

Abstract
Mutations in the E3 ubiquitin ligase parkin cause early onset autosomal recessive juvenile Parkinsonism (ARJP) presumably by having lack of function that alter the level, activity, aggregation or localization of its substrates. We recently reported that phospholipase Cγ1 (PLCγ1) is a substrate for parkin. Here, we show that parkin mutants and siRNA parkin knockdown cells have enhanced levels of PLCγ1 phosphorylation, basal phosphoinositide hydrolysis and intracellular Ca2+ ([Ca2+]i). The protein levels of Ca2+ regulated Protein Kinase C α (PKCα) were decreased in ARJP parkin mutant cells. Neomycin and dantrolene decreased [Ca2+]i levels in parkin mutants to those seen in wild-type (WT) parkin cells, suggesting that differences were a consequence of altered PLC activity. The protection of WT parkin against 6-hydroxydopamine (6OHDA) toxicity could also be established in ARJP mutants when pretreating with dantrolene, implying that balancing Ca2+ release from ryanodine-sensitive stores is decreasing the toxic effects from 6OHDA. Our findings suggests parkin as an important factor for maintaining Ca2+ homeostasis and that parkin deficiency leads to a PLC-dependent increase in [Ca2+]i levels that makes cells more vulnerable to neurotoxins such as 6OHDA.
Introduction
Parkinson's disease (PD) is a neurodegenerative disorder involving cell loss in various brain regions, especially dopamine neurons in the substantia nigra pars compacta (snpc) [1].
In recent years, mechanism of action studies on gene mutations causing rare familial forms of disease have given important new insights into PD pathogenesis. Of the familial PD genes, mutations in parkin are the most common cause of Autosomal Recessive Juvenile Parkinsonism (ARJP). Parkin is an E3 ubiquitin ligase in which mutations have been shown to alter the level, activity, aggregation or localization of its substrates.
Some studies have proposed parkin deficiency related consequences for intracellular signaling, including altered apoptotic stress activated protein kinase signaling [2]. Parkin has also been suggested to promote Akt signaling by preventing endocytosis and trafficking of the epidermal growth factor (EGF) receptor via proteasome independent ubiquitination of the parkin substrate Eps15 [3]. In the same signaling pathway, we have found that parkin interacts with and ubiquitylates phospholipase Cγ1 (PLCγ1). Although mutant parkin interacts with PLCγ1, it shows less potency to ubiquitinate this substrate compared to WT parkin and PLCγ1 levels are enhanced in brain homogenates from parkin knockout (KO) mice [4].
Parkin has also been shown to protect cells against damage induced by several agents, including dopamine [5], ceramide [6] and a mutant of α-synuclein [7], the major component of Lewy body inclusions, a pathological hallmark of PD. In parallel, a number of different ways have been proposed by which parkin mutations and deficiency can induce cellular toxicity [8]. Several studies have demonstrated that parkin is protective to oxidative stress [5] [9] is important for maintenance of mitochondrial morphology and function [10] [11] [6] [12] [13] [14] as well as mitophagy[15]. Whether the protective effects of parkin are due to its E3 ligase activity or to additional functions of the protein remains at the present unknown. In support of the first, parkin has been shown to protect against neurotoxicity induced by unfolded protein stress, suggesting that its function in the ubiquitination pathway may be to target for degradation of misfolded proteins derived from the endoplasmic reticulum (ER) [16, 17]. ER plays a pivotal role in the processing and folding of proteins and in the regulation of calcium (Ca2+) homeostasis. The ER stress response interferes with the role of ER as protein factory and a Ca2+ storage organelle, and excessively high intracellular Ca2+ can initiate apoptosis [18]. In contrast, low Ca2+ levels induce the ER stress response by promoting the accumulation of ER chaperones and Ca2+ transporting proteins [19]. One important phenotypic trait that distinguishes snpc dopaminergic neurons is that they are autonomously active and require a constant clearance of Ca2+ compared to other neurons that are activated by synaptic input. Also, dopaminergic neurons rely on L-type Ca2+ channels [20] whereas the activity of other neuron types mainly depends on Na+ channels. Thus snpc dopaminergic neurons have unique features that may make them more vulnerable to disrupted calcium homeostasis [21]. Indeed Ca2+ toxicity has been a subject of interest in neurodegenerative pathogenesis including PD for many years [22-24] and there is some evidence that use of Ca2+ channel blockers may even reduce the risk of disease [25]. Two of the parkin identified substrates, Parkin-associated endothelial-like receptor (Pael-R) [17] and PLCγ1 [4], are known to be involved in regulating intracellular Ca2+ concentrations [Ca2+]i [26]. It is therefore possible that an impairment of parkin substrates dependent regulation of [Ca2+]i could be part of the mechanisms by which parkin mutations lead to ARJP.
In the present study, we show that PLC signaling is altered in parkin deficient human neuroblastoma cell lines, resulting in a distrupted [Ca2+] homeostasis and increased vulnerability to 6OHDA. We further show that blocking of PLC activity or Ryanodine receptors (RyR) could reverse these effects.
Results
Effects of parkin deficiency on PLCγ1 activation, Phosphoinositide hydrolysis, and intracellular calcium
In order to address the functional role of parkin ubiquitination of PLCγ1, we investigated PLC activity in human neuroblastoma SH-SY5Y cell lines stably transfected with either WT, or mutant R42P or G328E parkin. We chose to utilize human neuroblastoma cell lines since parkin knockout in rodents does not result in the key pathological events seen in humans, such as dopaminergic cell death in snpc and substantial motor impairment [27]. Moreover, some of the experiments required stable expression of exogenous parkin. Protein levels of parkin and PLCγ1 were as published previously [4]. Treatment of cells with EGF is known to give a direct phosphorylation and activation of PLCγ1 via the EGF receptor [28]. In our hands, treatment of SH-SY5Y cells with EGF gave a significantly higher phosphorylation of PLCγ1 in R42P or G328E Parkin mutants, as compared to both non-transfected (NT) and WT parkin cells (Fig. 1). PLC activation leads to phosphoinositide (PI) hydrolysis resulting in diacylglycerol (DAG) and inositol 1,4,5 trisphosphate (IP3), the latter being an important intracellular second messenger for controlling ER calcium levels [29]. We therefore next determined whether the between cell type differences in PLCγ1 phosphorylation were reflected at the level of PI hydrolysis. Our results showed that basal PI hydrolysis was also significantly higher in both R42P and G328E parkin mutants as compared to NT and WT parkin cells (Fig. 2a). Also, siRNA knockdown of endogenous parkin in NT SH-SY5Y cells gave a similar increase in basal PI hydrolysis as to that seen in parkin mutant cells (Fig. 2b). In addition, we also tested for the consequences of knocking down c-Cbl, another known E3 ligases for PLCγ1 [30]. Similar to the effects seen with parkin knockdown, siRNA knockdown of c-Cbl also gave significantly increased basal PI hydrolysis (Fig. 2b). parkin and c-Cbl siRNA knockdown was verified by immunoblotting (Fig 2b).
