverse primer* | qRT-PCR | GGC CGG TAA AGG CCG AAT AG |
| qRT-PCR probe* | qRT-PCR | 6FAM ACC ATT GTA GTC CAC ACT GC NFQ MGB |
The 5'-fluorescently labeled probe [6-carboxyfluorescein (FAM)] were conjugated to a non-fluorescent quencher (NFQ) and a minor groove binder probe group (MGB) at the 3' end.
Denotes the space between F1c & F2 and B1c & B2.
RT-LAMP reaction conditions.
The RT-LAMP reaction contained the following components: 5 pmol of primers F3 and B3, 40 pmol of primers forward inner primer (FIP) and backward inner primer (BIP), 20 pmol loop primers loop forward (LF) and loop backward (LB) (Figure 1), 12.5 µL Buffer mix (Eiken, Japan), 1 U LAMP enzyme (Eiken), and 1 U of Fluorescent detection reagents (FDR) (Eiken), 1 U of reverse-transcriptase (I-script, Biorad), and 2 µL of extracted nucleic acid in a total volume of 25 µL. The reaction was incubated at 50°C for 10 minutes to activate the reverse-transcriptase enzyme; the reaction was then held at 64°C for 60 minutes for amplification and finally held at 80°C for 2 minutes to inactivate the reaction. Negative controls were included in every run to ensure no contamination was present. All RT-LAMP reactions were carried out at KIT-Biomedical Research in the Netherlands. The full function of the different primers is described and animated at (http://loopamp.eiken.co.jp/e/lamp/); primers LF and LB are added to increase the number of loops in the reaction thereby increasing the speed of reaction.
Reaction times were reduced experimentally in steps of 5 minutes: 60, 55, 50, 45, 40, 35, and 30. Reactions were carried out in a thermocycler (Bio-Rad); several were also carried out in a water bath to ensure amplification could be generated with basic equipment. Successful amplification was detected by the addition of 1 U FDR (Eiken) before the reaction took place, to avoid post-amplification handling. This was visualized with a UV lamp post-reaction.
Specificity of the LAMP reaction is achieved by the intricate design of three sets of primers. This was verified with melt curves of the LAMP products from the following species: L. donovani, L. guyanensis, L. major, L. tropica, L. mexicana, L. braziliensis, L. amazoniensis, L. aethipoia, and L. infantum (105 parasites per mL). SYBR green was added to the assay (pre-amplification) at 1.25 µL per 25 µL reaction; FDR reagent was excluded from these reactions because of interference. The curves were completed, after LAMP amplification, on the FAM channel using 0.5°C steps and a hold of 30 sec at each step from 68 to 95°C (Bio-Rad IQ5).
Additionally, specificity was confirmed by sequencing the LAMP product of species L. donovani (MHOM/SD/68/IS) and L. guyanensis (MHOM/BZ/75/M4147). One microliter (1 µL) of LAMP product was included in a PCR reaction with primers F2 and B2 (1 mM) (Table 1) dNTPs (0.4 mM), Hot start Taq (1U - Qiagen), and Buffer (10X) in final volume 25 µL. The reaction was run as follows: 10 minutes at 95°C, 30 cycles of 95°C 30 sec, 58°C for 30 sec, 72°C 45 sec, followed by 10 minutes extension at 72°C. The PCR product was run on an agarose gel (1%), purified (Base Clear) and sequenced directly (Macrogene). Resulting sequences were aligned using ClustalW (http://www.ebi.ac.uk/Tools/clustalw2/index.html) and BLAST searched.
Results
Reference DNA.
Eight primer sets were tested over a temperature range (58–66°C) with L. donovani 105 parasite per mL; only one primer set showed good amplification and was taken forward for further testing (Table 1). This primer set was active from 60 to 66°C showing suitable use for water bath amplification where the temperature may fluctuate. Further reactions were carried out at 64°C. The RT-LAMP assay was able to amplify all species of Leishmania tested, including all major Old and New world species capable of infecting humans (105 parasites per mL). In serial dilutions of L. donovani and L. guyanensis RNA/DNA, the RT-LAMP reaction could detect between 10 and 100 parasites per mL (Figure 2). Amplification could be achieved at 35 minutes for samples of high parasite concentration (around 104 per mL). However, samples of very low concentration (102 per mL) could only be detected at 50–60 minutes. A 60 minutes reaction time was used to ensure amplification of samples with low parasite concentrations as regularly found in the blood of patients infected with VL.
