

Abstract
In the presence of the N-heterocyclic carbene gold catalyst (NHC-AuIPr, 7), propargyl esters 1a–f and 13 undergo a [4C + 3C] cycloaddition reaction with cyclopentadiene and furan under mild conditions. The evidence suggests the formation of the seven-membered ring occurs by a direct cycloaddition process, rather than a stepwise cyclopropanation/Cope rearrangement sequence.
The potent antiangiogenesis natural product family of cortistatins contain a center seven-membered ring flanked by two six-membered rings.1,2 Our initial attempt to construct the tetracyclic ring system using a transannular [4C+3C] cycloaddition strategy was met with mixed results.3,4 In the same time, Mascarenas and co-workers reported an intramolecular [4C+3C] cycloaddition reaction, in which an allene functional group was selectively activated by Pt or Au catalyst.5 This type of allene-diene intramolecular [4C+3C] cycloaddition reactions was further improved to occur under milder conditions.6–8 Other reports using propargyl esters as reactants involved stepwise [4+3] cycloaddition reactions to prepare benzonorcaradienes and azepines.9,10 More recently, Harmata reported the treatment of 5-silyloxydioxins with 5 mol % AuCl3/AgSbF6 in the presence of cyclopentadiene or furan resulted in the formation of [4C+3C]-cycloadducts.11 In this report, we disclose an intermolecular version of gold-catalyzed formal [4C+3C] cycloaddition reactions. This discovery expands the employment of propargyl esters as precursors in gold-catalyzed [4+3] cycloaddition reactions.12
The likely mechanism for our recently reported gold-catalyzed transannular [4+3] cycloaddition could involve two possible pathways based on known examples in the literature.13–15 The first pathway involves a gold-stabilized allyl cation and the second involves a gold carbene intermediate. As shown in Scheme 1, the first pathway (I to II through A and B) includes (1) a 3,3-rearrangement of the propargyl ester to give an allenyl ester (A),16,17 (2) in situ activation by the same gold catalyst to generate an allyl cation B,18 and (3) a [4+3] cycloaddition followed by a 1,2-acetoxy migration and deauration to produce the tetracyclic ring system II. The second mechanism through intermediates C and D is depicted on the right side in Scheme 1. This pathway involves a 1,2-acetoxy migration followed by a cyclopropanation/Cope rearrangement to produce the same product. Diazoesters undergo cyclopropanation/Cope rearrangement reactions in the presence of rhodium catalysts and Davies and coworkers have studied these reactions extensively.19,20
Scheme 1.

Gold-catalyzed transannular [4+3] cycloaddition reaction: two possible pathways
Ohe and coworkers reported cyclopropanation of alkenes using propargylic carboxylates as vinylcarbene precursors and [RuCl2(CO)3]2 as the catalyst.21 More recently, Au(I)-catalyzed cyclopropanation of olefins using either propargyl esters or enynes as gold-(I)-carbene precursors were reported.22,23
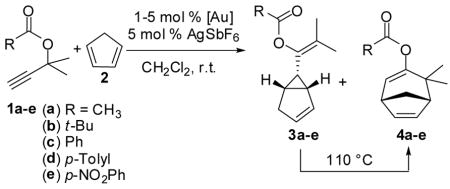
Because of their easy preparation, propargyl esters are convenient synthetic precursors. We are interested in expanding the usage of propargyl esters from transannular to inter- and intramolecular versions as the substrates in gold-catalyzed [4+3] cycloaddition reactions. The initial experiments were carried out using propargyl esters 1a–e and Au(III) catalyst PicAuCl2 6 (Table 1 and Fig. 1)24 which we have successfully employed in transannular [4+3] cycloaddition reactions.12 Other gold catalysts screened include the NHC-Au-IPr 7 and the R3PAu(I)Cl 8–10.
Table 1.
