

Abstract
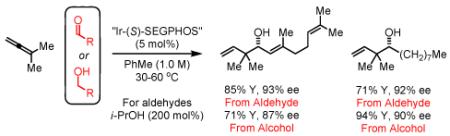
Enantioselective transfer hydrogenation of 1,1-dimethylallene 1a in the presence of aromatic, α,β-unsaturated or aliphatic aldehydes 2a-2i mediated by isopropanol and employing a cyclometallated iridium C,O-benzoate derived from allyl acetate, m-nitrobenzoic acid and (S)-SEGPHOS delivers products of reverse prenylation 4a-4i in good to excellent isolated yields (65-96%) and enantioselectivities (87-93% ee). In the absence of isopropanol, enantioselective carbonyl reverse prenylation is achieved directly from the alcohol oxidation level to furnish an equivalent set of adducts 4a-4i in good to excellent isolated yields (68-94%) and enantioselectivities (86-91% ee). Competition and isotopic labeling experiments suggest rapid alcohol-aldehyde redox equilibration in advance of carbonyl addition, and capture of the kinetically formed π-allyl complex at a rate faster than reversible β-hydride elimination-hydrometallation. This protocol represents an alternative to the use of allylboron reagents in enantioselective carbonyl reverse prenylation and represents the first use of allenes in enantioselective C-C bond forming transfer hydrogenation.
In the course of studies aimed at the development of C-C bond forming hydrogenations,1-6 highly enantioselective carbonyl allylation3d,e and crotylation3f employing allyl acetate and α-methyl allyl acetate as allyl and crotyl donors, respectively, were achieved under the conditions of iridium catalyzed transfer hydrogenation. For such reductive couplings, isopropanol is exploited as the hydrogen donor. Remarkably, primary alcohols may serve dually as hydrogen donors and aldehyde precursors, enabling carbonyl allylation or crotylation directly from the alcohol oxidation level.
With catalytic asymmetric methods for polyacetate and polypropionate construction in hand, an asymmetric prenylation protocol was sought. However, attempted use of allylic acetates as prenyl donors (2-methyl-3-butenyl acetate or 3-methyl-2-butenyl acetate) was unsuccessful. A potential solution to this problem resides in the use of 1,1-dimethylallene 1a as prenyl donor. However, in earlier studies on the iridium catalyzed reductive coupling of 1,1-dimethylallene 1a to aldehydes mediated by gaseous hydrogen, it was found that highly activated aldehydes were required and, despite an extensive assay of chiral phosphine ligands, only low levels of optical enrichment were observed.3a
Here, exploiting an ortho-cyclometallated iridium C,O-benzoate catalyst recently developed in our laboratory,3e we report an efficient protocol for enantioselective catalytic carbonyl reverse prenylation employing 1,1-dimethylallene 1a as the prenyl donor under transfer hydrogenation conditions.7 Unlike the related process employing elemental hydrogen,3a unactivated aldehydes couple efficiently. Additionally, the transfer hydrogenative protocol enables asymmetric carbonyl prenylation from the alcohol or aldehyde oxidation level, as demonstrated by the formation of identical sets of optically enriched adducts. This process constitutes an alternative to the use of stoichiometric metallic reagents in enantioselective carbonyl reverse prenylation8-12 and represents the first use of allenes in enantioselective C-C bond forming transfer hydrogenation.
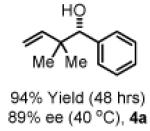
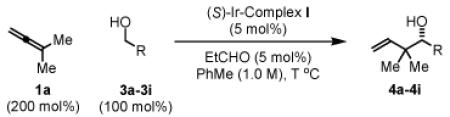
Initial studies focused on the coupling of 1,1-dimethylallene 1a to aldehyde 2a. After extensive study, which involved a nearly exhaustive assay of commercial chelating chiral phosphine ligands, optical results (4a, 94% yield, 89% ee, Table 1) were achieved upon exposure of a toluene solution of 1a and 2a to the cyclometallated iridium C,O-benzoate derived from allyl acetate, m-nitrobenzoic acid and (S)-SEGPHOS13 (eqn. 1), designated “(S)-Ir-Complex I,” in the presence of isopropanol (200 mol%). In contrast to related allyl acetate mediated allylations and crotylations, 1,1-dimethylallene 1a couples at relatively low temperature in the absence of basic additives.
