

Summary
A prototype handheld, compact, rapid thermocycler was developed for multiplex analysis of nucleic acids in an inexpensive, portable configuration. Instead of the commonly used Peltier heating/cooling element, electric thin-film resistive heater and a miniature fan enable rapid heating and cooling of glass capillaries leading to a simple, low-cost Thin-Film Resistive Thermocycler (TFRT). Computer-based pulse width modulation control yields heating rates of 6–7 K/s and cooling rates of 5 K/s. The four capillaries are closely coupled to the heater, resulting in low power consumption. The energy required by a nominal PCR cycle (20 s at each temperature) was found to be 57 ± 2 J yielding an average power of approximately 1.0 W (not including the computer and the control system). Thus the device can be powered by a standard 9 V alkaline battery (or other 9 V power supply). The prototype TFRT was demonstrated (in a benchtop configuration) for detection of three important food pathogens (E. coli ETEC, Shigella dysenteriae, and Salmonella enterica). PCR amplicons were analyzed by gel electrophoresis. The 35 cycle PCR protocol using a single channel was completed in less then 18 min. Simple and efficient heating/cooling, low cost, rapid amplification, and low power consumption make the device suitable for portable DNA amplification applications including clinical point of care diagnostics and field use.
Keywords: PCR, Thermocycler, Peltier, Thin-Film Resistive Heater, Data acquisition system (DAS), Proportional-derivative-integral (PID), DNA, Food pathogens, Food testing, Point of Care (POC)
1. Introduction
Polymerase chain reaction (PCR) is a powerful method for DNA amplification (1), which is routinely used for detection and analysis of DNA. Many commercial thermocyclers employ a Peltier chip (2–4), which is a solid-state technology that can be used for heating or cooling. Peltier systems are widespread and provide a very effective design in conjunction with standard commercially available PCR tubes. Peltier technology provides symmetric control of both heating and cooling in a single component and thus represents a preferred design. However, Peltier chips are made of semiconductor material that tends to be fairly massive. Thus, Peltier systems are not a likely candidate for high-speed cycling. Furthermore, standard PCR tubes have a fairly long thermal time constant (on the order of 10 s) and thus any attempt to achieve rapid PCR must start with a redesign of the reaction vessel to minimize that time constant. Air heating and cooling (5) is an alternative that has gained attention as a technique for real-time PCR, where the reaction is performed in a glass capillary (5–7). The capillary configuration provides a large surface area for heat transfer and a small cross sectional area, both of which minimize the thermal time constant. Such systems allow rapid cycling down to about one cycle per minute where the time constant of the reaction vessel (on the order of 4 s) again limits the minimum cycling rate. Some recent designs have employed infrared heating (8). This configuration requires lenses and filters, and also requires positioning of the reaction mixture at the appropriate focal distance of the optical system. Continuous flow PCR (CF-PCR) microfluidic devices, which consist of a single channel that is continuously looped through different temperature zones to carry out DNA denaturing, annealing, and extension, have also been reported (9–11). The PCRs are pumped through the channel with the cycle number typically determined by the number of loops through each isothermal zone required for the PCR. Microfabrication enabled the development of an electrokinetically driven PCR in a circular continuous flow format through three isothermal zones (12) employing a 300–400 V/cm electric field for DNA circulation between the zones. Each of these designs is more complex than the one that came before, and thus they do not address our design requirements that include low-cost, low-power, and rapid cycling.
Power use and integration of PCR amplification with amplicon analysis are critical elements in the design of portable thermocyclers. Several approaches for amplicon detection and analysis were developed including fluorogenic, capillary electrophoretic (CE), and electrochemical detection. Fluorogenic analysis of PCR amplicons (13–18) eliminates the need of post-PCR analysis such as electrophoretic separation. The integration of fluorogenic detection during PCR amplification enables real-time monitoring (17–20). PCR amplification and capillary electrophoretic (CE) analysis were coupled in a compact instrument for microbial detection (21). A microfabricated capillary electrophoresis chip was integrated with electrochemical detection of PCR amplicons (22). A similarly integrated system consisting of a microfabricated silicon-based PCR (microPCR) chamber and a microfabricated electrophoretic glass chip has been developed (23). On-chip amplification using TaqMan PCR in nanoliter volumes on a highly integrated silicon microchamber array was reported (24).
Rapid and robust detection of infectious disease agents, including food borne pathogens, is critical for public health security. An important group of microbial pathogens includes pathogenic E. coli, Shigella dysenteriae, and Salmonella enterica, which are some of the most common causes of food borne infectious disease worldwide, associated with a variety of diseases, including diarrhea, urinary tract infections, bacteremia, meningitis, and wound infections. These pathogens are commensal bacterial organisms ubiquitous in mammalian intestines as well as in the environment. Rapid differentiation of nonpathogenic, commensal strains of E. coli from pathogenic variants (pathovars) is a major clinical and public health concern.
PCR-based tests for detecting microorganisms are increasingly being implemented in clinical laboratories (25). Such tests offer high sensitivity, specificity, and enable better characterization of the microorganisms. PCR amplification followed by microarray analysis of the amplicons was shown to be a powerful method for microbial analysis (26–35). A recent example of DNA amplification and hybridization performed in an integrated plastic device (36) required approximately 1.5–2 h to complete one assay. Recent portable PCR instruments enable microbial detection to be performed outside the laboratory and completed in a matter of minutes (25, 37). This technology may enable the development of Point of Care (POC) diagnostics, reducing the need for (38) the traditional central laboratory approach for medical diagnostics. POC allows speedy identification of infectious disease resulting in better treatment and reduction of indiscriminate use of antibiotics, a major factor in the emergence of antibiotic-resistant pathogenic microorganisms.
