

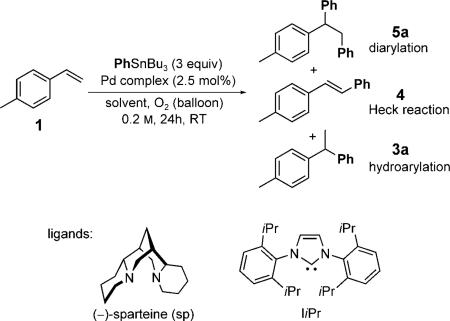
The Heck reaction is a widely used transformation in organic synthesis in which a terminal alkene and an organic halide are coupled in the presence of a palladium(0) catalyst.[1] Palladium(II)-catalyzed oxidative Heck reactions, in which a terminal oxidant (dioxygen or benzoquinone)[2] and an organometallic reagent are used, have also been developed to expand the scope of this transformation. In both types of Heck reaction, alkene insertion leads to a σ-alkyl palladium(II) intermediate D, which undergoes β-hydride elimination to form the product (Scheme 1).[3] Recently, significant effort has been invested in attempts to intercept related s-alkyl palladium(II) intermediates derived from alkenes in various processes to access diverse difunctionalized products.[4] However, there have been few successful intermolecular difunctionalization reactions initiated through a Heck insertion. A noteworthy example was recently reported by Sanford and Kalyani, who developed a 1,1-arylhalogenation of alkenes with an aryl stannane and a chloride source.[5] Other examples are mainly restricted to substrates that can not undergo β-hydride elimination.[6] Herein we report a new palladium(II)-catalyzed alkene difunctionalization reaction, which we presume is initiated by an oxidative Heck insertion. Two carbon–carbon single bonds are formed in a 1,2-difunctionalization of conjugated alkenes and a 1,1-difunctionalization of nonconjugated terminal alkenes with O2 as the terminal oxidant.
Scheme 1.

Proposed mechanism for the oxidative Heck reaction (a) and interception of the σ-alkyl palladium(II) intermediate by transmetalation (b).
Recently, our research group has been focused on the development of palladium-catalyzed alkene hydrofunctionalization[7] and difunctionalization[8] reactions that avoid products derived from β-hydride elimination. In successful hydro-functionalization reactions,[7] the proposed σ-alkyl palladium(II) intermediates, which are accessed by the insertion of a styrene derivative into a palladium hydride, are thought to be stabilized by a π-benzyl interaction prior to functionalization.[9] On the basis of this concept, it was proposed that a π-benzyl intermediate of type E, accessed through a Heck insertion, could slow β-hydride elimination and thus enable subsequent transmetalation to form F and reductive elimination to yield the product of diarylation (Scheme 1). Thus, the key issue to be addressed is the control of the relative rates of β-hydride elimination and transmetalation of the second equivalent of the organostannane. We believed that these rates could be controlled by tuning the ligand environment in the palladium complex.
Initially, the diarylation product 5a was observed as a side product under conditions originally used in the hydroarylation of 4-methylstyrene (Table 1, entry 1).[7b] Palladium(II)–N-heterocyclic carbene (NHC) complexes were selected early on in the optimization process, because they have been found to be robust catalysts for various aerobic oxidation[7d,10] and cross-coupling reactions.[11] The use of [Pd(IiPr)(OAc)2] led to a greater preference for diarylation over hydroarylation, although the oxidative Heck product was formed in higher yield (Table 1, entry 2). Enhancement of the cationic nature of the complex improved the selectivity for diarylation over the oxidative Heck reaction (Table 1, entry 3). This result suggests that the π-benzyl interaction is stronger with a more electrophilic catalyst (and β-hydride elimination is slower as a consequence), which is consistent with the reported isolation of π-benzyl complexes with cationic palladium species.[9]
Table 1.
Optimization for the diarylation of 4-methyl styrene.