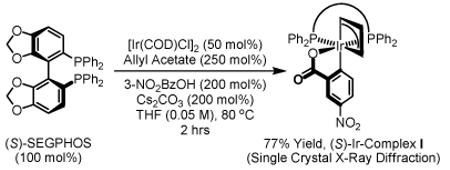 |
(eqn. 1) |
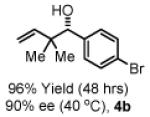
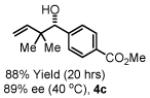
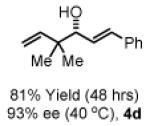
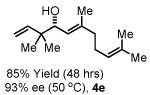
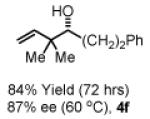
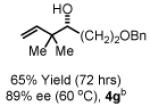
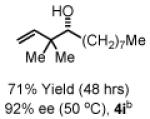
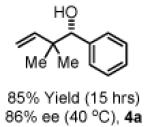
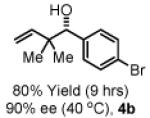
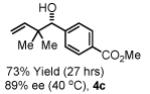
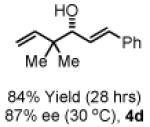
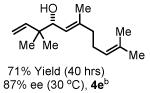
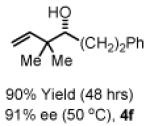
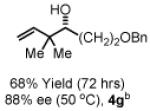
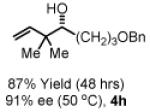
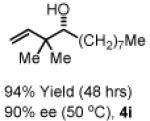
To explore the scope of this process, 1,1-dimethylallene 1a was coupled to a structurally diverse set of aldehydes 2a-2i (Table 1). Aromatic aldehydes 2a-2c, α,β-unsaturated aldehydes 2d and 2e, and aliphatic aldehydes 2f-2i are subject to reverse prenylation to furnish adducts 4a-4i in good to excellent isolated yields (65-96%) and enantioselectivities (87-93% ee). In the absence of isopropanol, enantioselective carbonyl reverse prenylation occurs directly from the alcohol oxidation level to furnish an identical set of adducts 4a-4i, once again with good to excellent isolated yields (68-94%) and enantioselectivities (86-91% ee). Especially noteworthy is the conversion of terpene 3e to sequiterpere 4e in the absence of any conventional preactivation of the reactants.
To gain further mechanistic insight, a crossover experiment was performed at 50 °C in the absence of isopropanol using the achiral cyclometallated iridium C,O-benzoate derived from allyl acetate, m-nitrobenzoic acid and BIPHEP. Exposure of 1,1-dimethylallene 1a to equimolar quantities of 2a and 3b under the aforementioned conditions provides 4a and 4b in 87% combined yield in a 1:1.2 ratio, respectively (eqn. 2, Scheme 1). Exposure of 1,1-dimethylallene 1a to equimolar quantities of 3a and 2b under otherwise identical conditions provides 4a and 4b in 95% yield in a 1:1.6 ratio, respectively (eqn. 3, Scheme 1). These experiments suggest rapid alcohol-aldehyde redox equilibration in advance of carbonyl addition.
Scheme 1. Isotopic labeling and competition experiments.a.

aYields are of material isolated by silica gel chromatography. See Supporting Information for detailed experimental procedures.
Isotopic labeling experiments also were performed at 50 °C using the achiral cyclometallated iridium C,O-benzoate derived from allyl acetate, m-nitrobenzoic acid and BIPHEP. Using d8-isopropanol as terminal reductant, aldehyde 2a is transformed to deuterio-4a, which incorporates deuterium at the interior vinylic position (97% 2H) and the terminal vinylic position (3% 2H) (eqn. 4, Scheme 1). Use of deuterio-3a in the absence of isopropanol delivers deuterio-4a, which incorporates deuterium at the interior vinylic position (95% 2H) and at the carbinol methine (>99% 2H) (eqn. 5, Scheme 1). High fidelity incorporation of deuterium suggests that capture of the kinetically formed π-allyl complex is faster than reversible β-hydride elimination-hydrometallation.