Au(III)-catalyzed cyclopropanation/Cope rearrangement reactions
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Golda catalyst | mol% | R | Time (h) | %Yield | Ratio (3 : 4) |
| 1 | 6 | 5 | Methyl (a) | 24 | 57 | 1 : 1.6 |
| 2 | 6 | 5 | t-Butyl (b) | 24 | 57 | 1 : 2.8 |
| 3 | 6 | 5 | Phenyl (c) | 24 | 90 | 1 : 2.1 |
| 4 | 6 | 5 | p-Tolyl (d) | 24 | 92 | 1 : 2.1 |
| 5 | 6 | 5 | p-nitrophenyl (e) | 24 | 63 | 1 : 1.3 |
| 6 | 7 | 5 | p-Tolyl (d) | 1 | 82 | 1 : 1.2 |
| 7 | 7 | 1 | p-Tolyl (d) | 23 | 92 | 1 : 1.6 |
| 8b | 8 | 5 | p-Tolyl (d) | 3 | n/a | n/a |
| 9b | 9 | 5 | p-Tolyl (d) | 2 | n/a | n/a |
| 10b | 10 | 5 | p-Tolyl (d) | 0.25 | n/a | n/a |
No silver co-catalyst was used for Au(III) catalyst (6).
Complex mixtures.
Fig. 1.
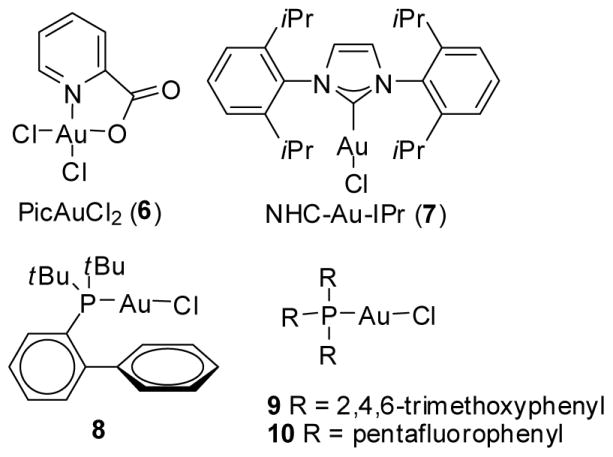
Au(III) catalyst 6 and Au(I) catalysts 7–10 were studied for their catalytic activity in the formal [4C + 3C] cycloaddition reactions.
The reactions of propargylic carboxylates 1a–e with cyclopentadiene were studied in CH2Cl2 at room temperature in the presence of gold catalysts 6–10. In the presence of the Au(III) catalyst 6, a cyclopropanation and a formal [4C + 3C] cycloaddition reaction occurred smoothly to afford a mixture of products 3 and 4. Although other Au(I) catalysts (8–10) were not effective (entries 8–10), we are pleased to find the gold catalyst with an N-heterocyclic carbene (NHC)25 ligand to be a highly effective catalyst for this transformation. With only 1% of the NHC-Au(I) catalyst 7, the reaction proceeded smoothly to give the corresponding products 3d and 4d (entry 7, Table 1). This reaction is very similar to the report by Ohe using [RuCl2(CO)3]2 as the catalyst,21 although a higher reaction temperature was required previously.
The reaction of 1a (R = Me) give the vinyl cyclopropane with endo stereochemistry, i.e., 3a, along with 3-acetoxy-4,4-dimethylbicyclo[3.2.1]-octa-2,6-diene (4a), both are known compounds from the previously reported study with Ru catalyst.21 The 1H and 13C NMR spectra of 3a and 4a are consistent with the reported data.21 No vinyl cyclopropane with exo configuration was observed.
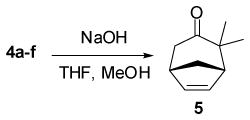
The vinyl cyclopropanes endo-3 can be converted to the corresponding bicyclo[3.2.1]octa-2,6-dienes (4) through a Cope rearrangement reaction by heating a solution of endo-3 in toluene at reflux for 12 h. The structure of the bicyclic enol esters 4 was further confirmed by convertion to the known ketone 526 with base catalyzed removal of the ester group, Eq (1).
 |
Eq. (1) |
With 5 mol% of the gold catalyst 6, several propargyl esters (1a–e) with different R group were compared for their reactivity (Table 1). The benzoate esters gave higher yields than the alkanoate esters (comparing entries 3 & 4 to 1 & 2). However, the low yields of 4a and 4b were most likely due to the volatility of the products.