Figure 1.
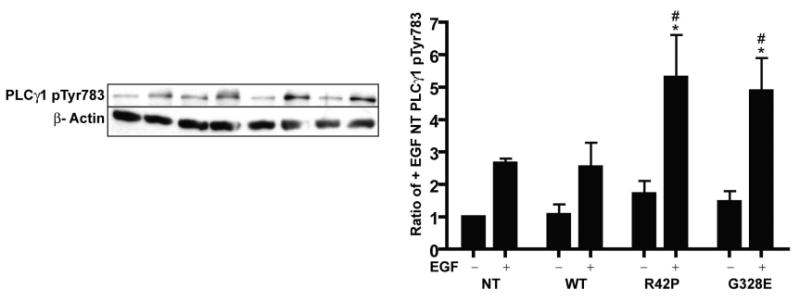
EGF-mediated PLCγ1 phosphorylation is increased in ARJP Parkin cell lines.
Immunoblotting of phosphoTyr783-PLCγ1, in NT and in stably transfected human SH-SY5Y neuroblastoma cells with WT parkin and the ARJP Parkin mutations R42P and G328E treated or untreated with EGF for 2 min. Histogram shows the quantification (mean±SEM) of phosphoTyr783-PLCγ1 normalized to actin from 5 independent experiments. **, p<0.01, ***, p<0.001 ANOVA, Fisher's post-hoc test for the comparison of treated vs. basal. #, p<0.05 ANOVA, Fisher's post-hoc test for the comparison of treated condition versus treated NT cells.
Figure 2.
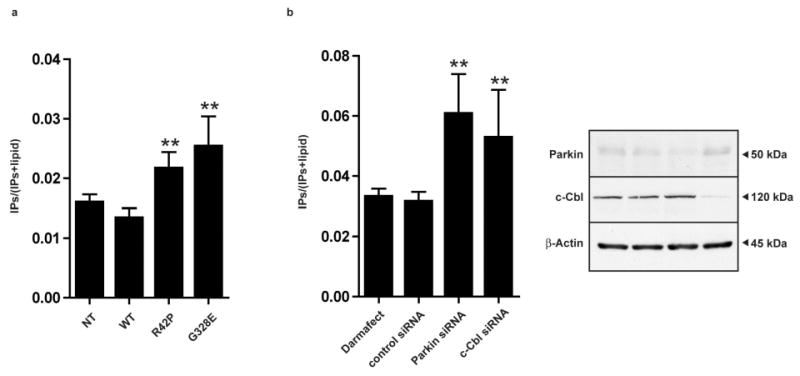
PI hydrolysis is enhanced in parkin deficient cell lines.
(a) Histograms show means ± SEM of PI hydrolysis measured in basal conditions of NT, Parkin WT, R42P and G328E transfected cells (n=5) and (b) in the different siRNA transfected cells (n=3). Parkin and c-Cbl protein levels were detected by Western blot analysis in SH-SY5Y neuroblastoma cells after siRNA knockdown of parkin and c-Cbl.
We next investigated the effects of parkin and parkin R42P and G328E mutations on [Ca2+]i. As seen in figure 3a, both R42P and G328E mutant cells had significantly higher basal [Ca2+]i as compared to NT and WT Parkin transfected cells. Similarly, knockdown of parkin or c-Cbl expression by siRNA also resulted in significantly higher basal [Ca2+]i as compared to control cells (Fig. 3b). These between cell differences were found when measurements were performed either in Ca2+-free PBS or in MEM buffer (Fig. 3a and 3b).
Figure 3.
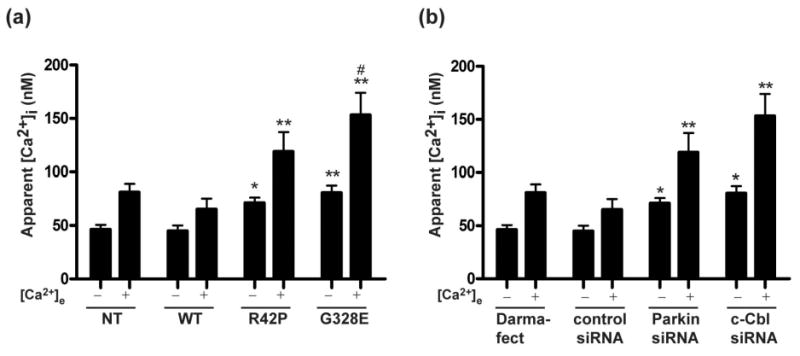
Parkin deficient cells have higher levels of [Ca2+].
Measurements were done in Ca2+-free PBS (-) and in MEM (+) containing normal (1 mM) Ca2+. Experiments were performed in (a) parkin transfected cells (n=12), and (b) siRNA transfected cells (n=3). For the siRNA experiments two groups were used as controls (NT cells treated with Darmafect and control siRNA). Statistical analyses of the results were carried out using ANOVA followed by Fisher's PLSD post-hoc test. *, p < 0.05; **, p < 0.01; ***: p < 0.001 against the respective value in both NT and WT. #, p < 0.05 against R42P.
Increased cytosolic Ca2+ levels in ARJP Parkin mutants are due to altered PLC activity
We next investigated whether the primary cause of increased basal [Ca2+]i levels seen in ARJP parkin mutant cells resulted from increased PLC activity or by altered Ca2+ influx from extracellular sources. Treatment with the PLC inhibitor neomycin (500 mM) reduced basal [Ca2+]i in R42P and G328E cells to estimated levels in WT parkin cells (Fig. 4a). Also, the RyR antagonist dantrolene (10 mM) gave a similar reversal of the R42P and G328E mutant Ca2+ levels, indicating that altered levels were due to a subsequent increased Ca2+-induced Ca2+ release response in these cells (Fig. 4a).
Figure 4.
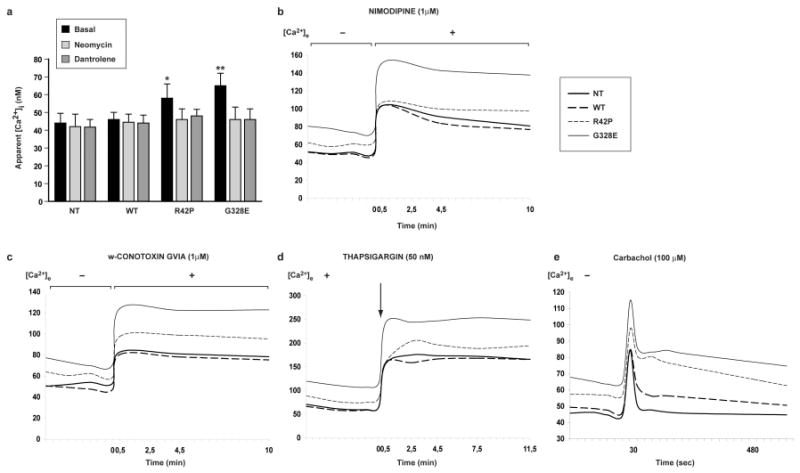
Altered Ca2+ homeostasis in parkin ARJP cell lines depends on PLC signaling.