In summary, we report the first use of allenes in enantioselective C-C bond forming transfer hydrogenation, as demonstrated by the development of an effective protocol for carbonyl reverse prenylation from the alcohol or aldehyde oxidation level. Future studies will focus on related alcohol-unsaturate C-C couplings, including diastereo- and enantioselective carbonyl crotylations employing 1,3-butadiene.
Table 1.
Enantioselective carbonyl reverse prenylation from the aldehyde oxidation level.a
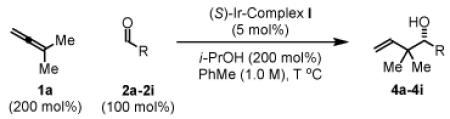 | ||
|---|---|---|
| 2a, R = Ph 2d, R = CH=CHPh 2g, R = (CH2)2OBn |
2b, R = p-BrPh 2e, R = Geranyl 2h, R = (CH2)3OBn |
2c, R = p-(CO2Me)Ph 2f, R = (CH2)2Ph 2i, R = (CH2)7Me |
|
|
|
|
|
|
|

|
|
Yields are of material isolated by silica gel chromatography. Enantiomeric excess was determined by chiral stationary phase HPLC analysis. Absolute stereochemistry was assigned by correlation of 4a, 4d and 4i to known compounds. See Supporting Information for details.
PhCl was used as solvent.
Table 2.
Enantioselective carbonyl reverse prenylation from the alchhol oxidation level.a
 | ||
|---|---|---|
| 3a, R = Ph 3d, R = CH=CHPh 3g, R = (CH2)2OBn |
3b, R = p-BrPh 3e, R = Geranyl 3h, R = (CH2)3OBn |
3c, R = p-(CO2Me)Ph 3f, R = (CH2)2Ph 3i, R = (CH2)7Me |
|
|
|
|
|
|
|
|
|
Yields are of material isolated by silica gel chromatography. Enantiomeric excess was determined by chiral stationary phase HPLC analysis. Absolute stereochemistry was assigned by correlation of 4a, 4d and 4i to known compounds. See Supporting Information for details.
PhCl was used as solvent.
Acknowledgments
Acknowledgment is made to the Robert A. Welch Foundation, the NIH-NIGMS (RO1-GM069445), the University of Texas Center for Green Chemistry and Catalysis, the ACS-GCI Pharmaceutical Roundtable and the Korea Research Foundation (KRF-2007-356-E00037) for partial support of this research. Dr. Oliver Briel of Umicore is thanked for the generous donation of [Ir(cod)Cl]2. Dr. Wataru Kuriyama and Dr. Yasunori Ino of Takasago are thanked for the generous donation of (S)SEGPHOS.
Footnotes
Supporting information available: Experimental procedures, HPLC data, and spectral data for new compounds. Single crystal X-ray diffraction data for (S)-Ir-Complex I. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.For selected reviews on C-C bond forming hydrogenation and transfer hydrogenation, see: Ngai M-Y, Kong JR, Krische MJ. J. Org. Chem. 2007;72:1063. doi: 10.1021/jo061895m. Skucas E, Ngai M-Y, Komanduri V, Krische M. J. Acc. Chem. Res. 2007;40:1394. doi: 10.1021/ar7001123. Bower JF, Kim IS, Patman RL, Krische M. J. Angew. Chem. Int. Ed. 2009;48:34. doi: 10.1002/anie.200802938.
- 2.For selected examples of enantioselective carbonyl and imine vinylation employing alkynes as vinyl donors under hydrogenation conditions, see: Kong J-R, Krische MJ. J. Am. Chem. Soc. 2006;128:16040. doi: 10.1021/ja0664786. Skucas E, Kong JR, Krische MJ. J. Am. Chem. Soc. 2007;129:7242. doi: 10.1021/ja0715896. Ngai M-Y, Barchuk A, Krische MJ. J. Am. Chem. Soc. 2007;129:12644. doi: 10.1021/ja075438e. Han SB, Kong J-R, Krische MJ. Org. Lett. 2008;10:4133. doi: 10.1021/ol8018874. Patman RL, Chaulagain MR, Williams VM, Krische MJ. J. Am. Chem. Soc. 2009;131:2066. doi: 10.1021/ja809456u.