The need for portable devices for detection of microbial pathogens led to the development of a new class of mobile, small, battery-powered instruments to perform real-time PCR in the field (37, 39). This was made possible by replacing or redesigning energy consuming components, such as the heating block and the Peltier elements, with new miniature energy efficient components such as thin-film resistive heaters, fans, and integrated LED, and silicon photodiode detectors for real time analysis of the amplicons.
One of the barriers to the spread of rapid amplification technology for POC clinical use is the cost of the instrumentation, which limits the use of the technology. To make biosensor technology more accessible, recently a manuscript was published (38) describing an inexpensive array biosensor. Here we describe a simple and inexpensive battery-powered thin-film resistive thermocycler (TFRT), which has potential to be the base of a portable thermocycler for rapid detection of microbial pathogens in the field or in the clinic.
2. Materials
Film heater, Minco HK913H.
Cooling fan, 25 mm diameter, Radio Shack 273–240.
Solid state relay, Omega SSRDC100VDC12.
Data acquisition and control board, INET 100.
Power supply, 12 VDC, HY1803D Sinometer.
Borosilicate glass capillary, 15 mm × 1 mm OD × 0.75 mm ID.
Type T thermocouple wire, 30 AWG (0.25 mm diameter), Omega TT-T-30.
Bacterial strains: Strains used in this study are E. coli ETEC E1881C, Shigella dysenteriae, and Salmonella enterica serovar Typhimurium. These strains were obtained from the FDA Center for Food Safety and Applied Nutrition (CFSAN) bacterial collection of Ms. Christine Keys and Dr. Farukh Khambaty.
Ultraspec 3000 spectrophotometer (Pharmacia, Peapack NJ).
BLAST (Basic Local Alignment Search Tool), National Institutes of Health http://www.ncbi.nlm.nih.gov/BLAST/
Oligo Design software, http://www.enme.umd.edu/bioengineering/
BSA (bovine serum albumin), powder, A2153, Sigma Aldrich, St. Louis, MO.
Ethidium bromide, 10 mg/mL, E1510, Sigma Aldrich, St. Louis, MO.
PCR grade mineral oil, DNase free, M8662, Sigma Aldrich, St. Louis, MO.
Gen AMP PCR System 2400 thermocycler.
AgaroseBP160–100, Fisher BioReagents, http://www.fishersci.com
TBE buffer, BP1396–86, Fisher BioReagents, http://www.fishersci.com
EDAS 290 digital camera/stand, Kodak, Rochester NY.
dNTP mix, PR-U1511, Promega Corp., Madison, WI.
Taq DNA polymerase, Sigma Aldrich St. Louis MO.
Oligonucleotide primers, custom sequence, Operon, Huntsville, AL.
3. Methods
3.1. Thin-Film Resistive Heating Thermocycler
The primary physical characteristic that limits the cycling speed is the thermal capacitance of the heated region. To achieve high-speed thermocycling, we used a low mass capillary cartridge coupled to a thin-film resistive heater. Two CPU cooling fans were used for fast and efficient cooling of the PCR capillaries. The result is the rapid low power thin-film resistive thermocycler (TFRT) benchtop prototype shown in Fig. 1. Referring to Fig. 1, the basic elements of the TFRT prototype are: a thin-film resistive heater (A) and two miniature fans (B1 and B2) used for fast and efficient cooling of the PCR capillaries (D). The capillaries used for the PCRs are assembled on a thin (0.2 mm) microscope cover slip with face area 15 × 15 mm placed on a layer of aluminum foil (for more even heating) and arranged side-by-side on the flat surface placed directly on the heating surface. The capillary cassette assembly includes a fine gage Type T thermocouple (C) used for temperature control.
Fig. 1.

Prototype thin-film resistive heating PCR thermocycler (TFRT). (I) Overall TFRT design schematic. (II) Schematic and views of the TFRT capillary cartridge assembly. The device components are: A Thin-film resistive heater; B1 and B2 fans; C thermocouple; D PCR capillaries; E1 and E2 relays; F computer interface; G control software; H power source (batteries or power supply).
The capillary cassette was placed directly on the heating surface of the thin-film resistive heater to minimize thermal capacitance. A Plexiglas frame was used to hold the assembly and allow access of the cooling air flow from the fans to the capillary cassette.
The prototype TFRT instrument consists of a thin-film resistive heater, two fans, thermocouple, controller, software, and a computer. The heating element has an effective face area of 15 × 15 mm and a resistance of 15 Ω (Fig. 1A). The PCR capillaries are coupled directly to the heater. For cooling, two fans are positioned on opposite sides of the test section. The fans and the heating element are powered by 9 VDC (see Note 1). The system temperature is sensed by a thermocouple (see Notes 2 and 3) mounted inside a capillary and mounted on the heater in a symmetrical fashion to the capillaries that hold the PCR mixtures. For prototype development, thermocouples were mounted in each of the capillaries to map the temperature distribution on the surface of the heating element.