(a) Both, PLC inhibitor neomycin (500 μM) and RyR antagonist dantrolene (10 μM) reduced basal [Ca2+]i in R42P and G328E mutants to those seen in WT parkin cells (n=3). [Ca2+]i measurements were performed after addition of (b) nimodipine (1μM) or (c) ω-conotoxin (1μM) respectively in PBS (-[Ca2+]e) and MEM (1 mM [Ca2+]e)as described in Material and Methods. (d) Basal and thapsigargin (TG) (50 nM)-stimulated Ca2+ measurements were made in MEM. Basal [Ca2+]i was higher in parkin mutants and TG induced similar responses in all cell types. (e) Basal and Carbachol (100 μM) stimulated measurements were made in PBS. In Figures b, c, d and e lines shows the average value of 3 independent experiments in each of which between 9 and 12 wells were analyzed per group. Statistical analysis was carried out using ANOVA followed by Fisher's PLSD post-hoc test. *, p < 0.05. **, p<0.01.
Blocking either L-type or N-type Ca2+ channels with nimodipine (1 mM) and ω-conotoxin (1 mM), did not change [Ca2+]i in any of the cell types (Fig. 4b and c), suggesting that differences arises from intracellular Ca2+ handling.
Treatment of cells with thapsigargin (TG) (50 nM), an inhibitor of the ER Ca2+ pump (SERCA) that depletes intracellular Ca2+ stores [31], gave a rapid increase in [Ca2+]i in all cells, followed by a slow increase that reached a plateau during the time of exposure. The shapes of the TG induced Ca2+ curves were parallel in all cell types, indicating that the between cell type differences types were attributable to differences in basal [Ca2+]i (Fig. 4d). We previously demonstrated that PLCβ levels were not change by overexpression of WT parkin or R42P and G328E parkin mutants[4]. Here we investigated if changes of PLCβ activity could contribute to the effects of these parkin mutants on [Ca2+]i. For that we treated cells with carbachol (100 μM), a specific agonist of muscarinic acetylcholine receptors that specifically activates PLCβ and not PLCγ.
Treatment of cells with carbachol gave rapid increases of [Ca2+]i. Peak increases were found approximately 30 sec after addition of carbachol and were significantly higher in both R42P and G328E mutants as compared to NT and WT parkin cells (Fig 4e). However, no significant differences were found when data were expressed as ratios (Peak / basal), suggesting that peak differences were due to basal [Ca2+]i differences among the cell types. In both R42P and G328E cells, the effects of carbachol on [Ca2+]i lasted longer time and a long tail-off effect was seen that was more pronounced in G328E mutants as compared with R42P cells (Fig 4e).
Protein kinase Cα protein levels in ARJP parkin mutations are decreased
PLC signaling regulates not only the release of ER calcium, but also leads to the formation of DAG, which results in activation of Protein kinase C (PKC). We next explored if the increased PLCγ activity and [Ca2+]i seen in ARJP parkin mutants have consequences for PKC. From the multiples PKC isoforms [32], we chose to investigate the effects on one Ca2+ dependent, PKCα and on one Ca2+ independent, PKCε. ARJP parkin mutant cells showed reduced protein levels of PKCα, while PKCε levels were unchanged (Fig 5a and b). PKC activity was determined by measurement of the translocation from soluble to particulate fractions as previously described [33]. There were no significant differences in the translocation of both PKC isoforms (Fig 5c and d).
Figure 5.
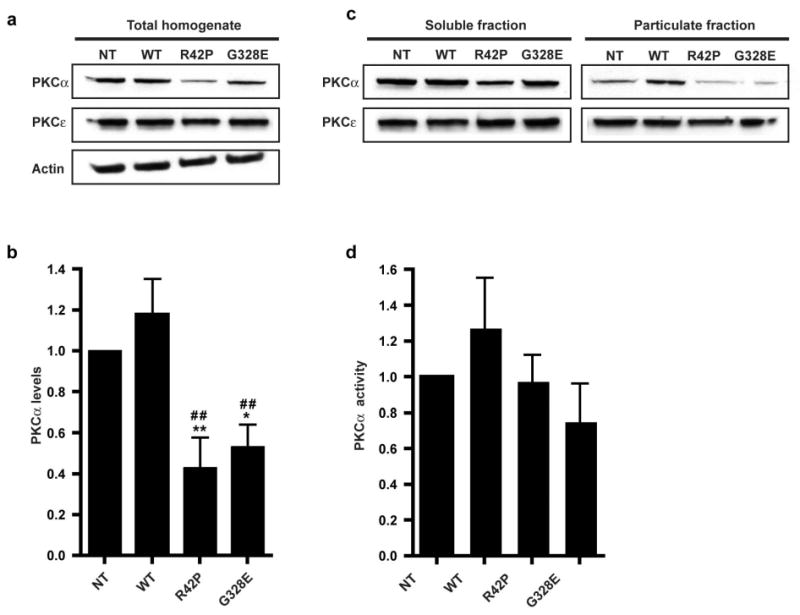
ARJP parkin mutant cells have lower protein levels of PKCα
(a) Immunoblotting of PKCα and PKCε levels in NT and in stably transfected human SH-SY5Y neuroblastoma cells with WT parkin and the ARJP parkin mutations R42P and G328E. (b) Representative immunoblots of PKCα and PKCε levels in soluble and particulate fractions. (c) Histogram shows total PKCα levels (mean±SEM, n=4) normalized to actin. (d) Histogram representing the ratio of particulate and total fractions of representing the relative activity of PKCα (mean±SEM, n= 4). Statistical analyses of the results were carried out using ANOVA followed by Fisher's PLSD post-hoc test. *, p < 0.05; **, p < 0.01; ***, p < 0.001 against the respective value in both NT * and WT #.
Dantrolene reverses the higher sensitivity to 6OHDA neurotoxicity seen in ARJP parkin mutans to the levels of WT parkin overexpressing cells
Both dopamine and its analogue 6OHDA have been shown to be toxic to SH-SY5Y cells with such toxicity being attenuated by overexpression of WT but not ARJP mutant forms of parkin [5]. We first confirmed these findings by measuring the effect of 6OHDA in our NT, WT parkin and parkin mutant (R42P, G328E) transfected cells. Treatment with 40 μM 6OHDA gave approximately a 15% decrease of cell viability with no significant differences among cell lines (Fig. 6a). However, as shown in figure 6a, and in agreement with the data of Jiang et al. [5], treatment of 120 μM 6OHDA was significantly less toxic in cells overexpressing WT parkin compared to NT and parkin mutant cells. Since we show that the RyR antagonist dantrolene reversed the increase of [Ca2+]i seen in R42P and G328E Parkin mutants, we next studied the effect of dantrolene on toxicity due to 6OHDA (120 μM). Cells were pretreated with dantrolene (10 μM) for 30 min, prior co-incubation with 6OHDA and dantrolene for 6 h. Blocking of RyR significantly reduced the amount of cell death in parkin mutant cells when exposed to 6OHDA, resulting in a equal cell viability level as WT parkin cells with or without Dantrolene (Fig. 6b). We also explored weather compromised mitochondrial Ca2+ buffering could be a participating factor in 6OHDA mediated toxicity in ARJP parkin mutant cells, we blocked the mitochondrial permeability transition pore (mPTP) with cyclosporine A prior to and during the 6OHDA treatment. However this treatment failed to rescue ARJP parkin mutant cells from 6OHDA provoked cell death (data not shown) and these results were therefore in accordance with previous studies [34, 35].