Figure 2.
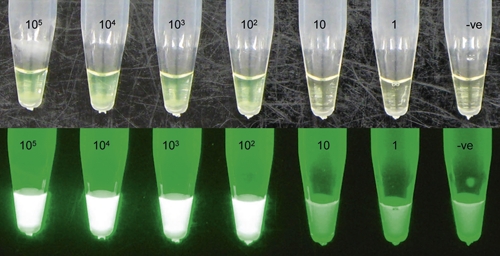
Serial dilution of Leishmania donovani (MHOM/SD/68/IS) amplified reverse transcriptase-loop-mediated isothermal amplification (RT-LAMP) (Serial dilution suspended in TE buffer). A, reaction held under normal light; B, reaction held under a simple UV light. Negative control included. This figure appears in color at www.ajtmh.org.
Melt curves of the following species were completed to ensure the specificity of the reaction. All isolates had a melting temperature (Tm) of 80.5°C, indicating the amplification of identical sequences and amplicons. Three species had a melting temperature of 81°C; this probably relates to base pair substitutions in the 18S rDNA in this region between species.
In addition, sequence analysis was completed of the LAMP product F2 to B2 regions; this revealed a band of 135 bp; two isolates (L. donovani [MHOM/SD/68/IS] and L. guyanensis [MHOM/BZ/75/M4147]) were sequenced revealing 100% sequence identity with the reference DNA (Accession nos.: FN396548 and FN396549).
Clinical samples.
Of the 30 blood samples tested from VL patients, 25 were positive with RT-LAMP and 27 with qRT-PCR (Table 2), giving a sensitivity of 83% (CI: 65–94%) for RT-LAMP and 90% (CI: 72–97%) for qRT-PCR compared with microscopy of bone marrow and lymph-node aspirates. Of the 50 healthy endemic controls 49 were negative with qRT-PCR and RT-LAMP giving a specificity of 98% (CI: 88–99.9%) for both techniques, the positive samples were different for each test. It is possible that this false positive reaction is an asymptomatic carrier of L. donovani; however, DAT results were negative.
Table 2.
Results of quantitative reverse transcriptase-polymerase chain reaction (RT-PCR) vs. reverse transcriptase-loop-mediated isothermal amplification (RT-LAMP) results. Visceral leishmaniasis (VL), cutaneous leishmaniasis (CL), and healthy endemic controls (HEC) are shown
| VL parasite/mL | CL parasites/mL | HEC | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| > 10,000 | > 1,000 | > 100 | > 10 | 0 | > 10,000 | > 1,000 | > 100 | > 10 | 0 | 0 | |
| RT-LAMP- positive | 8 | 9 | 5 | 3 | 0 | 32 | 8 | 1 | 1 | 0 | 1 |
| RT-LAMP- negative | 0 | 0 | 0 | 2 | 3 | 0 | 0 | 0 | 1 | 0 | 49 |
| Total | 8 | 9 | 5 | 5 | 3 | 32 | 8 | 1 | 2 | 0 | 50 |
| 30 | 43 | 50 | |||||||||
Of the 43 CL samples from Suriname, 42 were positive with RT-LAMP against the gold standard, qRT-PCR in this case, sensitivity is 98% (CI: 86–99.9%). The qRT-PCR was used as the gold standard here as conventional microscopy results were only available on Day 1 of treatment. At Day 1 and 1 week after treatment, many samples contained extremely high levels of parasitemia > 10,000 per mL (Table 2). The one LAMP negative sample was collected from a patient during week 4 of follow-up after treatment; according to qRT-PCR this sample contained 16 parasites per mL (Table 2). We have used follow-up samples from patients with low parasitemia to show the limits of LAMP analytical sensitivity at approximately 10–100 parasites per mL.