 | ||||
|---|---|---|---|---|
| Entry | Pd complex | Solvent | Conv. [%]a | Yield of 5a [%b (5a/4/3a) |
| 1c | Pd(sp)Cl2 | IPA | 90 | 32 (1.6:0.8:1) |
| 2d | Pd(IiPr)(OAc)2 | IPA | 99 | 41 (8.2:8.6:1) |
| 3d | Pd(IiPr)(OCOCF3)2 | IPA | 99 | 55 (4.2:0.9:1) |
| 4d | Pd(IiPr)(OTs)2 | IPA | 30 | 26 (26:2.0:1) |
| 5d | Pd(IiPr)(OTs)2 | DCE | 99 | 55 (3.0:0.3:1) |
| 6d | Pd(IiPr)(OTs)2 | dioxane | 99 | 58 (4.1:0.6:1) |
| 7d | Pd(IiPr)(OTs)2 | DMA | 99 | 60 (7.0:2.4:1) |
| 8 | Pd(IiPr)(OTs)2 | DMA | 46 | 36 (20:4.5:1) |
| 9e | Pd(IiPr)(OTs)2 | DMA | 78 | 50 (17:5.3:1) |
| 10ef | Pd(IiPr)(OTs)2 | DMA | 91 | 63 (16:0.3:1) |
| 11efg | Pd(IiPr)(OTs)2 | DMA | >99 | 90 (22:2.3:1) |
| 12efgh | Pd(IiPr)(OTs)2 | DMA | >99 | 92 (42:1.5:1) |
The conversion was measured by GC with an internal standard.
The yield was determined by GC.
CuCl2 (7.5 mol%) was used.
The reaction was performed at 45 °C.
Pd complex: 6 mol%.
Activated molecular sieves (3 Å, 100 mg) were added.
Cu(OTf)2 (25 mol %) was used.
Concentration of the reaction mixture (with respect to 1): 0.1 m.
Tf = trifluoromethanesulfonyl, Ts = p-toluenesulfonyl.
A dramatic change in selectivity for the diarylation product over the oxidative Heck product was observed when the counterion was changed from trifluoroacetate to tosylate. Unfortunately, the more cationic complex [PdII-(IiPr)(OTs)2] was unstable under these conditions (Table 1, entry 4). However, a change of solvent from isopropyl alcohol (IPA) to dichloroethane (DCE), dioxane, or N,N-dimethylacetamide (DMA) resulted in improved catalyst stability (Table 1, entries 5–7), whereby the use of DMA led to the most promising result.[12] Further optimization led to a decrease in temperature (Table 1, entry 8) and an increase in catalyst loading (Table 1, entry 9). Three final changes were needed: 1) the addition of molecular sieves[13] (Table 1, entry 10), 2) the addition of Cu(OTf)2 (Table 1, entry 11), which has been shown to facilitate transmetalation,[14] and 3) a decrease in concentration (Table 1, entry 12). These conditions led to excellent conversion into 5a (92 % yield, as determined by GC). A greater than 10:1 ratio of the 1,2- to the 1,1-diarylation product was mainly observed for reactions carried out under these conditions (see below).
The generality of the PdII-catalyzed difunctionalization of styrenes was explored under the optimized conditions, initially by the evaluation of different organostannanes (Table 2, entries 1–5). The electronic nature of the aryl stannane had little effect on the cross-coupling reaction, with the exception of a decrease in selectivity for the 1,2-diarylation product with the electron-rich stannane 2c (Table 2, entry 3). Electron-rich styrenes, including those with ortho substitution, were found to undergo the diarylation reaction successfully with PhSnBu3 in good yields (Table 2, entries 6–8). The good reactivity of an organostannane derived from a cyclic enol ether indicates that a wide range of alternative organostannanes can be anticipated as substrates (Table 2, entry 9). Terminal 1,3-dienes were also evaluated. With these substrates, a π-allyl species[15] can be formed rather than a π-benzyl-stabilized intermediate (Table 2, entries 10–12). To our delight, the 1,2-diarylation of 1,3-dienes yielded the desired product as a single isomer. Finally, the difunctionalization of a 1,3-diene with a non-aryl organostannane was successful, albeit relatively low yielding (Table 2, entry 13).
Table 2.
Scope of the palladium-catalyzed 1,2-diarylation of styrene derivatives and 1,3-dienes with organostannanes.
 | ||||
|---|---|---|---|---|
| Entry | R1 | R2 | Product | Yield [%]a |
| 1 | p-MeC6H4 | Ph (2a) | 5a | 78 |
| 2 | p-MeC6H4 | p-FC6H4 (2b) | 5b | 64 |
| 3 | p-MeC6H4 | p-MeOC6H4 (2c) | 5c | 65b |
| 4 | p-MeC6H4 | m,m-(MeO)2C6H3 (2d) | 5d | 65 |
| 5 | p-MeC6H4 | p-CF3C6H4 (2e) | 5e | 68 |
| 6 | p-MeOC6H4 | Ph | 5f | 85 |
| 7 | o-MeC6H4 | Ph | 5g | 73 |
| 8 | o-MeOC6H4 | Ph | 5h | 73 |
| 9 | p-MeC6H4 |
|
5i | 57 |
| 10c |
|
m,m-(MeO)2C6H3 | 5j | 55 |
| 11c | p-FC6H4 | 5k | 59 | |
| 12d |
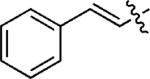
|
p-FC6H4 | 5l | 62 |
| 13d |
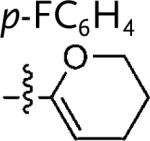
|
5m | 37 | |
Average yield of the isolated product in at least two experiments.