Figure 6.
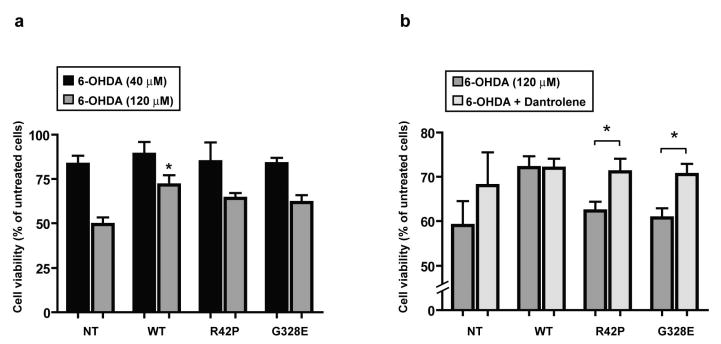
ARJP parkin mutations confer a higher sensitivity to 6OHDA neurotoxicity that was reversed by dantrolene.
(a) Effects of 6OHDA on MTT reduction in NT, WT parkin and in ARJP parkin mutations R42P and G328E cells. Cells were treated with 40 or 120 μM 6OHDA for 6 h (n=6). (b) Dantrolene reverses 6OHDA toxicity in ARJP mutants to the levels seen in WT parkin. Cells were treated with 120 μM 6OHDA with or without 10 μM dantrolene for 6 h. In dantrolene treated cells, an additional pretreatment for 30 min was also performed. Untreated cells were used as a control. Cell viability was analyzed by the MTT assay (n=3). Data (mean ± S.E.M.) are expressed as percentage of values in untreated NT cells (*, p < 0.05; ANOVA followed by Fisher's PLSD post-hoc test).
Discussion
The identification of parkin as an E3 ligase suggested that a deficient protein ubiquitination and/or degradation of substrates are behind the pathological mechanisms linking parkin mutations with ARJP. We previously demonstrated that PLCγ1 is ubiquitinated by parkin and that R42P and G328E parkin generate significantly lower levels of ubiquitinated PLCγ1 compared to WT in vitro. WT parkin expression significantly reduced the levels of PLCγ1 in human neuroblastoma. We further showed that PLCγ1 levels were increased in parkin KO mice brain homogenates [4]. PLCγ1 has been implicated in multiple signaling pathways that control cell division, differentiation, motility, and apoptosis and is activated upon stimulation of receptors for growth factors including the EGFR [36]. Activation of EGFR results in PLCγ1 phosphorylation at several sites, Tyr783 being the most crucial [37]. In addition, PLCγ1, together with other PLC isoforms and PI3K, hydrolyze phosphoinositides, resulting in the formation of two major second messengers, IP3 and DAG. IP3 releases Ca2+ from the ER, through activation of IP3 receptors and subsequently also RyR [36]. In the present study, we demonstrate that ARJP parkin mutations (R42P and G328E) and partial knockdown of parkin by siRNA results in increased phosphorylation of PLCγ1 after EGF treatment, as well as in enhanced basal PI hydrolysis and [Ca2+]i (summarized in Figure 7).
Figure 7.
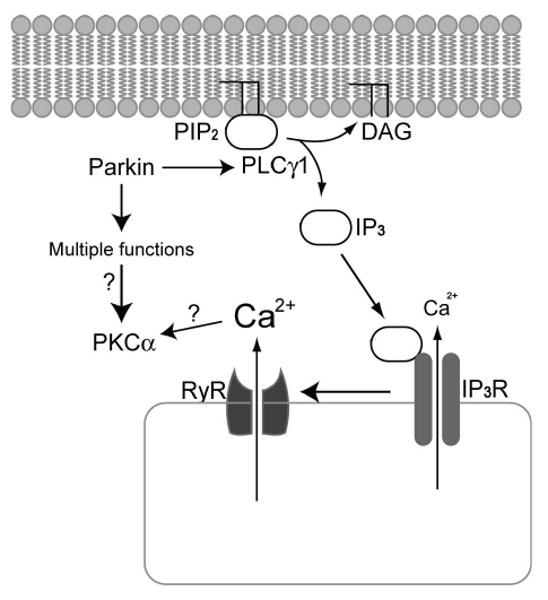
Summary of proposed mechanism for PLCγ1 induced calcium toxicity in ARJP parkin cells. Parkin was shown previously to ubiquitylate PLCγ1. In this paper we show that ARJP parkin mutant cells and parkin siRNA leads to enhanced PI hydrolysis and increased release of Ca2+ from intracellular stores, increasing sensitivity to cell death induced by 6OHDA. Disrupted Ca2+ homeostasis and/or other parkin related functions, ultimately alters PKCα protein levels.
PLC isoforms are known to control, independently of its lipase activity, the size and the duration of PLCβ mediated Ca2+ signals by regulating a secondary Ca2+ entry via ionotropic channels [38, 39]. Both WT and ARJP parkin mutants had similar responses to carbachol (that activates PI hydrolysis via G-protein-coupled acetylcholine muscarinic receptors and PLCβ), suggesting no differences in PLCβ activity among these cells types. However, compared to WT parkin cells, ARJP parkin mutants have longer lasting responses to carbachol, seen as long tail-off effects after Ca2+ peaks. These long tail-off effects were also blocked by neomycin and dantrolene, which is also consistent with effects being mediated by PLC isoenzymes and by RyR, respectively. A secondary Ca2+ entry after depletion of Ca2+ stores by TG or the Ca2+ ionophore ionomycin that is independent of PLCγ, has been previously described [39]. We investigated contribution of this mechanism to the differences seen in [Ca2+]i. Both WT and ARJP parkin mutants responded similarly to TG and to ionomycin (data not shown), indicating no differences in secondary Ca2+ entry among these cells.
To better define the mechanism responsible for the enhanced [Ca2+]i, seen in ARJP parkin mutants, we used specific blockers of PLC, RyR and of different Ca2+ channels. Both the PLC inhibitor neomycin and the RyR antagonist dantrolene reversed the high basal [Ca2+]i seen in R42P and G328E parkin cells to those seen in WT parkin cells. In contrast, blocking plasma membrane L-type and N-type Ca2+ channels with nimodipine and w-conotoxin, respectively, was without effect.
Together, these results indicate that the increased basal [Ca2+]i seen in R42P and G328E parkin cells are due to enhanced PLC activity and are mediated via RyR. The fact that siRNA knockdown of both parkin and c-Cbl also resulted in higher PI hydrolysis and [Ca2+]i levels confirms that these effects are consequence of a loss of parkin function leading to deregulated PLCγ1 ubiquitination. Rare mutations in c-Cbl have been associated with myeloid leukemia [40] [41] however there is to date no correlation between c-Cbl mutations and any neuronal diseases.