Discussion
This study shows the use of an RT-LAMP system as a pan-Leishmania diagnostic test. This assay can be used for the sensitive amplification of Leishmania parasites from purified RNA/DNA and from patient samples taken in the field from endemic areas. A pan-Leishmania assay was designed so all species could be amplified in one test; the differential diagnosis between VL and CL can be made with clinical symptoms, and by producing one test greater standardization can be achieved. The diagnostic sensitivity of the test was 83% (CI: 65–94%) from the circulating blood of VL patients compared with microscopic examination of bone marrow and lymph-node aspirates. For CL patients, the test showed 98% sensitivity (CI: 86–99.9%); the increase in sensitivity is caused by the high parasite numbers in CL infections as shown by the qRT-PCR results. This shows that RT-LAMP has a comparable sensitivity to other molecular diagnostics yet amplification can be achieved quickly, with basic equipment and without post-amplification handling.
A sensitivity of 83% for VL samples compared with microscopy of bone marrow samples compares with studies of PCR results on blood samples, e.g., 70% sensitivity,27 92% sensitivity,13 and 90% (CI: 72–97%) sensitivity of qRT-PCR from this study. Of the five RT-LAMP negative samples, three were also negative with qRT-PCR, the other two samples showed an infection with only 15 and 6 parasites per mL with qRT-PCR, respectively. This shows that RT-LAMP is able to amplify samples with 93% (CI: 74–99%) sensitivity compared with qRT-PCR in the circulating blood; blood sampling is a less invasive method of detection than spleen, bone marrow and lymph-node aspirates and is, therefore, safer for the patient. These results also compare favorably with another LAMP assay designed for the specific amplification of L. donovani; this assay was able to amplify 8 of 10 blood samples from parasitologically confirmed patients, a comparable sensitivity to nested PCR.28
For suspected VL patients, the RT-LAMP assay has the potential to be used as an initial screen of suspected patients with DAT/rK39 positive serology; only patients with negative LAMP will then require an aspirate, saving time, money, and potential infection from aspirate sampling. It is of vital importance that this assay is now tested with suspected VL cases to assess the real sensitivity and specificity as a diagnostic tool.
The test performed with high sensitivity (98%) for CL, this observed increase is caused by high numbers of parasites in biopsy samples. This compares positively with other molecular diagnostics such as PCR Oligo-chromatography (74% sensitivity on CL aspirates and 92% on skin scrapings29). The RT-LAMP could not amplify post-treatment patients when numbers of parasites fell to extremely low levels of around 10 parasites per mL. Because of such high parasitemia, the amplification time could be reduced from 60 to 40 minutes. The reverse transcriptase step increases the sensitivity by at least 10-fold; when high parasitemia is present; this step may be omitted, which would save on cost and one step in the amplification procedure. This is true for the species found in Suriname, predominantly L. guyanensis, but must be checked for other species of Leishmania parasite that cause CL as not all contain such high numbers of parasites in biopsy samples such as L. major (El Safi S, personal communication). Again, the assay should now be tested on CL suspected patients to assess the sensitivity as a diagnostic tool.
In a brief cost evaluation, the RT-LAMP assay compares favorably with other molecular diagnostic tests. The cost (including transportation and FDR but excluding staffing costs) per 25 μL reaction is $3.5; in addition it is possible to reduce the volume of the reaction by half to $1.75 with no adverse consequences (Adams E, unpublished data). This is less or comparable to other molecular diagnostics such as real time-PCR, estimated at $12 and PCR-restriction fragment length polymorphism (PCR-RFLP) estimated at $2.5 per reaction.16 In this study the FDR reagent (Eiken) was used as the detection method of choice as it can be added pre-reaction to avoid post-amplification handling, a contamination prone step. Other detection methods for LAMP amplification include the addition of ethidium bromide, which is carcinogenic, and the use of a tubidimeter, an expensive piece of laboratory equipment. We believe that the FDR reagent is currently the best method available; however, the cost of this reagent may hinder its use in endemic regions.
The RT-LAMP reaction was tested for cross-reactivity with other diseases with differential diagnosis to VL and CL; these included DNA/RNA samples from Malaria, Typhoid, Brucellosis, Leprosy, Tuberculosis, Leprosy, other a-typical mycobacterial infections, and Leptospirosis (common in late stage patients) and no cross reaction was observed. However, cross reaction of this assay was observed with Trypanosoma brucei and T. cruzi because of the similarity of the 18S region between the Kinetoplastida. A one hundred-fold difference between the amplification of Trypanosoma and Leishmania was detected. In practice the symptoms of sleeping sickness and Chagas disease are different to leishmaniasis; however, it should be the aim of future research to eliminate this cross reaction; this can be achieved by the design of species-specific LAMP reactions, or degenerate primer sets. It is not expected that this assay could amplify patient samples, especially from Chagas patients where the parasitemia is so low. The differential diagnoses of VL and CL do not overlap; in fact, the clinical symptoms of these diseases are quite different. The distinction between the different forms of leishmaniasis (VL, PKDL, CL, and MCL) is not possible with this assay; here differential diagnosis must be made on clinical symptoms.