A 3:1 mixture of 1,2- and 1,1-diarylation products was formed.
The reaction was performed at 45 °C.
The reaction was performed at 40 °C.
Conspicuously absent from our discussion of the reaction scope so far are electron-poor styrene derivatives. The treatment of various electron-poor styrene derivatives with PhSnBu3 under the optimized conditions described above resulted in a mixture of 1,2- and 1,1-diarylation products (Figure 1). Of significance is a clear relationship between the electronic nature of the styrene substrate and the resulting ratio of 1,2- to 1,1-diarylation products, whereby the most electron poor substrate, with an NO2 substituent in the meta position, led to the lowest ratio of the 1,2- to the 1,1-diarylation product (1,2/1,1). When the Hammett s values were plotted against log(1,2/1,1), a linear free-energy relationship was observed with a ρ value of –0.88. This observation is consistent with the destabilization of the cationic π-benzyl palladium complex E by an electron-withdrawing group to enable β-hydride elimination and reinsertion of the coordinated alkene with formation of the more stable π-benzyl palladium complex H. In other words, the ratio is dependent on the relative rates of β-hydride elimination and transmetalation of the second equivalent of PhSnBu3.
Figure 1.

1,2-Diarylation versus 1,1-diarylation of styrenes and resulting Hammett analysis.
On the basis of these findings, we turned our attention towards terminal alkene substrates, as we believed that with these substrates β-hydride elimination and reinsertion would lead to a stable π-benzyl palladium complex. This complex could then undergo a second transmetalation and subsequent reductive elimination to yield the 1,1-diarylation product.[16] Indeed, the treatment of 1-nonene with several aryl stannanes yielded the 1,1-diarylation products exclusively (Scheme 2). We carried out several mechanistic experiments to explore this process further. When the isotopically labeled alkene [D2]6 was used as a substrate in the reaction with 2b, both deuterium atoms were conserved in the product. This result is consistent with the mechanistic proposal outlined above. Furthermore, no crossover was observed when [D2]6 and 1-undecene were used as substrate, which suggests that the coordinated alkene does not dissociate prior to formation of the 1,1-diarylation product.
Scheme 2.

1,1-Diarylation of terminal alkenes (a) and related mechanistic experiments (b).
In summary, we have disclosed a unique difunctionalization reaction of terminal alkenes, whereby conjugated alkenes undergo the 1,2-addition of organostannanes, and simple terminal alkenes undergo 1,1-addition. Two carbon–carbon bonds are formed in this transformation, which provides facile access to diaryl methine compounds, a common pharmacophore.[17] The outcome of the reaction is controlled both by the stability of the π-benzyl or π-allyl intermediate formed and by the unique cationic catalyst employed. Mechanistic experiments suggest that the regioselectivity of the reaction is determined by the relative rates of the second transmetalation versus β-hydride elimination. These concepts will guide the development of enantioselective variants of this transformation and new reactions in which two different groups can be added to a terminal alkene.
Footnotes
This research was supported by the National Institutes of Health (NIGMS RO1 GM3540). M.S.S. thanks the Dreyfus Foundation (Teacher-Scholar Award) and Pfizer for their support. We are grateful to Johnson Matthey for the gift of various palladium salts. We thank Keith Gligorich for initial experiments.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.200900218.
References
- 1.For reviews of the Heck reaction, see: Heck RF. Org. React. 1982;27:345–390.; Beletskaya IP, Cheprakov AV. Chem. Rev. 2000;100:3009–3066. doi: 10.1021/cr9903048.; Whitcombe NJ, Hii KK, Gibson SE. Tetrahedron. 2001;57:7449–7476.; Kondolff I, Doucet H, Santelli M. Tetrahedron Lett. 2003;44:8487–8491.