A proper regulation of Ca2+ homeostasis is crucial for maintaining balanced concentrations in the cell, so during normal conditions changes are transient and do not cause adverse effects. However, when components that are influencing the Ca2+ homeostasis are altered, transient increases from normal activity can lead to toxicity, which has been suggested to be part of the pathogenesis in several neurodegenerative diseases [23, 24, 42, 43]. Moreover, the pathological cell death in snpc of PD patients has been proposed to be caused by the increased vulnerability of these cells due to their higher metabolic activity and Ca2+ load [24]. In a recent report, transfections with mutated PTEN induced Kinase-1 (PINK1), another ARJP causative gene, was shown to increase cytosolic Ca2+, which was associated with mitochondrial impairement [44]. Ca2+ is a powerful secondary messenger that when present in excess activates degrading caspases and calpains which disrupts cytoskeletal proteins, membrane receptors and metabolic enzymes [45, 46]. Furthermore, disrupted Ca2+ homeostasis causes oxidative stress [47, 48] and is inducing apoptosis by mechanisms that perturb the mitochondrial function [49], leading to energetic deficiency and release of pro apoptotic proteins [18] and reactive oxygen species [50, 51].
In agreement with others[2, 5], we showed that overexpression of parkin WT was partially protective when challenging the cells with the dopamine metabolite 6OHDA and that this protective effect was attenuated in the ARJP parkin mutants. We also showed that this lack of protection in parkin mutants was reversed by the RyR antagonist dantrolene, suggesting that higher sensitivity to 6OHDA seen in ARJP parkin mutants is due to altered IP3 / Ca2+ signaling.
PLC activation has two major outcomes, release of calcium from ER and activation of PKC. The conventional subclass of PKC isoforms are regulated both by DAG and Ca. Our results show that parkin deficiency leads to enhanced [Ca2+]i levels which could have an impact on PKC activity. Therefore we investigated the protein levels and activity of the [Ca2+]i dependent and independent PKCα and PKCε respectively. We detected reduced protein levels of PKCα in the ARJP parkin mutant cells, however the relative activity was similar as in WT parkin and NT cells. It has been consistently reported that an overactivation of PKC results in a downregulation of the enzyme levels [52]. Another possibility is that parkin regulates the gene transcription of PKCα. This has been reported previously for other genes [53-55] but further experiments have to be performed to determine the link between PKCα and parkin.
In view of the vast number of identified parkin substrates [56], the vulnerability to toxic insults in parkin ARJP must be a combination of an imbalance in many systems, of which some might be overlapping. One possible other parkin substrate that may coincide to the same toxic pathway as we have described here, is the G-protein coupled receptor Pael-R1 that has been shown to regulate PLC activity [57] and subsequently mobilize [Ca2+]i [26]. Our results would suggest that the accumulation of Pael-R1 in ARJP parkin mutants and knockdown by parkin siRNA would also contribute to an unbalanced Ca2+ homeostasis and thus a higher sensitivity to toxic agents. Therefore we suggest that PLCγ1 may not act alone to change Ca2+ responses but in concert with additional substrates of parkin.
In summary we demonstrated that ARJP parkin mutants have enhanced PLCγ1 activity and consequently increased basal levels of PI hydrolysis and disturbances in PLC-mediated Ca2+ homeostasis. We also demonstrated that the increased [Ca2+]i seen in ARJP parkin mutants conferred a higher sensitivity to the toxicity of 6OHDA that could be reversed by blocking RyR. Our findings suggest that disruption of PLCγ1 signaling/Ca2+ homeostasis could be one of the mechanisms by which ARJP parkin mutations mediate neuronal death.
Materials and Methods
Materials
EGF, Dowex 1×8-200 (chloride form), 3-(4,5-dimethyl-thiazol-3-yl)-2,5-diphenyltetrazoliumbromide (MTT), probenecid, neomycin, dantrolene, and 6OHDA were purchased from Sigma-Aldrich (Germany). Fluo-3acetoxymethyl (Fluo3-AM) ester and PluronicF-127 were purchased from Molecular Probes (The Netherlands). Myo-[2-3H]inositol (10 Ci/mmol) from NEM (USA). Nimodipine and ω–conotoxin were from Alomon labs (Israel). All other chemicals were standard laboratory reagents.
DNA-constructs, transfections and cell culture
Human dopaminergic SH-SY5Y neuroblastoma cells were stably transfected with WT, R42P and G328E parkin constructs as described previously [4]. Cells were cultured at 37°C, 5% CO2, in Eagle's Minimum essential medium with Glutamax containing 10% Foetal Calf Serum (FCS). Transfected cells were supplemented with 200 μg/ml geneticin. All cell culture supplies were purchased from Invitrogen (Sweden). Parkin, c-Cbl and control siRNA knockdown were performed in SH-SY5Y cells by transfecting 30nM siRNA with DarmaFECT (Darmacon, Sweden) following the manufacturer's instructions. In all cases, transfections were performed for 72 h. The knockdown of the different proteins were confirmed by western blotting.
Immunoblot analysis
EGF treatment of SHSY5Y cells was performed at a concentration of 100 ng/ml for 2 minutes and at 37°C. Prior EGF treatment, cells were 2 hours in serum free conditions. Cells were lysed in lysis buffer (20 mM Tris-HCl, 137 mM Na Cl, 2 mM EDTA, 2% Nonidet P-40, 2% Triton-×100 and protease and phosphatase inhibitor cocktails) (Sigma-Aldrich, Germany). Cell extract protein amounts were quantified using the BCA protein assay kit (Pierce, USA). Equivalent amounts of protein were separated using 10% acrylamide gels. Proteins were transferred to a nitrocellulose membrane (Schleicher & Schuell, Germany). Western immunoblotting was performed using either anti phospho-PLCγ1 (Tyr783) rabbit polyclonal IgG (Upstate, USA), anti-parkin (Cell signaling, USA), anti-PLCγ1 or anti-c-Cbl, (BD transduction laboratories, Germany) with overnight incubations at a 1:1000 dilution. The secondary antibodies were anti-rabbit or anti-mouse horseradish peroxidase-linked (Amersham, UK), and were used at 1:2000 dilution for 1 h in room temperature. Detection was made by the ECL method (Amersham, UK) and exposure to Hyper film MP (Amersham, UK).