3.2. Measurement and Control Hardware
Figure 1A is a schematic of the temperature measurement and control circuit. A type T thermocouple (see Note 2), inserted in a glass capillary, measures the temperature of a capillary with similar characteristics to capillaries holding the reaction mixture. The thermocouple is connected to an A/D channel of the data acquisition system (DAS) in a differential configuration (see Note 3). The DAS system is also used to communicate output commands (D/A channels) from the computer to the thermocycler. The DAS system employed in this study has eight channels available that can be used as either inputs or outputs. The fans and the heater are connected to the output channels of the board via solid state relays.
The computer interface employs an off-the-shelf data acquisition system and two switching relays (Fig. 1E), one for the heater and one for the fans. The system was powered by a 9 V power supply or batteries.
24.3.3. Measurement and Control Software
The control system was written in VisualBasic to read the temperature of the PCR zone and adjust the heater and fans to follow a user-specified PCR temperature profile. The control system logic includes PID (proportional-derivative-integral) functionality (see Note 4). In addition, the logic provides features to anticipate over or under-shoot following the temperature transitions to maximize control fidelity. The control program, implemented on a dedicated PC computer, drives the heater and fans via the DAS. The heater control system is based on PWM (pulse width modulation) running at 0.5 Hz, which represents a compromise between the desire for a high frequency to provide better control and a low frequency to allow the computer to complete overhead tasks without disrupting control (see Note 5).
3.4. TFRT Performance
The temperature and the setpoint for a typical PCR run consisting of 40 cycles are shown in Fig. 2A. This plot shows the repeatability of the control system from cycle to cycle. An expanded view of a typical single cycle is shown in Fig. 2B where the heating and cooling rates can be determined.
Fig. 2.

Thermal performance of the prototype thin-film resistive heating thermocycler (TFRT). (A) Initial denaturation followed by 35 cycle run. (B) Data from a single cycle.
Rapid amplification was a key design goal for the TFRT system. For the case shown in Fig. 2, the time required to heat the test section from 54 to 72°C is 2.45 s corresponding to a heating rate of 7.3 K/s. For heating from 72 to 94°C the time is 3.60 s, corresponding to a heating rate of 6.1 K/s. For cooling from 94 to 54°C it takes 7.46 s corresponding to cooling rate of 5.4 K/s. The heating and cooling rates are functions of the device design and the control system settings. The largest slope of the temperature traces represents the maximum capability of the system in this configuration (the maximum slopes of both heating curves are similar). As the temperature approaches the setpoint, the control system reduces the power input to avoid overshoot (heating) or undershoot (cooling). The best performance of the system is 2 s to heat from 72 to 94°C, 2 s to heat from 54 to 72°C and cooling from 94 to 54°C in 5.5 s in a single capillary mode indicating a maximum heating rate of 9 K/s and a maximum cooling rate of 7.3 K/s.
A five capillary TFRT cassette was used for amplification of four targets simultaneously (one capillary holds the control thermocouple). Temperature uniformity, which is an essential element of PCR reproducibility, was measured in a simulated PCR run. For this test, the capillaries were filled with mineral oil to simulate the PCR mixture, and each capillary carried a thermocouple similar to the thermocouple used for temperature control. Figure 3A shows the simulated PCR run consisting of ten cycles. Data analysis suggests that, in general, the temperature among the capillaries is uniform within an error band: for the 72 and 54°C levels the temperature varied by less than ±1 K around the set point and for DNA melting, the temperature is 92°C ± 2 K. Insulating the cassette was found to reduce the variability among capillaries (not shown) but PCR was found to be repeatable at the indicated variability so the insulation was not included in this study. In PCR, the most critical temperatures are the annealing and polymerization, where significant deviation especially for the annealing temperature will substantially reduce the PCR yield. The melting temperature, in general, is more tolerant to a broader range of temperatures.
Fig. 3.

Temperature uniformity of five capillary cartridge. (A) Initial denaturation followed by ten cycle run. (B) Data from a single cycle.
3.5. System Energy Requirements
Low energy consumption is a critical design feature for operating a portable PCR instrument in the field. The energy consumption for the TFRT benchtop prototype (not including the computer and the control system) was measured at voltage inputs of 8, 9, and 10 V, and the energy consumed for heating, cooling, and for the relays was determined by integrating the power over time as the control system varied the input power.
The relays were found to dissipate only 16 mW at 0.5 A. This is negligible compared with the nominal 5 W heater power. The fans dissipate approximately 1 W each but they are on for only a brief time (approximately 4 s each cycle) so that the fan energy use is also secondary to the primary energy needs of the heating element. The pulse width control system turns the heater on at full power for a fraction of each control period with the size of the fraction modulated to maintain temperature control. Thus, to determine the energy use over a PCR cycle, it is sufficient to determine the heater on-time. At a fixed power supply voltage (V) and a fixed heater resistance (R) (the heater resistance does not measurably change over the temperature range of interest), the energy use is just the on-time multiplied by the power determined as V2/R. The energy required by a nominal PCR cycle (20 s at each temperature) was found to be 57 ± 2 J for all three power supply voltages. The control system adjusts the power input to only that needed to heat the system, which is independent of voltage. If the power is averaged over the cycle and the fan power is included, it yields an average power of 1.08 W.
The majority of TFRT tests were run at a power supply voltage of 9 V with the plan to utilize a 9 V battery in a portable version. Tests were also run using a battery and the system worked well as shown in Fig. 4. A standard 9 V alkaline battery is rated at approximately 4.5 W-h. On the basis of an average power of 1.08 W, this implies a battery life of 4.2 h which is consistent with what we observed. It is interesting to note that a typical lithium ion laptop battery has a rating on the order of 65 W-h, so long term battery operation of this PCR device is practical using existing battery technology, and it appears feasible to power the PCR device and the computer controller from the same battery.