In other LAMP studies the DNA/RNA preparation procedure has been minimized because of the high sensitivity of these assays. For example, in Malaria detection, direct amplification from parasites in boiled blood was successful20; and for the Trypanosoma, amplification was possible from native sera, buffy-coat and heat-treated blood, serum, and cerebrospinal fluid (CSF).23 For CL infections the parasite loads should be high enough for direct amplification from swab samples and boiled skin scrapings. However, the parasite load of VL is low, as shown by qRT-PCR, therefore, amplification with such crude preparation of nucleic acids may not be possible. Further work in selection of the best DNA/RNA preparation methods is needed to ensure minimal time and equipment required.
In conclusion, we have demonstrated the use of a sensitive and specific RT-LAMP assay for the detection of Leishmania species in purified DNA/RNA preparations and, importantly, from clinical samples collected in Suriname and Sudan. The next step for this assay is to use it in endemic settings to ensure that results can be achieved with the same sensitivity and specificity, especially for clinically suspected patients. The sensitivity and specificity of the assay compares favorably with other molecular diagnostics, yet has the advantages of a low technology tool with fast amplification and a simple readout system. Improvements in the bottleneck of DNA/RNA preparations where there is little pre-handling of samples in combination with this assay will enable the development of an effective diagnostic tool, suitable for field settings.
Acknowledgments
We thank Linda Oskam, Indra Bergval, Theresia Abdoel, Marcel Belt (KIT-BR), and Wendy Gibson (University of Bristol, UK) for DNA and Richard Anthony (KIT-BR), Peter de Vries, and Henry de Vries (Academic Medical Centre, Netherlands) for advice on differential diagnostics. We also thank Kimberly Boer, Mariska Leeflang (KIT-BR), and Mark Perkins (FIND) for epidemiological statistics and critical appraisal of the manuscript. Thanks must go to the patients and staff involved in this study, especially to the Department of Dermatology, Academic Hospital Paramaribo and the Dermatological Service, Paramaribo.
Footnotes
Financial support: KIT. Clinical material from Sudan was collected within the framework of a collaborative research project sponsored by the European community FP6 call - priority INCO-DEV - TRYLEIDIAG, contract 015379. Collection of samples from Suriname was funded by the Netherlands Organization for Scientific Research – WOTRO Science for Global Development contract W96-210.
Authors' addresses: Emily R. Adams, Gerard J. Schoone, and Henk D. F. H. Schallig, Koninklijk Instituut voor de Tropen (KIT), KIT Biomedical Research, Amsterdam, The Netherlands, E-mails: e.adams@kit.nl, g.schoone@kit.nl, and h.schallig@kit.nl. Al Farazdag Ageed and Sayda El Safi, Faculty of Medicine, University of Khartoum, Khartoum, Sudan, E-mails: elfarazdega@yahoo.com and shelsafi@yahoo.com.