Phosphoinositides hydrolysis Assay
Cells were cultured to 75-80% confluence in 10 cm Petri dishes. One day prior to the experiment, cells were changed to serum-free media containing 5 μCi/ml myo-[2-3H]inositol and incubated for 24 h. Basal PI hydrolysis was measured as described previously [58, 59]. Cells were harvested by scraping with a rubber policeman in 4 ml of PBS. Contents were centrifuged at 500 × g for 15 min. Pellets were washed twice with 37 °C PBS and resuspended in 3 ml of 37 °C Krebs-Henseleit bicarbonate buffer containing 10 mM LiCl(KHB/Li), gassed with 5% CO2, 95% O2 and centrifuged again (4300 × g, 15 min). Cell pellets were re-suspended in 210 μl of KHB/Li, regassed, and 50 μl added to glass centrifuge tubes containing 250 μl of KHB/Li buffer. The tubes were incubated at 37 °C under an atmosphere of 5% CO2, 95% O2 with gentle agitation for 25 min. Incubations were stopped by adding 940 μl of chloroform:methanol (1:2). Tubes were incubated on ice for 30 min and phases separated by adding 310 μl of chloroform and 310 μl water followed by vortexing and centrifugation. 750 μl of the aqueous phase were removed and labelled IPs separated from myo-[2-3H]inositol by Dowex chromatography. The chloroform phase was removed, placed into scintillation vials, and allowed to evaporate before determination of ‘lipid dpm’ by scintillation spectroscopy. Results were expressed as dpm IPs/(dpm IPs + dpm lipid).
PKC translocation
PKC translocation was determined as described previously [33]. Approximately 5 × 106 cells were washed with ice cold PBS and harvested by scraping in lysis buffer containing 20 mM Tris HCl pH 7.4, 0.32 mM sucrose, 2 mM EDTA, 50 mM β-mercaptoethanol (Sigma-Aldrich, Germany), protease inhibitor cocktail and sonicated (12 s., 22 μ) on ice. A sample from this fraction was saved for total lysate analysis. The lysates were then ultracentrifuged at 100 000 × g for 30 min at +4 °C. Supernatants were designated as soluble fractions. The samples were analyzed by western blotting and the ratio of particulate and total fractions is referred to as translocation between cytosol and membrane compartments.
Intracellular Ca2+ measurements
Intracellular Ca2+ concentrations ([Ca2+]i) were essentially determined as previously described [58, 59]. In brief, the cells cultured in 96-well plates were loaded with minimum essential medium without phenol red containing 5 μM Fluo-3 AM ester, 0.5% (v/v) Pluronic F-127, and 1 mM probenecid (90 min in the dark at room temperature). After loading, the cells were incubated for 120 min in minimum essential medium without phenol red with 1 mM probenecid in the dark at room temperature to allow intracellular esterases to decompose the Fluo-3 AM ester. Basal [Ca2+]i was measured in both Ca2+-free PBS and phenol red-free MEM (containing 1 mM Ca2+). In the experiments including nimodipine (1 μM) or ω-conotoxin (1 μM), ([Ca2+]i was measured first in PBS. The PBS was removed and MEM added to the cells, with the respective blocker being present during all the measurements. TG was used at 50 nM and [Ca2+]i was measured in phenol red-free MEM. Measurements with carbachol was performed after measuring basal apparent [Ca2+]i, then the PBS was removed, and 100 μM carbachol in PBS solution at 37 °C was added. Carbachol was present for all subsequent apparent [Ca2+]i measures. For the experiments with neomycin or dantrolene, the agents were included in MEM without phenol red and used during the 120 min of incubation period and also in the PBS for the 10 min incubation when basal [Ca2+]i were measured
MTT assay
Cell viability was determined by the MTT assay. MTT powder was dissolved in MEM without phenol red at 0.3 mg/ml and then added to the cells. After 1 h at 37°C, the medium was removed and the formazan crystals were dissolved in isopropanol. Aliquots were moved to a 96-well plate and optical densities read at 540 nm in a Molecular Devices Spectra MAX 250 plate reader. For the experiments with dantrolene, cells were pretreated with 120 μM dantrolene for 30 min, followed by treatment with 120 μM 6OHDA for 6 h. Control cells received the equivalent amount of vehicle. Results were expressed as a percentage of values obtained for non-treated cells.
Statistical analyses
Analyses of differences were carried out by ANOVA followed by Fisher's PLSD post-hoc test. A value of p<0.05 was considered statistically significant.
Acknowledgments
This work was supported by grants from the following Swedish foundations: Swedish Brain Power, Parkinsonsfonden, Riskbankens Jubileum fond, Karolinska Institutets foundation for geriatric research, Loo and Hans Ostermans foundation, Gun and Bertil Stohnes foundation, K.A: Wallenberg, Stiftelsen för Gamla Tjänarinnor and Åke Wibergs foundation. This research was also supported (in part) by the Intramural Research Program of the National Institute on Aging, National Institutes of Health, Ayudas postdoctorales, Gobierno de Navarra and LIONS foundation for research of age related disorders.
Abbreviations
- 6OHDA
6-hydroxydopamine
- ARJP
Autosomal recessive juvenile Parkinsonism
- [Ca2+]I
Intracellular Ca2+
- DAG
Diacylglycerol
- EGF
Epidermal growth factor
- ER
Endoplasmic reticulum
- FAD
Familial Alzheimer's disease
- FCS
Foetal Calf Serum
- Fluo-AM
Fluo-3acetoxymethyl
- IP3
Inositol 1,4,5 trisphosphate
- KHB/Li
Krebs-Henseleit bicarbonate buffer/LiCl
- KO
Knockout
- MTT
3-(4,5-dimethyl-thiazol-3-yl)-2,5-diphenyltetrazoliumbromide
- NT
Non-transfected
- Pael-R
Parkin-associated endothelial-like receptor
- PD
Parkinson's disease
- PI
Phosphoinositide
- PINK1
PTEN induced Kinase-1
- PKC
Protein kinase C
- PLCγ1
Phospholipase Cγ1
- PS1
Presenilin 1
- RyR
Ryanodine receptors
- Snpc
Substantia nigra pars compacta
- TG
Thapsigargin
- WT
Wild-type
References
- 1.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 2.Hasegawa T, Treis A, Patenge N, Fiesel FC, Springer W, Kahle PJ. Parkin protects against tyrosinase-mediated dopamine neurotoxicity by suppressing stress-activated protein kinase pathways. Journal of neurochemistry. 2008;105:1700–1715. doi: 10.1111/j.1471-4159.2008.05277.x. [DOI] [PubMed] [Google Scholar]