- 2.For examples of oxidative Heck reactions, see: Du X, Suguro M, Hirabayashi K, Mori A, Nishikata T, Hagiwara N, Kawata K, Okeda T, Wang HF, Fugami K, Kosugi M. Org. Lett. 2001;3:3313–3316. doi: 10.1021/ol016529y.; Parrish JP, Jung YC, Shin SI, Jung KW. J. Org. Chem. 2002;67:7127–7130. doi: 10.1021/jo020159p.; Jung YC, Mishra RK, Yoon CH, Jung KW. Org. Lett. 2003;5:2231–2234. doi: 10.1021/ol034458s.; Andappan MMS, Nilsson P, von Schenck H, Larhed M. J. Org. Chem. 2004;69:5212–5218. doi: 10.1021/jo049434t.; Yoo KS, Yoon CH, Jung KW. J. Am. Chem. Soc. 2006;128:16384–16393. doi: 10.1021/ja063710z.; Delcamp JH, Brucks AP, White MC. J. Am. Chem. Soc. 2008;130:11270–11271. doi: 10.1021/ja804120r.; for an example in which oxygen and base-free conditions are used, see: Ruan J, Li X, Saidi O, Xiao J. J. Am. Chem. Soc. 2008;130:2424–2425. doi: 10.1021/ja0782955.
- 3.Organ MG, Chass GA, Fang DC, Hopkinson AC, Valentea C. Synthesis. 2008:2776–2797. [Google Scholar]
- 4.For an overview of alkene difunctionalization reactions under palladium catalysis, see: Jensen KH, Sigman MS. Org. Biomol. Chem. 2008;6:4083–4088. doi: 10.1039/b813246a.; for examples of alkene diamination, see: Muniz K, Hovelmann CH, Streuff J. J. Am. Chem. Soc. 2008;130:763–773. doi: 10.1021/ja075041a.; Muniz K, Streuff J, Hovelmann CH, Nunez A. Angew. Chem. 2007;119:7255–7258. doi: 10.1002/anie.200702160.; Angew. Chem. Int. Ed. 2007;46:7125–7127. doi: 10.1002/anie.200702160.; for an example of aminooxygenation, see: Desai LV, Melanie SS. Angew. Chem. 2007;119:5839–5842.; Angew. Chem. Int. Ed. 2007;46:5737–5740. doi: 10.1002/anie.200701454.; for an example of aminoacetoxylation, see: Liu G, Stahl SS. J. Am. Chem. Soc. 2006;128:7179–7181. doi: 10.1021/ja061706h.
- 5.Kalyani D, Sanford MS. J. Am. Chem. Soc. 2008;130:2150–2151. doi: 10.1021/ja0782798. [DOI] [PubMed] [Google Scholar]
- 6.For examples of alkene difunctionalization reactions with norbornene-type compounds, see: Shaulis KM, Hoskin BL, Townsend JR, Goodson FE, Incarvito CD, Rheingold AL. J. Org. Chem. 2002;67:5860–5863. doi: 10.1021/jo016212b.; Fugami K, Hagiwara S, Oda H, Kosugi M. Synlett. 1998:477–478.; Oda H, Ito K, Kosugi M, Migita T. Chem. Lett. 1994:1443–1444.; for examples of alkene difunctionalization reactions in which the alkyl palladium intermediate can not undergo β-hydride elimination, see: Fretwell P, Grigg R, Sansano JM, Sridharan V, Sukirthalingam S, Wilson D, Redpath J. Tetrahedron. 2000;56:7525–7539.; Poli G, Giambastiani G, Heumann A. Tetrahedron. 2000;56:5959–5989.; Grigg R, Sridharan V. J. Organomet. Chem. 1999;576:65–87.; for examples of alkene difunctionalization reactions with allenes, see: Jeganmohan M, Cheng CH. Chem. Commun. 2008:3101–3117. doi: 10.1039/b800440d.; Jeganmohan M, Shanmugasundaram M, Cheng CH. Chem. Commun. 2003:1746–1747.; for an example of alkyne difunctionalization, see: Zhou C, Larock RC. J. Org. Chem. 2005;70:3765–3777. doi: 10.1021/jo048265+.