References
- 1.Pearson RD, Sousa AQ. Clinical spectrum of leishmaniasis. Clin Infect Dis. 1996;22:1–13. doi: 10.1093/clinids/22.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Zijlstra EE, Musa AM, Khalil EA, el-Hassan IM, el-Hassan AM. Post-kala-azar dermal leishmaniasis. Lancet Infect Dis. 2003;3:87–98. doi: 10.1016/s1473-3099(03)00517-6. [DOI] [PubMed] [Google Scholar]
- 3.Salotra P, Singh R. Challenges in the diagnosis of post kala-azar dermal leishmaniasis. Indian J Med Res. 2006;123:295–310. [PubMed] [Google Scholar]
- 4.Desjeux P. Leishmaniasis: current situation and new perspectives. Comp Immunol Microbiol Infect Dis. 2004;27:305–318. doi: 10.1016/j.cimid.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 5.Berman JD. Human leishmaniasis: clinical, diagnostic, and chemotherapeutic developments in the last 10 years. Clin Infect Dis. 1997;24:684–703. doi: 10.1093/clind/24.4.684. [DOI] [PubMed] [Google Scholar]
- 6.Faber WR, Oskam L, van Gool T, Kroon NC, Knegt-Junk KJ, Hofwegen H, van der Wal AC, Kager PA. Value of diagnostic techniques for cutaneous leishmaniasis. J Am Acad Dermatol. 2003;49:70–74. doi: 10.1067/mjd.2003.492. [DOI] [PubMed] [Google Scholar]
- 7.Zijlstra EE, el-Hassan AM. Leishmaniasis in Sudan. Visceral leishmaniasis. Trans R Soc Trop Med Hyg. 2001;95((Suppl 1)):S27–S58. doi: 10.1016/s0035-9203(01)90218-4. [DOI] [PubMed] [Google Scholar]
- 8.Singh S, Sivakumar R. Recent advances in the diagnosis of leishmaniasis. J Postgrad Med. 2003;49:55–60. doi: 10.4103/0022-3859.927. [DOI] [PubMed] [Google Scholar]
- 9.Boelaert M, Rijal S, Regmi S, Singh R, Karki B, Jacquet D, Chappuis F, Campino L, Desjeux P, Le RD, Koirala S, Van der Stuyft P. A comparative study of the effectiveness of diagnostic tests for visceral leishmaniasis. Am J Trop Med Hyg. 2004;70:72–77. [PubMed] [Google Scholar]
- 10.Sundar S, Mondal D, Rijal S, Bhattacharya S, Ghalib H, Kroeger A, Boelaert M, Desjeux P, Richter-Airijoki H, Harms G. Implementation research to support the initiative on the elimination of kala azar from Bangladesh, India and Nepal–the challenges for diagnosis and treatment. Trop Med Int Health. 2008;13:2–5. doi: 10.1111/j.1365-3156.2007.01974.x. [DOI] [PubMed] [Google Scholar]
- 11.Mondal D, Singh SP, Kumar N, Joshi A, Sundar S, Das P, Siddhivinayak H, Kroeger A, Boelaert M. Visceral leishmaniasis elimination programme in India, bangladesh, and Nepal: reshaping the case finding/case management strategy. PLoS Negl Trop Dis. 2009;3:e355. doi: 10.1371/journal.pntd.0000355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kar K. Serodiagnosis of leishmaniasis. Crit Rev Microbiol. 1995;21:123–152. doi: 10.3109/10408419509113537. [DOI] [PubMed] [Google Scholar]
- 13.Deborggraeve S, Boelaert M, Rijal S, De DS, Dujardin JC, Herdewijn P, Buscher P. Diagnostic accuracy of a new Leishmania PCR for clinical visceral leishmaniasis in Nepal and its role in diagnosis of disease. Trop Med Int Health. 2008;13:1378–1383. doi: 10.1111/j.1365-3156.2008.02154.x. [DOI] [PubMed] [Google Scholar]
- 14.Van der Meide W, Guerra J, Schoone G, Farenhorst M, Coelho L, Faber W, Peekel I, Schallig H. Comparison between quantitative nucleic acid sequence-based amplification, real-time reverse transcriptase PCR, and real-time PCR for quantification of Leishmania parasites. J Clin Microbiol. 2008;46:73–78. doi: 10.1128/JCM.01416-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Vries PJ, van der Meide WF, Godfried MH, Schallig HD, Dinant HJ, Faber WR. Quantification of the response to miltefosine treatment for visceral leishmaniasis by QT-NASBA. Trans R Soc Trop Med Hyg. 2006;100:1183–1186. doi: 10.1016/j.trstmh.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 16.Reithinger R, Dujardin JC. Molecular diagnosis of leishmaniasis: current status and future applications. J Clin Microbiol. 