

Abstract
A new competitive electrochemiluminescence-based immunoassay for the type-2 brevetoxins in oyster extracts was developed. The assay was verified by spiking known amounts of PbTx-3 into 80% methanol extracts of Gulf Coast oysters. We also provide preliminary data demonstrating that 100% acetone extracts, aqueous homogenates, and the clinical matrixes urine and serum can also be analyzed without significant matrix interferences. The assay offers the advantages of speed (≃2h analysis time); simplicity (only 2 additions, one incubation period, and no wash steps before analysis); low limit of quantitation (conservatively, 50 pg/mL = 1 ng/g tissue equivalents); and a stable, nonradioactive label. Due to the variety of brevetoxin metabolites present and the lack of certified reference standards for liquid chromatography–mass spectrometry confirmation, a true validation of brevetoxins in shellfish extracts is not possible at this time. However, our assay correlated well with another brevetoxin immunoassay currently in use in the United States. We believe this assay could be useful as a regulatory screening tool and could support pharmacokinetic studies in animals and clinical evaluation of neurotoxic shellfish poisoning victims.
Brevetoxins, neurotoxins produced by the dinoflagellate Karenia brevis (formerly Gymnodinium breve, Ptychodiscus brevis), can cause fish kills, human respiratory discomfort, and economic damage to commercial fisheries and tourist industries during coastal bloom conditions known as red tides. Ingestion of brevetoxin-contaminated shellfish causes neurotoxic shellfish poisoning (NSP), which results in a complex of symptoms including nausea, oral paresthesias, ataxia, myalgia, and fatigue. In severe cases, tachycardia, seizures, and loss of consciousness can occur. Although a fatal case of NSP has yet to be reported, this potential certainly exists for susceptible subpopulations in the absence of medical intervention. This may be especially true with small children (1). Aerosol exposure to brevetoxins follows fragmentation of algal cells in the surf and may cause respiratory irritation, coughing, and watery eyes, which rapidly resolve upon cessation of exposure (2). Good pharmacokinetic data in humans are lacking, and the human lethal dose is not known.
For many years, the mouse bioassay has remained the gold standard assay method for screening for brevetoxins in shellfish. Recently, other more sensitive assays have been evaluated as potential alternatives for the mouse bioassay. Immunoassays, such as the radioimmunoassay (RIA) and the enzyme-linked immunosorbent assay (ELISA), have consistently shown to be the most sensitive (3-5). However, the RIA suffers from the requirement for a radioactive label and, thus, the implications of radioactive waste disposal. The ELISA method of Naar et al. (4) avoids the complications of radioisotope use but requires a 2-step amplification procedure and a total assay time of 4 h. Another ELISA, developed in New Zealand by Garthwaite et al. (5), offers reasonable sensitivity but is not currently used for regulatory screening.
Receptor-based assays (1, 6) can also be used to screen for brevetoxins. These assays are based upon the pharmacologically relevant binding of toxin to the endogenous sodium channel receptor in rat brain membranes. While not as sensitive as immunoassays, they can nonetheless detect toxins well below the current action level of 80 μg/100 g shellfish. Unfortunately, they also suffer from the drawbacks of radioisotope use and the requirement of preparing membranes from animal tissues. However, an important characteristic of receptor-based assays is that they measure total pharmacological activity rather than the structural components recognized by immunoassays. Recent work (7-9) suggests a role of molluscan metabolites rather than parent brevetoxins as the primary causative agents in NSP. Initial experiments suggest that these metabolites bind to the receptor with affinities that may differ significantly from the parent molecules, yet are recognized equally by antibodies recognizing the parent molecules. This difference in activity may cause receptor-based assays to potentially underestimate the total toxin concentration in a sample compared to immunoassays when parent toxins are used to construct the standard curve. Until we have a better understanding of the metabolic fate of these molluscan metabolites in humans, it seems that immunoassays may offer the best primary screening assay for brevetoxins.