Fig. 4.

TFRT performance using a 9 V alkaline battery.
3.6. Design of PCR Primers
Table 1 lists the primers used to amplify genes from three bacterial pathogens. Genes selected for the detection of bacteria were uid (E. coli), ipaH (Shigella), and pfkB-thrS intergenic region (Salmonella). The NCBI database was used to find and retrieve the gene DNA sequence from a complete genome of the target organism. That sequence was then used as a search sequence against the entire database (using BLAST) to gain some understanding of its uniqueness. The sequences with high similarity were then aligned using CLUSTAL W (40–42) to identify unique conserved regions. Primers were then designed, based on those regions using Oligo Design 1.621 software (43), and are listed in Table 1.
Table 1.
Primers for PCR amplification of UID (E. coli ETEC E1881C), ipaH (Shigella dysenteriae), and Salmonella specific region (Salmonella enterica serovar Typhimurium)
| Gene | Name | Sequence | Length | T (°C) | Amplicon size |
|---|---|---|---|---|---|
| uid | UID-F | TCACTGTAGCCATATGTCATGAGAG | 25 | 54.0 | 351 |
| UID-R | CCAGACTGAATGCCCACAGG | 20 | 54.8 | ||
| ipaH | IpaH-F | ACAATCTGATGTATCACAGATATGGCAT | 28 | 54.7 | 515 |
| IpaH-R | GCAGTGCGGAGGTCATTTGCTGT | 23 | 60.2 | ||
| pfkB-thrS region | Sal-F | TGTAGCGTTGGCTGTCCTCA | 20 | 55.6 | 534 |
| Sal-R | CAGCGTAATTTCCTTCACTTTTTGA | 25 | 52.9 | ||
Protocol (described for the uid gene in E. coli):
Search Pubmed for nucleotide sequences with the search terms “uid AND coli”. Numerous whole genomes are returned. Chose Escherichia coli HS (gi:157065147).
Once the Genbank sequence is displayed, search (using the broswer search feature) for “uid”. The uid region, including uidA and uidR, spans 2,793 bp and is chosen as the target.
Using the target sequence, run BLAST against the entire nucleotide database. The result is 17 closely identical matches with greater than 98% identity. Of these, 11 are E. coli complete genomes and 6 are Shigella complete genomes.
The unique E. coli sequences were then copied to a file for multiple alignment using Clustal. It was determined that the uid region was strongly conserved among all the sequences with only single point differences distributed among long stretches of identical sequence. These differences could be SNP’s or sequencing errors. In any case, they are not expected to be significant in choosing the primers.
The primers were chosen using Oligo Design, which has a search algorithm with multiple inputs. The primary inputs are the target melting temperature for the primers. In this case, a melting temperature of 54.5°C was selected resulting in the primer set in Table 1. The algorithm steps through the entire sequence and determines, at each location, if there is a sequence length that meets the input criteria (including melting temperature, length, hairpin potential, presence of repeats, 3′ end annealing strength, self annealing and primer dimer potential). It then steps through in the opposite direction. Candidate primer pairs that satisfy the amplicon length input specification are then displayed.
3.7. PCR Amplification of Individual Toxin Genes
The standard PCR mixture (50 μL) contained five units of Taq DNA polymerase, 1× PCR buffer supplemented with 3.5 mM MgCl2, 200 nM of forward and reverse primers, 400 mM each of dNTP (dATP, dGTP, dCTP, and dTTP), 1 mg/mL BSA (see Note 6), 0.2 μg/mL ethidium bromide, and 100 ng of DNA template (see Note 7). Typically, 3–5 μL of PCR mixture was loaded in each capillary. To prevent evaporation during thermal cycling, both ends of the capillaries were sealed with 1–2 μL of PCR-grade mineral oil. The control for the PCR amplification was performed using a Gene AMP PCR System 2400 thermocycler with the following cycle conditions: initial activation at 94°C for 5 min; 35–40 cycles at 94°C for 20 s, 54°C for 30 s, 72°C for 40 s; and the final extension at 72°C for 1 min. It is noted that the ramping times are not included in these specifications; when ramping times are included the total cycle time is 3.5 min resulting in a total time of 122 min for the control reaction (see Note 8). The nominal TFRT PCR runs were performed with the following conditions: 92°C – 40 s, 40 cycles of (92°C - 5 s, 54°C - 15 s, 71°C - 15 s), 71°C - 1 min. These specifications do include the ramping time such that the total time is 25 min. The cycle times were adjusted for longer and shorter total times, as described below.
The presence of amplified PCR products was detected in two ways. Ethidium bromide included in the reaction mixture allowed rapid determination of PCR products in the capillaries by visualization of the capillaries under UV illumination (see Note 9). After that determination, the reaction was passed through a 1.5% agarose gel by electrophoresis in 1× TBE buffer. Gels were stained with ethidium bromide and photographed in UV light with a Kodak digital camera EDAS 290.