- 3.Fallon L, Belanger CM, Corera AT, Kontogiannea M, Regan-Klapisz E, Moreau F, Voortman J, Haber M, Rouleau G, Thorarinsdottir T, et al. A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI(3)K-Akt signalling. Nat Cell Biol. 2006;8:834–842. doi: 10.1038/ncb1441. [DOI] [PubMed] [Google Scholar]
- 4.Dehvari N, Sandebring A, Flores-Morales A, Mateos L, Chuan YC, Goldberg MS, Cookson MR, Cowburn RF, Cedazo-Minguez A. Parkin-mediated Ubiquitination Regulates Phospholipase C-gamma1. Journal of cellular and molecular medicine. 2008 doi: 10.1111/j.1582-4934.2008.00443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang H, Ren Y, Zhao J, Feng J. Parkin protects human dopaminergic neuroblastoma cells against dopamine-induced apoptosis. Hum Mol Genet. 2004;13:1745–1754. doi: 10.1093/hmg/ddh180. [DOI] [PubMed] [Google Scholar]
- 6.Darios F, Corti O, Lucking CB, Hampe C, Muriel MP, Abbas N, Gu WJ, Hirsch EC, Rooney T, Ruberg M, et al. Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum Mol Genet. 2003;12:517–526. doi: 10.1093/hmg/ddg044. [DOI] [PubMed] [Google Scholar]
- 7.Petrucelli L, O'Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Hardy J, et al. Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron. 2002;36:1007–1019. doi: 10.1016/s0896-6273(02)01125-x. [DOI] [PubMed] [Google Scholar]
- 8.Moore DJ, West AB, Dikeman DA, Dawson VL, Dawson TM. Parkin mediates the degradation-independent ubiquitination of Hsp70. Journal of neurochemistry. 2008;105:1806–1819. doi: 10.1111/j.1471-4159.2008.05261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosen KM, Veereshwarayya V, Moussa CE, Fu Q, Goldberg MS, Schlossmacher MG, Shen J, Querfurth HW. Parkin protects against mitochondrial toxins and beta-amyloid accumulation in skeletal muscle cells. J Biol Chem. 2006;281:12809–12816. doi: 10.1074/jbc.M512649200. [DOI] [PubMed] [Google Scholar]
- 10.Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 11.Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuroda Y, Mitsui T, Kunishige M, Matsumoto T. Parkin affects mitochondrial function and apoptosis in neuronal and myogenic cells. Biochem Biophys Res Commun. 2006;348:787–793. doi: 10.1016/j.bbrc.2006.06.201. [DOI] [PubMed] [Google Scholar]
- 13.Kuroda Y, Mitsui T, Kunishige M, Shono M, Akaike M, Azuma H, Matsumoto T. Parkin enhances mitochondrial biogenesis in proliferating cells. Hum Mol Genet. 2006;15:883–895. doi: 10.1093/hmg/ddl006. [DOI] [PubMed] [Google Scholar]
- 14.Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- 15.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy. 2009;5 doi: 10.4161/auto.5.5.8505. 8505 [pii] [DOI] [PubMed] [Google Scholar]
- 16.Imai Y, Soda M, Takahashi R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. The Journal of biological chemistry. 2000;275:35661–35664. doi: 10.1074/jbc.C000447200. [DOI] [PubMed] [Google Scholar]
- 17.Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001;105:891–902. doi: 10.1016/s0092-8674(01)00407-x. [DOI] [PubMed] [Google Scholar]
- 18.Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- 19.Paschen W. Endoplasmic reticulum dysfunction in brain pathology: critical role of protein synthesis. Curr Neurovasc Res. 2004;1:173–181. doi: 10.2174/1567202043480125. [DOI] [PubMed] [Google Scholar]
- 20.Ping HX, Shepard PD. Apamin-sensitive Ca(2+)-activated K+ channels regulate pacemaker activity in nigral dopamine neurons. Neuroreport. 1996;7:809–814. doi: 10.1097/00001756-199602290-00031. [DOI] [PubMed] [Google Scholar]
- 21.Wilson CJ, Callaway JC. Coupled oscillator model of the dopaminergic neuron of the substantia nigra. J Neurophysiol. 2000;83:3084–3100. doi: 10.1152/jn.2000.83.5.3084. [DOI] [PubMed] [Google Scholar]
- 22.Paschen W. Mechanisms of neuronal cell death: diverse roles of calcium in the various subcellular compartments. Cell Calcium. 2003;34:305–310. doi: 10.1016/s0143-4160(03)00138-6. [DOI] [PubMed] [Google Scholar]
- 23.Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6:337–350. doi: 10.1111/j.1474-9726.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- 24.Surmeier DJ. Calcium, ageing, and neuronal vulnerability in Parkinson's disease. Lancet Neurol. 2007;6:933–938. doi: 10.1016/S1474-4422(07)70246-6. [DOI] [PubMed] [Google Scholar]
- 25.Becker C, Jick SS, Meier CR. Use of antihypertensives and the risk of Parkinson disease. Neurology. 2008;70:1438–1444. doi: 10.1212/01.wnl.0000303818.38960.44. [DOI] [PubMed] [Google Scholar]
- 26.Goldman RS, Finkbeiner SM, Smith SJ. Endothelin induces a sustained rise in intracellular calcium in hippocampal astrocytes. Neuroscience letters. 1991;123:4–8. doi: 10.1016/0304-3940(91)90144-i. [DOI] [PubMed] [Google Scholar]
- 27.Terzioglu M, Galter D. Parkinson's disease: genetic versus toxin-induced rodent models. Febs J. 2008;275:1384–1391. doi: 10.1111/j.1742-4658.2008.06302.x. [DOI] [PubMed] [Google Scholar]
- 28.Wahl MI, Nishibe S, Kim JW, Kim H, Rhee SG, Carpenter G. Identification of two epidermal growth factor-sensitive tyrosine phosphorylation sites of phospholipase C-gamma in intact HSC-1 cells. The Journal of biological chemistry. 1990;265:3944–3948. [PubMed] [Google Scholar]
- 29.Berridge MJ, Heslop JP, Irvine RF, Brown KD. Inositol trisphosphate formation and calcium mobilization in Swiss 3T3 cells in response to platelet-derived growth factor. The Biochemical journal. 1984;222:195–201. doi: 10.1042/bj2220195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tvorogov D, Carpenter G. EGF-dependent association of phospholipase C-gamma1 with c-Cbl. Experimental cell research. 2002;277:86–94. doi: 10.1006/excr.2002.5545. [DOI] [PubMed] [Google Scholar]
- 31.Parekh AB, Penner R. Store depletion and calcium influx. Physiological reviews. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- 32.Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev. 2008;88:1341–1378. doi: 10.1152/physrev.00034.2007. 88/4/1341 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cedazo-Minguez A, Bonecchi L, Winblad B, Post C, Wong EH, Cowburn RF, Benatti L. Nicergoline stimulates protein kinase C mediated alpha-secretase processing of the amyloid precursor protein in cultured human neuroblastoma SH-SY5Y cells. Neurochem Int. 1999;35:307–315. doi: 10.1016/s0197-0186(99)00074-1. S0197-0186(99)00074-1 [pii] [DOI] [PubMed] [Google Scholar]