Liquid chromatography coupled to mass spectrometry (LC/MS) has also been developed as a viable screening tool in analyzing for brevetoxins and metabolites in shellfish (8). However, until purified reference standards become available for the major brevetoxin metabolites, quantitation by LC/MS will remain problematic.
We present here the development of a competitive immunoassay for PbTx-2-type brevetoxins based upon electrochemiluminescence (ECL) and a flow-through paramagnetic bead format. This format eliminates the need for radiolabeled reagents and offers similar sensitivity, greater simplicity, and reduced assay time as compared to the current ELISA.
Experimental
Apparatus
Concentrator.—Speed Vac Model SVC-100H (Savant Instruments, Holbrook, NY).
Solid-phase extraction (SPE) columns.—SepPak Plus C18, No. WAT020515 (Waters Corp., Milford, MA); Bond Elut, LRC-C18, 500 mg (Varian, Harbor City, CA); Strata X, 60 mg/3 mL (Phenomenex, Torrance, CA).
Liquid chromatograph.—Surveyor quaternary pump with autosampler (Thermo Finnigan, Franklin, MA).
Mass spectrometer.—LCQ Deca XP (quadrupole) with electrospray interface and Xcalibur software (Thermo Finnigan).
LC column.—Luna C18(2), 150 × 4.6 mm, 3 μ, No. 00F-4251-E0 (Phenomenex).
ECL detector.—BioVeris M-SERIES® M8 Analyzer (BioVeris, Inc., Gaithersburg, MD).
Microtiter plates.—Costar round bottom, polypropylene, 96-well, No. 4080 (Corning, Inc., Corning, NY).
Rotating mixer.—Lab Quake (J.T. Baker, Phillipsburg, NJ).
Tissue homogenizer.—TX3160A(Daigger, Vernon, IL).
Reagents
PbTx-3.—Center for Marine Science, University of North Carolina at Wilmington (Wilmington, NC).
Anti-PbTx antibodies.—Center for Marine Science, University of North Carolina at Wilmington.
EZ-Link Sulfo-NHS biotinylation kit.—No. 21420 (Pierce, Rockford, IL).
DynaBeads™ M270 streptavidin.—No. 653.03 (Dynal Biotech ASA, Oslo, Norway).
BV-TAG™ Plus amine.—No. 140013 (BioVeris, Inc.).
Serum control.—No. 14-402E (Cambrex Bio Science, Walkersville, MD).
Urine control.—No. U9631 (Sigma, St. Louis, MO).
Cyanogen bromide activated Sepharose 4B.—No. C9142 (Sigma).
Assay buffer.—Phosphate-buffered saline (PBS; 60 mM, pH 7.4) containing 0.1% Tween 20 and 1 mg/mL bovine serum albumin.
Toxin Standards
PbTx-3 was purchased as preweighed dry amounts in vacuum-sealed ampoules. A stock solution was prepared at 1 mg/mL in methanol and kept at −20°C. Appropriate dilutions were made into assay buffer to prepare standard curves at competitor concentrations of 0.0015–33 ng/mL.
Antibodies
Goat polyclonal antibrevetoxin serum was a gift of Daniel G. Baden, (University of North Carolina at Wilmington). Antibodies were affinity-purified at the United States Army Medical Research Institute of Infectious Diseases (USAMRIID) by passage over a column containing PbTx-3 covalently coupled to Sepharose 4B. After being washed with PBS to eliminate unbound material, the brevetoxin-specific antibodies were eluted with 0.1 M glycine, pH 3.5. The eluted fraction was then dialyzed extensively against distilled water to remove the glycine and lyophilized. The antibodies had been previously characterized with regards to cross-reactivity with other Type-2 brevetoxins sharing the same backbone structure (4, 10). The brevetoxins PbTx-3, PbTx-2, and PbTx-9 are all recognized equally; cross-reactivity with the PbTx-1 (Type-1) class of brevetoxins was minimal.