Microbial DNA was amplified using the TFRT prototype PCR machine described above. Runs were made using a single capillary (Fig. 5) and with a cassette of five capillaries (Fig. 6). Single capillary amplification of the ipaH gene from Shigella dysenteriae was performed using the primers shown in Table 1. The gel results are shown in Fig. 5a along with the direct visualization in the capillaries in Fig. 5b. Lane 1 is a negative control with no target DNA. As a positive control, a reaction was performed in a conventional Peltier based thermocycler (Fig. 5, lane 5). The control reaction was run in a conventional thin-wall 200 μL PCR tube and was transferred to a capillary for visualization as shown in Fig. 5b. The remaining lanes (lanes 2–4) in Fig. 5 are PCRs run in the TFRT prototype in a single capillary mode with varying cycle time. The cycle times for lanes 2–4 were 37 min (20 s at 94°C, 20 s at 54°C, and 20 s at 72°C), 20 min (10, 12, 10 s), and 17.5 min (5, 12, 10 s), respectively.
Fig. 5.
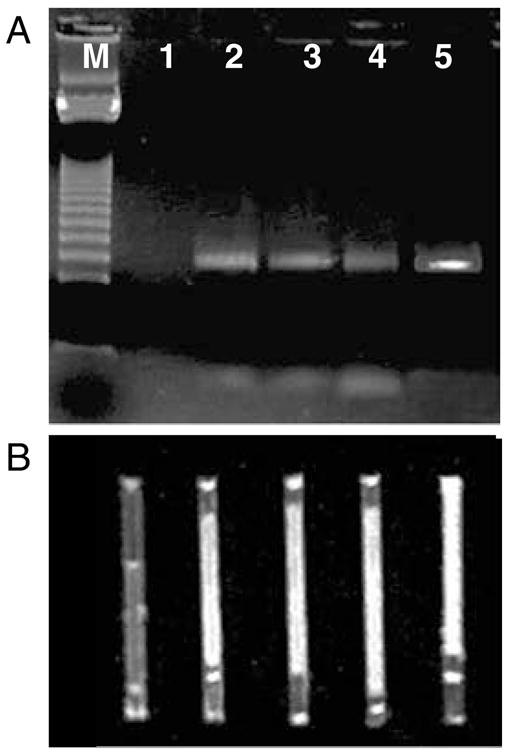
Amplification of Shigella dysenteriae DNA using the single channel TFRT. PCR amplification (35 cycles) of ipaH gene from Shigella dysenteriae on a conventional Peltier-based thermocycler and thin-film resistive thermocycler (TFRT) in a single-capillary mode. (A) gel, (B) capillaries. M – 123 bp DNA ladder (see Note 10), 1 – negative control, 2 – TFRT (37 min), 3 – TFRT (20 min), 4 – TFRT (17.5 min), 5 – Conventional thermocycler (122 min).
Fig. 6.
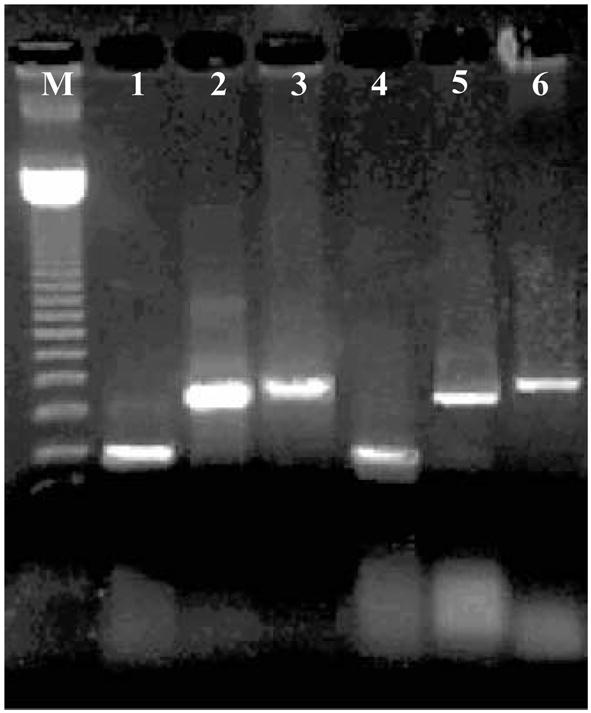
DNA amplification of three foodborne pathogens using TFRT. DNA amplified using Peltier-based thermocycler lanes 1–3 and DNA amplified using the prototype thin-film resistive thermocycler (TFRT) lanes 4–6. M – 123 bp DNA ladder (see Note 9), 1 – uid (E. coli ETEC E1881C), 2 – ipaH (Shigella dysenteriae), 3 – Salmonella specific region (Salmonella enterica serovar Typhimurium), 4, 5, 6 same on the TFRT thermocycler (total time of amplification (40 cycles) was 122 and 25 min for the Peltier and TFRT thermocyclers, respectively).
To enable multiple simultaneous PCRs, a TFRT cassette of five capillaries (four PCRs plus the temperature sensor) was designed and tested. Amplification using a Peltier-based thermocycler is compared with the TFRT results in Fig. 6. For both devices the thermal protocol was similar; initial denaturation at 92°C - 40 s followed by 40 cycles of 92°C - 5 s, 54°C - 15 s, 71°C - 15 s and final elongation of 71°C - 1 min. Although both thermocyclers used the same parameters, the ramping rate of the commercial instrument is slower resulting in slower amplification (see Note 8) Three PCRs were run simultaneously for detection of three microbial pathogen DNA sequences as shown in Table 1. The amplicon size range in these experiments is 349–720bp.
As shown in Fig. 6, all target sequences were amplified in the TFRT (lanes 4–6) with comparable signals to that obtained from the commercial cycler (lanes 1–3). However, the TFRT is significantly faster (25 min) than the amplification using the commercial instrument (80 min). These results demonstrate the potential use of the TFRT system for rapid identification of multiple microbial pathogens simultaneously.