Encouraged by the catalytic efficiency of the NHC-Au-IPr catalyst 7, the study was expanded to include furan as a diene substrate. In the presence of 1% of 7, propargyl ester 1f was allowed to react with 5 eqv. of furan, Scheme 2. Interestingly, the effect of the gold catalyst 7 parallels the ruthenium catalyst ([RuCl2(CO)3]2)27 and the triene aldehyde 11 was obtained as the exclusive product after 2 hours at room temperature in the solvent of CH2Cl2. However, when the same reaction was conducted in pentane a significant amount of the formal [4+3] cycloaddition product 4f was also isolated.
Scheme 2.

Reactions of furan with 1f in the presence of catalyst 7
The structure of 4f was confirmed by converting to the known ketone 12.28 The structure of the propargyl ester was examined by using 13 as a reactant in the presence of catalyst 7, Scheme 3.
Scheme 3.

Reactions of secondary propargyl ester 13 in the presence of catalyst 7.
To our delight, the reaction proceeded smoothly to afford a mixture in 95% yield with one diminant product. The major product, tentatively assigned as 14, was isolated along with minor isomers that have similar polarity and are difficult to separate. To expedite the isolation and structure identification, this mixture was subjected to the usual base-catalyzed removal of the ester group. This led to a clean separation of the major product 15a and its diastereomer 15b along with small amount of unidentified isomers. Product 15a is a known compound which was previously prepared using a classical oxyallyl cation addition to cyclopentadiene.29,30
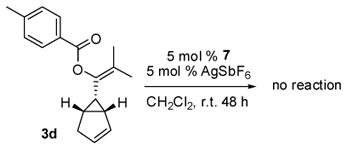
Under catalysis of the NHC-AuIPr catalyst 7, the results are dramatically different for propargyl esters 1a–f and 13. The former gave a nearly 1:1 mixture of cyclopropanation product 3 and the formal [4+3] cycloaddition product 4, while the latter produced predominantly the [4+3] cycloaddition product 14. It is likely that compound 14 is produced directly from an intermolecular [4+3] cycloaddition process. Evidence in support of a direct [4+3] cycloaddition lies in the mild conditions of the reaction. High temperature (refluxing in toluene for 12 h) was required for converting the cyclopropanation products 3a–e to 4a–e while compound 14 was obtained at room temperature from an overnight reaction. To further explore the pathways for the formation of the [4C+3C] cycloaddition products, isolated compound 3d was recommitted to the reaction conditions with fresh gold catalyst 7 for two days at rt, Eq (2). No reaction was observed. This strongly suggests that the formation of the products 4a–e came from a direct [4C+3C] cycloaddition mechanism.
 |
Eq. (2) |
We have shown that gold catalyst 7 is capable of initiating an intermolecular [4+3] cycloaddition reaction. Based on the evidence, the formation of the seven-membered rings occurs by a direct [4+3] cycloaddition mechanism, rather than a stepwise cyclopropanation/Cope rearrangement sequence. Further study on ligand effects on the product ratio is underway in our laboratories.
Supplementary Material
Acknowledgments
Financial support from the National Institutes of Health (GM069441) is gratefully acknowledged.
Footnotes
Supplementary data associated with this article (experimental procedures, NMR spectra) can be found in the online version at
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Watanabe Y, Aoki S, Tanabe D, Setiawan A, Kobayashi M. Tetrahedron. 2007;63:4074–4079. [Google Scholar]
- 2.Aoki S, Watanabe Y, Sanagawa M, Setiawan A, Kotoku N, Kobayashi M. J Am Chem Soc. 2006;128:3148–3149. doi: 10.1021/ja057404h. [DOI] [PubMed] [Google Scholar]