2007;45:21–25. doi: 10.1128/JCM.02029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van der Meide WF, Schoone GJ, Faber WR, Zeegelaar JE, de Vries HJ, Ozbel Y, Lai AFR, Coelho LI, Kassi M, Schallig HD. Quantitative nucleic acid sequence-based assay as a new molecular tool for detection and quantification of Leishmania parasites in skin biopsy samples. J Clin Microbiol. 2005;43:5560–5566. doi: 10.1128/JCM.43.11.5560-5566.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000;28:E63. doi: 10.1093/nar/28.12.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thekisoe OM, Bazie RS, Coronel-Servian AM, Sugimoto C, Kawazu S, Inoue N. Stability of Loop-Mediated Isothermal Amplification (LAMP) reagents and its amplification efficiency on crude trypanosome DNA templates. J Vet Med Sci. 2009;71:471–475. doi: 10.1292/jvms.71.471. [DOI] [PubMed] [Google Scholar]
- 20.Poon LL, Wong BW, Ma EH, Chan KH, Chow LM, Abeyewickreme W, Tangpukdee N, Yuen KY, Guan Y, Looareesuwan S, Peiris JS. Sensitive and inexpensive molecular test for falciparum malaria: detecting Plasmodium falciparum DNA directly from heat-treated blood by loop-mediated isothermal amplification. Clin Chem. 2006;52:303–306. doi: 10.1373/clinchem.2005.057901. [DOI] [PubMed] [Google Scholar]
- 21.Han ET, Watanabe R, Sattabongkot J, Khuntirat B, Sirichaisinthop J, Iriko H, Jin L, Takeo S, Tsuboi T. Detection of four Plasmodium species by genus- and species-specific loop-mediated isothermal amplification for clinical diagnosis. J Clin Microbiol. 2007;45:2521–2528. doi: 10.1128/JCM.02117-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Njiru ZK, Mikosza AS, Matovu E, Enyaru JC, Ouma JO, Kibona SN, Thompson RC, Ndung'u JM. African trypanosomiasis: sensitive and rapid detection of the sub-genus Trypanozoon by loop-mediated isothermal amplification (LAMP) of parasite DNA. Int J Parasitol. 2008;38:589–599. doi: 10.1016/j.ijpara.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Njiru ZK, Mikosza AS, Armstrong T, Enyaru JC, Ndung'u JM, Thompson AR. Loop-mediated isothermal amplification (LAMP) method for rapid detection of Trypanosoma brucei rhodesiense. PLoS Negl Trop Dis. 2008;2:e147. doi: 10.1371/journal.pntd.0000147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Curtis KA, Rudolph DL, Owen SM. Rapid detection of HIV-1 by reverse-transcription, loop-mediated isothermal amplification (RT-LAMP) J Virol Methods. 2008;151:264–270. doi: 10.1016/j.jviromet.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 25.Li QM, Ma XJ, Gao HC, Zhou R, Kuang ZZ, Hou YD. Evaluation of reverse transcription loop-mediated isothermal amplification for detection of avian influenza A H5N1 virus. Bing Du Xue Bao. 2008;24:178–184. [PubMed] [Google Scholar]
- 26.Boom R, Sol CJ, Salimans MM, Jansen CL, Wertheim-van Dillen PM, van der Noordaa J. Rapid and simple method for purification of nucleic acids. J Clin Microbiol. 1990;28:495–503. doi: 10.1128/jcm.28.3.495-503.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Osman OF, Oskam L, Zijlstra EE, Kroon NC, Schoone GJ, Khalil ET, el-Hassan AM, Kager PA. Evaluation of PCR for diagnosis of visceral leishmaniasis. J Clin Microbiol. 1997;35:2454–2457. doi: 10.1128/jcm.35.10.2454-2457.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takagi H, Itoh M, Islam MZ, Razzaque A, Ekram ARM, Hashighuchi Y, Noiri E, Kimura E. Sensitive, specific and rapid detection of Leishmania donovani DNA by loop-mediated isothermal amplification. Am J Trop Med Hyg. 2009;81:578–582. doi: 10.4269/ajtmh.2009.09-0145. [DOI] [PubMed] [Google Scholar]
- 29.Espinosa D, Boggild AK, Deborggraeve S, Laurent T, Valencia C, Pacheco R, Miranda-Verastegui C, Llanos-Cuentas A, Leclipteux T, Dujardin JC, Buscher P, Arevalo J. Leishmania OligoC-TesT as a simple, rapid and standardized tool for the molecular diagnosis of cutaneous leishmaniasis in Peru. J Clin Microbiol. 2009;47:2560–2563. doi: 10.1128/JCM.00259-09. [DOI] [PMC free article] [PubMed] [Google Scholar]