- 3.For carbonyl allylation via iridium catalyzed hydrogenative and transfer hydrogenative coupling of dienes, allenes and allyl acetates, see: Skucas E, Bower JF, Krische MJ. J. Am. Chem. Soc. 2007;129:12678. doi: 10.1021/ja075971u. Bower JF, Skucas E, Patman RL, Krische MJ. J. Am. Chem. Soc. 2007;129:15134. doi: 10.1021/ja077389b. Bower JF, Patman RL, Krische MJ. Org. Lett. 2008;10:1033. doi: 10.1021/ol800159w. Kim IS, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2008;130:6340. doi: 10.1021/ja802001b. Kim IS, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2008;130:14891. doi: 10.1021/ja805722e. Kim IS, Han S-B, Krische MJ. J. Am. Chem. Soc. 2009;131:2514. doi: 10.1021/ja808857w.
- 4.For carbonyl allylation via ruthenium catalyzed transfer hydrogenative coupling of dienes and allenes, see: Shibahara F, Bower JF, Krische MJ. J. Am. Chem. Soc. 2008;130:6338. doi: 10.1021/ja801213x. Shibahara F, Bower JF, Krische MJ. J. Am. Chem. Soc. 2008;130:14120. doi: 10.1021/ja805356j. Ngai M-Y, Skucas E, Krische M. J. Org. Lett. 2008;10:2705. doi: 10.1021/ol800836v.
- 5.For carbonyl propargylation via ruthenium catalyzed transfer hydrogenative coupling of 1,3-enynes, see: Patman RL, Williams VM, Bower JF, Krische MJ. Angew. Chem. Int. Ed. 2008;47:5220. doi: 10.1002/anie.200801359.
- 6.For a recent review covering intermolecular aldol and Mannich addition via rhodium catalyzed hydrogenative coupling of enones, see: Han SB, Hassan A, Krische MJ. Synthesis. 2008:2669. doi: 10.1055/s-2008-1067220.
- 7.Palladium catalyzed reductive coupling of 1,1-dimethylallene to carbonyl compounds mediated by SnCl2 provides racemic products of reverse prenylation, see: Chang H-M, Cheng C-H. Org. Lett. 2000;2:3439. doi: 10.1021/ol0064563.
- 8.For enantioselective carbonyl reverse prenylation employing allylboron reagents, see: Brown HC, Jadhav PK. Tetrahedron Lett. 1984;25:1215. Jadhav PK, Bhat KS, Perumal T, Brown HC. J. Org. Chem. 1986;51:432.
- 9.For enantioselective carbonyl reverse prenylation employing allylindium reagents, see: Loh T-P, Zhou JR, Li X-R. Tetrahedron Lett. 1999;40:9333. Loh T-P, Zhou JR, Yin Z. Org. Lett. 1999;1:1855.
- 10.For enantioselective carbonyl reverse prenylation employing allylsilicon reagents, see: Nakajima M, Saito M, Shiro M, Hashimoto S-I. J. Am. Chem. Soc. 1998;120:6419. Nakajima M, Kotani S, Ishizuka T, Hashimoto S. Tetrahedron Lett. 2005;46:157. Denmark SE, Fu J. J. Am. Chem. Soc. 2001;123:9488. doi: 10.1021/ja016552e. Denmark SE, Fu J, Lawler MJ. J. Org. Chem. 2006;71:1523. doi: 10.1021/jo052203h.
- 11.For enantioselective carbonyl reverse prenylation employing allyltin reagents, see: Boldrini GP, Tagliavini E, Trombini C, Umani-Ronchi A. J. Chem. Soc., Chem. Commun. 1986 Boldrini GP, Lodi L, Tagliavini E, Tarasco C, Trombini C, Umani-Ronchi A. J. Org. Chem. 1987;52:5447.
- 12.For enantioselective carbonyl “α-prenylation,” see: Hong B-C, Hong J-H, Tsai Y-C. Angew. Chem. Int. Ed. 1998;37:468. doi: 10.1002/(SICI)1521-3773(19980302)37:4<468::AID-ANIE468>3.0.CO;2-Z. Cheng H-S, Loh T-P. J. Am. Chem. Soc. 2003;125:4990. doi: 10.1021/ja029700p.
- 13.Saito T, Yokozawa T, Ishizaki T, Moroi T, Sayo N, Miura T, Kumobayashi H. Adv. Synth. Catal. 2001;343:264. [Google Scholar]