- 3.Craft DT, Gung BW. Tetrahedron Lett. 2008;49:5931–5934. doi: 10.1016/j.tetlet.2008.07.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gung BW, Craft DT. Tetrahedron Lett. 2009;50:2685–2687. [Google Scholar]
- 5.Trillo B, Lopez F, Gulias M, Castedo L, Mascarenas JL. Angew Chem Int Edit. 2008;47:951–954. doi: 10.1002/anie.200704566. [DOI] [PubMed] [Google Scholar]
- 6.Benitez D, Tkatchouk E, Gonzalez AZ, Goddard WA, Toste FD. Org Lett. 2009;11:4798–4801. doi: 10.1021/ol9018002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alonso I, Trillo B, Lopez F, Montserrat S, Ujaque G, Castedo L, Lledos A, Mascarenas JL. J Am Chem Soc. 2009;131:13020. doi: 10.1021/ja905415r. [DOI] [PubMed] [Google Scholar]
- 8.Mauleon P, Zeldin RM, Gonzalez AZ, Toste FD. J Am Chem Soc. 2009;131:6348–6349. doi: 10.1021/ja901649s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gorin DJ, Dube P, Toste FD. J Am Chem Soc. 2006;128:14480–14481. doi: 10.1021/ja066694e. [DOI] [PubMed] [Google Scholar]
- 10.Shapiro ND, Toste FD. J Am Chem Soc. 2008;130:9244. doi: 10.1021/ja803890t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harmata M, Huang CF. Tetrahedron Lett. 2009;50:5701–5703. [Google Scholar]
- 12.Gung BW, Craft DT, Bailey LN, Kirschbaum K. Chem Eur J. 2010;16:639. doi: 10.1002/chem.200902185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furstner A, Morency L. Angew Chem Int Edit. 2008;47:5030–5033. doi: 10.1002/anie.200800934. [DOI] [PubMed] [Google Scholar]
- 14.Echavarren AM. Nature Chemistry. 2009;1:431–433. doi: 10.1038/nchem.344. [DOI] [PubMed] [Google Scholar]
- 15.Benitez D, Shapiro ND, Tkatchouk E, Wang YM, Goddard WA, Toste FD. Nature Chemistry. 2009;1:482–486. doi: 10.1038/nchem.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang LM. J Am Chem Soc. 2005;127:16804–16805. doi: 10.1021/ja056419c. [DOI] [PubMed] [Google Scholar]
- 17.Marion N, Nolan SP. Angew Chem Int Ed. 2007;46:2750–2752. doi: 10.1002/anie.200604773. [DOI] [PubMed] [Google Scholar]
- 18.Trillo B, Lopez F, Montserrat S, Ujaque G, Castedo L, Lledos A, Mascarenas JL. Chem-Eur J. 2009;15:3336–3339. doi: 10.1002/chem.200900164. [DOI] [PubMed] [Google Scholar]
- 19.Olson JP, Davies HML. Org Lett. 2008;10:573–576. doi: 10.1021/ol702844g. [DOI] [PubMed] [Google Scholar]
- 20.Reddy RP, Davies HML. J Am Chem Soc. 2007;129:10312. doi: 10.1021/ja072936e. [DOI] [PubMed] [Google Scholar]
- 21.Miki K, Ohe K, Uemura S. J Org Chem. 2003;68:8505–8513. doi: 10.1021/jo034841a. [DOI] [PubMed] [Google Scholar]
- 22.Johansson MJ, Gorin DJ, Staben ST, Toste FD. J Am Chem Soc. 2005;127:18002–18003. doi: 10.1021/ja0552500. [DOI] [PubMed] [Google Scholar]
- 23.Lopez S, Herrero-Gomez E, Perez-Galan P, Nieto-Oberhuber C, Echavarren AM. Angew Chem Int Edit. 2006;45:6029–6032. doi: 10.1002/anie.200602448. [DOI] [PubMed] [Google Scholar]
- 24.Hashmi ASK, Weyrauch JP, Rudolph M, Kurpejovic E. Angew Chem Int Edit. 2004;43:6545–6547. doi: 10.1002/anie.200460232. [DOI] [PubMed] [Google Scholar]
- 25.Marion N, Nolan SP. Chem Soc Rev. 2008;37:1776–1782. doi: 10.1039/b711132k. [DOI] [PubMed] [Google Scholar]
- 26.Turro NJ, Edelson SS, Williams JR, Darling TR, Hammond WB. J Am Chem Soc. 1969;91:2283. [Google Scholar]
- 27.Miki K, Fujita M, Uemura S, Ohe K. Org Lett. 2006;8:1741–1743. doi: 10.1021/ol0604769. [DOI] [PubMed] [Google Scholar]
- 28.Fohlisch B, Gehrlach E, Herter R. Angew Chem Int Edi Engl. 1982;21:137–137. [Google Scholar]
- 29.Chan TH, Li MP, Mychajlowskij W, Harpp DN. Tetrahedron Lett. 1974:3511–14. [Google Scholar]
- 30.Ong BS, Chan TH. Heterocycles. 1977;7:913–18. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.