Protocol:
Flush the capillaries with isopropyl alcohol, followed by DI water. After flushing, dry capillaries at 50°C for 30 min prior to use.
Load capillaries with PCR solution by dipping one end in the previously mixed solution. Capillary forces draw the solution into the capillary. The total reaction volume in the capillary was approximately 5 μL (11.3 mm of capillary length).
Load 1–2 mm of mineral oil into each end of the capillary to seal the reaction against evaporation. The oil can be drawn into the capillary in a manner similar to loading the solution (i.e., by capillary forces).
Lay the capillaries on the TFRT heater surface. For multiple capillaries, load the capillaries in a cassette frame prior to placing on the heater surface.
Run the thermocycler software to execute the PCR thermal cycles. The software allows the user to specify time and temperature for each portion of the cycle.
After the reaction is complete, detect the amplicon in one of two ways: (1) direct visualization of the fluorescence from the capillary, or (2) extract the PCR products from the capillary and run gel electrophoresis.
Acknowledgments
This work was supported in part by the FDA Office of Science and by USDA grant # 2003-35201-13784.
Footnotes
During testing, the system was powered by a 9 VDC power supply. The system was also tested using a standard 9 V battery. Although the battery was found to have sufficient energy to power an entire PCR run, a practical device would need a larger battery to provide longer operational life.
Thermocouples were fabricated from commercial thermocouple wire by stripping the insulation approximately 1 cm from the end. The two wires were then twisted together, doubled over, and soldered. After fabrication, the thermocouple was tested for electrical continuity and for temperature measurement fidelity. Thermocouples made from the same spool of wire were found to provide closely identical signals giving a relative temperature measurement error of less than ±0.05 K. Thermocouples were made from 0.25 mm diameter wire to allow insertion of the thermocouple in the capillaries. Because of the fine wire, all operations described in this note require considerable care to avoid breaking the wire.
Thermocouples were attached to the data acquisition board in a differential configuration with one lead grounded to the board. Because of the exposed nature of the thermocouple junction, care must be exercised to avoid electrical grounding of the junction, which can result in a ground loop that can cause significant temperature measurement errors (due to the low signal level of the thermocouple).
The control algorithm used is a modified proportional-integral-derivative (PID) algorithm. Because of the frequent setpoint changes associated with the three temperature levels in the PCR protocol, the algorithm is designed to anticipate these. In particular, the algorithm allows the user to achieve rapid heating while avoiding overshoot after temperature increases by setting an offset temperature difference. Prior to closing to within that difference of the setpoint, the heater is turned on full power. After passing the offset level, the proportional controller takes over. Once the temperature either passes the setpoint or plateaus, the integral action is turned on.
The control signal calculated by the PID algorithm (see Note 3) is translated into a heating profile by way of pulse width modulation. The heater receives a pulse train (frequency of 0.5 Hz) with variable on time. During the on segment of each 0.5 s pulse, the heater is on at full power. The integrated power over the full pulse is controlled to achieve the desired temperature. The thermal capacitance of the capillaries and the heater itself is sufficient to damp out any temperature fluctuations such that the temperature measured inside the capillaries is steady (does not show any 0.5 Hz ripple).
It was found (results not shown) that the addition of bovine serum albumen (BSA, 1 mg/mL) to the reaction significantly improved PCR yield. This is thought to be because BSA adsorbs to the glass preferentially and blocks adsorption of DNA and the polymerase on the capillary wall.
DNA from freshly grown cells was extracted using a phenol–chloroform extraction. The presence, concentration and purity of genomic DNA in the prepared samples were detected by measuring the absorbance at 260 and 280 nm using a spectrophotometer.
It is noted that, unlike the conventional thermocycler, the control system for the TFRT prototype includes the ramp time in the time at each temperature. Thus, although the system is set to a given temperature for a fixed time (e.g., 10 s for the rapid cycling case), the prototype takes as much as 7 s to make temperature transitions so the total time at the setpoint temperature is less than the value given in the protocol.
The PCR recipe included intercalating dye, ethidium bromide (0.2 μg/mL), to enable in-capillary detection of the amplification. Direct detection of PCR amplicons was achieved by placing the capillaries on a UV transilluminator (Fig. 5b), which enabled rapid analysis and demonstrated the potential of the platform for real-time detection.
On both Figs. 5 and 6, the image of the first two bars of the ladder are obscured such that the first easily visible bar is 369 bp. Thus, the gel results in Fig. 5 (ipaH amplicon is 515 bp) line up close to the second bar of the ladder which is 492 bp. Figure 6 has a similar interpretation. The ladder bars were obscured by the gel loading buffer dye.