- 34.Lee CS, Park WJ, Ko HH, Han ES. Differential involvement of mitochondrial permeability transition in cytotoxicity of 1-methyl-4-phenylpyridinium and 6-hydroxydopamine. Mol Cell Biochem. 2006;289:193–200. doi: 10.1007/s11010-006-9164-0. [DOI] [PubMed] [Google Scholar]
- 35.Fujita H, Ogino T, Kobuchi H, Fujiwara T, Yano H, Akiyama J, Utsumi K, Sasaki J. Cell-permeable cAMP analog suppresses 6-hydroxydopamine-induced apoptosis in PC12 cells through the activation of the Akt pathway. Brain Res. 2006;1113:10–23. doi: 10.1016/j.brainres.2006.06.079. S0006-8993(06)01877-4 [pii] [DOI] [PubMed] [Google Scholar]
- 36.Rebecchi MJ, Pentyala SN. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiological reviews. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- 37.Kim HK, Kim JW, Zilberstein A, Margolis B, Kim JG, Schlessinger J, Rhee SG. PDGF stimulation of inositol phospholipid hydrolysis requires PLC-gamma 1 phosphorylation on tyrosine residues 783 and 1254. Cell. 1991;65:435–441. doi: 10.1016/0092-8674(91)90461-7. 0092-8674(91)90461-7 [pii] [DOI] [PubMed] [Google Scholar]
- 38.Bird GS, Aziz O, Lievremont JP, Wedel BJ, Trebak M, Vazquez G, Putney JW., Jr Mechanisms of phospholipase C-regulated calcium entry. Curr Mol Med. 2004;4:291–301. doi: 10.2174/1566524043360681. [DOI] [PubMed] [Google Scholar]
- 39.Patterson RL, van Rossum DB, Ford DL, Hurt KJ, Bae SS, Suh PG, Kurosaki T, Snyder SH, Gill DL. Phospholipase C-gamma is required for agonist-induced Ca2+ entry. Cell. 2002;111:529–541. doi: 10.1016/s0092-8674(02)01045-0. [DOI] [PubMed] [Google Scholar]
- 40.Caligiuri MA, Briesewitz R, Yu J, Wang L, Wei M, Arnoczky KJ, Marburger TB, Wen J, Perrotti D, Bloomfield CD, et al. Novel c-CBL and CBL-b ubiquitin ligase mutations in human acute myeloid leukemia. Blood. 2007;110:1022–1024. doi: 10.1182/blood-2006-12-061176. blood-2006-12-061176 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dunbar AJ, Gondek LP, O'Keefe CL, Makishima H, Rataul MS, Szpurka H, Sekeres MA, Wang XF, McDevitt MA, Maciejewski JP. 250K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res. 2008;68:10349–10357. doi: 10.1158/0008-5472.CAN-08-2754. 68/24/10349 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Foster TC. Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell. 2007;6:319–325. doi: 10.1111/j.1474-9726.2007.00283.x. [DOI] [PubMed] [Google Scholar]
- 43.Bojarski L, Herms J, Kuznicki J. Calcium dysregulation in Alzheimer's disease. Neurochem Int. 2008;52:621–633. doi: 10.1016/j.neuint.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 44.Marongiu R, Spencer B, Crews L, Adame A, Patrick C, Trejo M, Dallapiccola B, Valente EM, Masliah E. Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson's disease by disturbing calcium flux. J Neurochem. 2009;108:1561–1574. doi: 10.1111/j.1471-4159.2009.05932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chan SL, Mattson MP. Caspase and calpain substrates: roles in synaptic plasticity and cell death. Journal of neuroscience research. 1999;58:167–190. [PubMed] [Google Scholar]
- 46.Nixon RA. The calpains in aging and aging-related diseases. Ageing research reviews. 2003;2:407–418. doi: 10.1016/s1568-1637(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 47.Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- 48.Mattson MP, Chan SL. Calcium orchestrates apoptosis. Nat Cell Biol. 2003;5:1041–1043. doi: 10.1038/ncb1203-1041. [DOI] [PubMed] [Google Scholar]
- 49.Graier WF, Frieden M, Malli R. Mitochondria and Ca(2+) signaling: old guests, new functions. Pflugers Arch. 2007;455:375–396. doi: 10.1007/s00424-007-0296-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lipton SA, Singel DJ, Stamler JS. Nitric oxide in the central nervous system. Progress in brain research. 1994;103:359–364. doi: 10.1016/s0079-6123(08)61149-8. [DOI] [PubMed] [Google Scholar]
- 51.Mattson MP. Modification of ion homeostasis by lipid peroxidation: roles in neuronal degeneration and adaptive plasticity. Trends in neurosciences. 1998;21:53–57. doi: 10.1016/s0166-2236(97)01188-0. [DOI] [PubMed] [Google Scholar]
- 52.Dehvari N, Cedazo-Minguez A, Isacsson O, Nilsson T, Winblad B, Karlstrom H, Benedikz E, Cowburn RF. Presenilin dependence of phospholipase C and protein kinase C signaling. J Neurochem. 2007;102:848–857. doi: 10.1111/j.1471-4159.2007.04571.x. JNC4571 [pii] [DOI] [PubMed] [Google Scholar]
- 53.Liu M, Aneja R, Sun X, Xie S, Wang H, Wu X, Dong JT, Li M, Joshi HC, Zhou J. Parkin regulates Eg5 expression by Hsp70 ubiquitination-dependent inactivation of c-Jun NH2-terminal kinase. J Biol Chem. 2008;283:35783–35788. doi: 10.1074/jbc.M806860200. M806860200 [pii] [DOI] [PubMed] [Google Scholar]
- 54.Henn IH, Bouman L, Schlehe JS, Schlierf A, Schramm JE, Wegener E, Nakaso K, Culmsee C, Berninger B, Krappmann D, et al. Parkin mediates neuroprotection through activation of IkappaB kinase/nuclear factor-kappaB signaling. J Neurosci. 2007;27:1868–1878. doi: 10.1523/JNEUROSCI.5537-06.2007. 27/8/1868 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Unschuld PG, Dachsel J, Darios F, Kohlmann A, Casademunt E, Lehmann-Horn K, Dichgans M, Ruberg M, Brice A, Gasser T, et al. Parkin modulates gene expression in control and ceramide-treated PC12 cells. Mol Biol Rep. 2006;33:13–32. doi: 10.1007/s11033-005-3961-5. [DOI] [PubMed] [Google Scholar]
- 56.Cookson MR. Parkin's substrates and the pathways leading to neuronal damage. Neuromolecular medicine. 2003;3:1–13. doi: 10.1385/NMM:3:1:1. [DOI] [PubMed] [Google Scholar]
- 57.Ambar I, Sokolovsky M. Endothelin receptors stimulate both phospholipase C and phospholipase D activities in different cell lines. European journal of pharmacology. 1993;245:31–41. doi: 10.1016/0922-4106(93)90166-7. [DOI] [PubMed] [Google Scholar]
- 58.Cedazo-Minguez A, Popescu BO, Ankarcrona M, Nishimura T, Cowburn RF. The presenilin 1 deltaE9 mutation gives enhanced basal phospholipase C activity and a resultant increase in intracellular calcium concentrations. The Journal of biological chemistry. 2002;277:36646–36655. doi: 10.1074/jbc.M112117200. [DOI] [PubMed] [Google Scholar]
- 59.Popescu BO, Cedazo-Minguez A, Benedikz E, Nishimura T, Winblad B, Ankarcrona M, Cowburn RF. Gamma-secretase activity of presenilin 1 regulates acetylcholine muscarinic receptor-mediated signal transduction. The Journal of biological chemistry. 2004;279:6455–6464. doi: 10.1074/jbc.M306041200. [DOI] [PubMed] [Google Scholar]