- 7.a Gligorich KM, Schultz MJ, Sigman MS. J. Am. Chem. Soc. 2006;128:2794–2795. doi: 10.1021/ja0585533. [DOI] [PubMed] [Google Scholar]; b Gligorich KM, Cummings SA, Sigman MS. J. Am. Chem. Soc. 2007;129:14193–14195. doi: 10.1021/ja076746f. [DOI] [PubMed] [Google Scholar]; c Podhajsky SM, Sigman MS. Organometallics. 2007;26:5680–5686. doi: 10.1021/om700675z. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Iwai Y, Gligorich KM, Sigman MS. Angew. Chem. 2008;120:3263–3266. doi: 10.1002/anie.200705317. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:3219–3222. doi: 10.1002/anie.200705317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a Schultz MJ, Sigman MS. J. Am. Chem. Soc. 2006;128:1460–1461. doi: 10.1021/ja0579053. [DOI] [PubMed] [Google Scholar]; b Zhang Y, Sigman MS. J. Am. Chem. Soc. 2007;129:3076–3077. doi: 10.1021/ja070263u. [DOI] [PubMed] [Google Scholar]
- 9.a Becker Y, Stille JK. J. Am. Chem. Soc. 1978;100:845–850. [Google Scholar]; b Lin YS, Yamamoto A. Organometallics. 1998;17:3466–3478. [Google Scholar]; c Lin YS, Yamamoto A. Bull. Chem. Soc. Jpn. 1998;71:723–734. [Google Scholar]; d Johns AM, Utsunomiya M, Incarvito CD, Hartwig JF. J. Am. Chem. Soc. 2006;128:1828–1839. doi: 10.1021/ja056003z. [DOI] [PubMed] [Google Scholar]; e Johns AM, Tye JW, Hartwig JF. J. Am. Chem. Soc. 2006;128:16010–16011. doi: 10.1021/ja067084h. [DOI] [PubMed] [Google Scholar]
- 10.a Jensen DR, Schultz MJ, Mueller JA, Sigman MS. Angew. Chem. 2003;115:3940–3943. doi: 10.1002/anie.200351997. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2003;42:3810–3813. doi: 10.1002/anie.200351997. [DOI] [PubMed] [Google Scholar]; b Sigman MS, Jensen DR. Acc. Chem. Res. 2006;39:221–229. doi: 10.1021/ar040243m. [DOI] [PubMed] [Google Scholar]
- 11.a Kantchev EAB, O'Brien CJ, Organ MG. Angew. Chem. 2007;119:2824–2870. [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:2768–2813. doi: 10.1002/anie.200601663. [DOI] [PubMed] [Google Scholar]; b Scott NM, Nolan SP. In: N-Heterocyclic Carbenes in Synthesis. Nolan SP, editor. Wiley-VCH; Weinheim: 2006. pp. 55–72. [Google Scholar]; c Viciu MS, Nolan SP. Topics in Organometallic Chemistry. Vol. 14. Springer; Berlin: 2005. pp. 241–278. [Google Scholar]
- 12.The hydroarylation product is thought to be formed from a palladium hydride generated from the Heck reaction. However, in some cases in Table 1, the yield of the hydroarylation product is greater than that of the Heck product. The additional palladium hydride may be formed by transmetalation of an n-butyl group from PhSnBu3, followed by β-hydride elimination.
- 13.Steinhoff BA, King AE, Stahl SS. J. Org. Chem. 2006;71:1861–1868. doi: 10.1021/jo052192s. [DOI] [PubMed] [Google Scholar]
- 14.For reviews of the Stille reaction and the effect of copper salts, see: Espinet P, Echavarren AM. Angew. Chem. 2004;116:4808–4839. doi: 10.1002/anie.200300638.; Angew. Chem. Int. Ed. 2004;43:4704–4734. doi: 10.1002/anie.200300638.; Farina V, Krishnamurthy V, Scott WJ. Org. React. 1997;50:1–652.
- 15.Löber O, Kawatsura M, Hartwig JF. J. Am. Chem. Soc. 2001;123:4366–4367. doi: 10.1021/ja005881o. [DOI] [PubMed] [Google Scholar]
- 16.For an example of 1,1-diarylation, see: Thiery E, Harakat D, Le Bras J, Muzart J. Organometallics. 2008;27:3996–4004.
- 17.For recent examples of biologically active diaryl methine derivatives, see: Moriconi A, Cesta MC, Cervellera MN, Aramini A, Coniglio S, Colagioia S, Beccari AR, Bizzarri C, Cavicchia MR, Locati M, Galliera E, Di Benedetto P, Vigilante P, Bertini R, Allegretti M. J. Med. Chem. 2007;50:3984–4002. doi: 10.1021/jm061469t.; Chen J-J, Chen P-H, Liao C-H, Huang S-Y, Chen I-S. J. Nat. Prod. 2007;70:1444–1448. doi: 10.1021/np070186g.; Liang H, Wu X, Yalowich JC, Hasinoff BB. Mol. Pharmacol. 2008;73:686–696. doi: 10.1124/mol.107.041624.