References
- 1.Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, Mullis KB, Erlich HA. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. 1988;239(4839):487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- 2.Wittbrodt J, Erhardt W. An inexpensive and versatile computer-controlled PCR machine using a Peltier element as a thermoelectric heat pump. Trends Genet. 1989;5(7):202–203. doi: 10.1016/0168-9525(89)90081-4. [DOI] [PubMed] [Google Scholar]
- 3.Wilding P, Shoffner MA, Kricka LJ. PCR in a silicon microstructure. Clin Chem. 1994;40(9):1815–1818. [PubMed] [Google Scholar]
- 4.Khandurina J, McKnight TE, Jacobson SC, Waters LC, Foote RS, Ramsey JM. Integrated system for rapid PCR-based DNA analysis in microfluidic devices. Anal Chem. 2000;72(13):2995–3000. doi: 10.1021/ac991471a. [DOI] [PubMed] [Google Scholar]
- 5.Chapin K, Lauderdale TL. Evaluation of a rapid air thermal cycler for detection of Mycobacterium tuberculosis. J Clin Microbiol. 1997;35(8):2157–2159. doi: 10.1128/jcm.35.8.2157-2159.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedman NA, Meldrum DR. Capillary tube resistive thermal cycling. Anal Chem. 1998;70(14):2997–3002. doi: 10.1021/ac971303n. [DOI] [PubMed] [Google Scholar]
- 7.Lagally ET, Emrich CA, Mathies RA. Fully integrated PCR-capillary electrophoresis microsystem for DNA analysis. Lab Chip. 2001;1(2):102–107. doi: 10.1039/b109031n. [DOI] [PubMed] [Google Scholar]
- 8.Oda RP, Strausbauch MA, Huhmer AF, Borson N, Jurrens SR, Craighead J, Wettstein PJ, Eckloff B, Kline B, Landers JP. Infrared-mediated thermocycling for ultrafast polymerase chain reaction amplification of DNA. Anal Chem. 1998;70(20):4361–4368. doi: 10.1021/ac980452i. [DOI] [PubMed] [Google Scholar]
- 9.Kopp MU, Mello AJ, Manz A. Chemical amplification: continuous-flow PCR on a chip. Science. 1998;280(5366):1046–1048. doi: 10.1126/science.280.5366.1046. [DOI] [PubMed] [Google Scholar]
- 10.Park N, Kim S, Hahn JH. Cylindrical compact thermal-cycling device for continuous-flow polymerase chain reaction. Anal Chem. 2003;75(21):6029–6033. doi: 10.1021/ac0346959. [DOI] [PubMed] [Google Scholar]
- 11.Obeid PJ, Christopoulos TK, Crabtree HJ, Backhouse CJ. Microfabricated device for DNA and RNA amplification by continuous-flow polymerase chain reaction and reverse transcription-polymerase chain reaction with cycle number selection. Anal Chem. 2003;75(2):288–295. doi: 10.1021/ac0260239. [DOI] [PubMed] [Google Scholar]
- 12.Chen J, Wabuyele M, Chen H, Patterson D, Hupert M, Shadpour H, Nikitopoulos D, Soper SA. Electrokinetically synchronized polymerase chain reaction microchip fabricated in polycarbonate. Anal Chem. 2005;77(2):658–666. doi: 10.1021/ac048758e. [DOI] [PubMed] [Google Scholar]
- 13.Eggerding FA. A one-step coupled amplification and oligonucleotide ligation procedure for multiplex genetic typing. PCR Methods Appl. 1995;4(6):337–345. doi: 10.1101/gr.4.6.337. [DOI] [PubMed] [Google Scholar]
- 14.Tyagi S, Kramer FR. Molecular beacons: probes that fluoresce upon hybridization. Nat Biotechnol. 1996;14(3):303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 15.Tyagi S, Marras SA, Kramer FR. Wavelength-shifting molecular beacons. Nat Biotechnol. 2000;18(11):1191–1196. doi: 10.1038/81192. [DOI] [PubMed] [Google Scholar]
- 16.Nazarenko IA, Bhatnagar SK, Hohman RJ. A closed tube format for amplification and detection of DNA based on energy transfer. Nucleic Acids Res. 1997;25(12):2516–2521. doi: 10.1093/nar/25.12.2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ririe KM, Rasmussen RP, Wittwer CT. Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal Biochem. 1997;245(2):154–160. doi: 10.1006/abio.1996.9916. [DOI] [PubMed] [Google Scholar]
- 18.Wittwer CT, Ririe KM, Andrew RV, David DA, Gundry RA, Balis UJ. The LightCycler: a microvolume multisample fluorimeter with rapid temperature control. Biotechniques. 1997;22(1):176–181. doi: 10.2144/97221pf02. [DOI] [PubMed] [Google Scholar]
- 19.Northrup MA, Benett B, Hadley D, Landre P, Lehew S, Richards J, Stratton P. A miniature analytical instrument for nucleic acids based on micromachined silicon reaction chambers. Anal Chem. 1998;70(5):918–922. doi: 10.1021/ac970486a. [DOI] [PubMed] [Google Scholar]
- 20.Wittwer CT, Herrmann MG, Gundry CN, Elenitoba-Johnson KS. Real-time multiplex PCR assays. Methods. 2001;25(4):430–442. doi: 10.1006/meth.2001.1265. [DOI] [PubMed] [Google Scholar]
- 21.Lagally ET, Scherer JR, Blazej RG, Toriello NM, Diep BA, Ramchandani M, Sensabaugh GF, Riley LW, Mathies RA. Integrated portable genetic analysis microsystem for pathogen/infectious disease detection. Anal Chem. 2004;76(11):3162–3170. doi: 10.1021/ac035310p. [DOI] [PubMed] [Google Scholar]
- 22.Ertl P, Emrich CA, Singhal P, Mathies RA. Capillary electrophoresis chips with a sheath-flow supported electrochemical detection system. Anal Chem. 2004;76(13):3749–3755. doi: 10.1021/ac035282a. [DOI] [PubMed] [Google Scholar]
- 23.Rodriguez I, Lesaicherre M, Tie Y, Zou Q, Yu C, Singh J, Meng LT, Uppili S, Li SF, Gopalakrishnakone P, Selvanayagam ZE. Practical integration of polymerase chain reaction amplification and electrophoretic analysis in microfluidic devices for genetic analysis. Electrophoresis. 2003;24(1–2):172–178. doi: 10.1002/elps.200390010. [DOI] [PubMed] [Google Scholar]
- 24.Matsubara Y, Kerman K, Kobayashi M, Yamamura S, Morita Y, Takamura Y, Tamiya E. On-chip nanoliter-volume multiplex TaqMan polymerase chain reaction from a single copy based on counting fluorescence released microchambers. Anal Chem. 2004;76(21):6434–6439. doi: 10.1021/ac0497149. [DOI] [PubMed] [Google Scholar]
- 25.Tang YW, Procop GW, Persing DH. Molecular diagnostics of infectious diseases. Clin Chem. 1997;43(11):2021–2038. [PubMed] [Google Scholar]
- 26.Sergeev N, Distler M, Courtney S, Al-Khaldi SF, Volokhov D, Chizhikov V, Rasooly A. Multipathogen oligonucleotide microarray for environmental and biodefense applications. Biosens Bioelectron. 2004;20(4):684–698. doi: 10.1016/j.bios.2004.04.030. [DOI] [PubMed] [Google Scholar]
- 27.Al-Khaldi SF, Myers KM, Rasooly A, Chizhikov V. Genotyping of Clostridium perfringens toxins using multiple oligonucleotide microarray hybridization. Mol Cell Probes. 2004;18(6):359–367. doi: 10.1016/j.mcp.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 28.Volokhov D, Pomerantsev A, Kivovich V, Rasooly A, Chizhikov V. Identification of Bacillus anthracis by multiprobe microarray hybridization. Diagn Microbiol Infect Dis. 2004;49(3):163–171. doi: 10.1016/j.diagmicrobio.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 29.Johnson J, Jinneman K, Stelma G, Smith BG, Lye D, Messer J, Ulaszek J, Evsen L, Gendel S, Bennett RW, Swaminathan B, Pruckler J, Steigerwalt A, Kathariou S, Yildirim S, Volokhov D, Rasooly A, Chizhikov V, Wiedmann M, Fortes E, Duvall RE, Hitchins AD. Natural atypical Listeria innocua strains with Listeria monocytogenes pathogenicity island 1 genes. Appl Environ Microbiol. 2004;70(7):4256–4266. doi: 10.1128/AEM.70.7.4256-4266.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sergeev N, Volokhov D, Chizhikov V, Rasooly A. Simultaneous analysis of multiple staphylococcal enterotoxin genes by an oligonucleotide microarray assay. J Clin Microbiol. 2004;42(5):2134–2143. doi: 10.1128/JCM.42.5.2134-2143.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Volokhov D, Chizhikov V, Chumakov K, Rasooly A. Microarray analysis of erythromycin resistance determinants. J Appl Microbiol. 2003;95(4):787–798. doi: 10.1046/j.1365-2672.2003.02046.x. [DOI] [PubMed] [Google Scholar]
- 32.Volokhov D, Chizhikov V, Chumakov K, Rasooly A. Microarray-based identification of thermophilic Campylobacter jejuni, C. coli, C. lari, and C. upsaliensis. J Clin Microbiol. 2003;41(9):4071–4080. doi: 10.1128/JCM.41.9.4071-4080.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Volokhov D, Rasooly A, Chumakov K, Chizhikov V. Identification of Listeria species by microarray-based assay. J Clin Microbiol. 2002;40(12):4720–4728. doi: 10.1128/JCM.40.12.4720-4728.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Al-Khaldi SF, Martin SA, Rasooly A, Evans JD. DNA microarray technology used for studying foodborne pathogens and microbial habitats: minireview. J AOAC Int. 2002;85(4):906–910. [PubMed] [Google Scholar]
- 35.Chizhikov V, Rasooly A, Chumakov K, Levy DD. Microarray analysis of microbial virulence factors. Appl Environ Microbiol. 2001;67(7):3258–3263. doi: 10.1128/AEM.67.7.3258-3263.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y, Rauch CB, Stevens RL, Lenigk R, Yang J, Rhine DB, Grodzinski P. DNA amplification and hybridization assays in integrated plastic monolithic devices. Anal Chem. 2002;74(13):3063–3070. doi: 10.1021/ac020094q. [DOI] [PubMed] [Google Scholar]
- 37.Higgins JA, Nasarabadi S, Karns JS, Shelton DR, Cooper M, Gbakima A, Koopman RP. A handheld real time thermal cycler for bacterial pathogen detection. Biosens Bioelectron. 2003;18(9):1115–1123. doi: 10.1016/s0956-5663(02)00252-x. [DOI] [PubMed] [Google Scholar]
- 38.Golden J, Shriver-Lake L, Sapsford K, Ligler F. A “do-it-yourself” array biosensor. Methods. 2005;37(1):65–72. doi: 10.1016/j.ymeth.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 39.Belgrader P, Young S, Yuan B, Primeau M, Christel LA, Pourahmadi F, Northrup MA. A battery-powered notebook thermal cycler for rapid multiplex real-time PCR analysis. Anal Chem. 2001;73(2):286–289. doi: 10.1021/ac000905v. [DOI] [PubMed] [Google Scholar]
- 40.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31(13):3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25(24):4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herold KE, Rasooly A. Oligo design: a computer program for development of probes for oligonucleotide microarrays. Biotechniques. 2003;35(6):1216–1221. doi: 10.2144/03356bc02. [DOI] [PubMed] [Google Scholar]