Antibody/Bead Preparation
Affinity-purified goat anti-PbTx antibodies were biotinylated using commercial reagents according to the manufacturer’s directions at a biotin/antibody molar ratio of 5:1. Biotinylated antibody (5 mg, lyophilized) was reconstituted with 1 mL streptavidin-coated magnetic beads, which had been prewashed 5 times with 1 mL assay buffer. The antibody/bead mixture was mixed for 30 min at room temperature on a rotating end-over-end mixer, then collected on a magnet and washed 5 times with 1 mL PBS. The washed beads were reconstituted with 1 mL assay buffer and stored at 4°C in the presence of 0.1% sodium azide. This preparation has been held in excess of 1 year with no apparent loss of activity.
Ruthenylated PbTx-3 (Ru-PbTx-3)
Ru-PbTx-3 was synthesized according to the scheme in Figure 1. PbTx-3 (100 μg, 0.11 μmol) was dissolved in 50 μL dried ethyl acetate. This solution was added to 16 mg (99 μmol) carbonyldiimidazole (CDI) in a nuclear magnetic resonance (NMR) tube. Dry dimethylformamide (400 μL) was added to the tube. The tube was flushed with argon, capped, and incubated at room temperature for 4 h with occasional shaking. Excess CDI was quenched by adding 1.8 μL (100 μmol) water to the reaction mixture, mixing briefly, and incubating for an additional 30 min at room temperature. The BV-TAG™ ruthenylating reagent (1.0 mg, 0.85 μmol) and diisopropylethylamine (1.5 μL, 8.6 μmol) were then added sequentially to the reaction mixture. The tube was mixed vigorously and then incubated at 80°C for 2.5 h and 60°C for an additional 16 h. The solvent was removed from the crude product by evaporation under reduced pressure in a SpeedVac concentrator. The crude product was suspended in about 200 μL water to which was added the minimum volume of ethanol necessary to dissolve the material.
Figure 1.

Synthesis of Ru-PbTx-3. Compound 1: PbTx-3. Compound 2: BV-TAG™ Plus NHS Ester. Compound 3: Ru-PbTx-3.
Two SepPak C18 reversed-phase chromatography cartridges were attached in series. The cartridges were washed with methanol and then equilibrated with water. The crude product solution was loaded on the SepPaks and washed with water, and the product was eluted with a discontinuous gradient of ethanol in water (5 mL each of 5, 10, 20, 30%, etc. solutions of ethanol in water). Fractions containing the desired product were identified by electrospray ionization (ESI)-MS. The product fractions were combined and concentrated to dryness on a SpeedVac, then stored in aliquots of 100 nmol at −20°C. The yield was approximately 90% as determined by absorbance measurements (the extinction coefficient for the ruthenium complex at 455 nm is 15 400 M/cm).
The product was characterized by reversed-phase LC/MS on an LCQ Deca system. Separation was carried out using a C18 column and isocratic elution conditions (45 mM of product gave one major peak at 455 nm). The positive electrospray mass spectrum of this peak gave a mass of 1049.4 amu, which is consistent with the expected [M/2]2+ parent ion of 1049.3 amu.
For use in the ECL assay, each aliquot was reconstituted with 210 μL methanol (MeOH) and stored at −20°C; they have retained activity for over 2 years.
Shellfish Extracts
Extracts were prepared from eastern oysters (Crassostrea virginica) collected from the Gulf of Mexico. Acetone extracts were prepared according to ref. 11. Oyster homogenates (1 g) were extracted twice with 2 mL acetone by sonication, mixing on a Vortex mixer, and centrifugation at 2800 × g for 5 min at 5°C. Supernatants were combined and evaporated under a stream of nitrogen. The dry residue was resolubilized in 2 mL of 80% MeOH and extracted twice with 2 mL of 95% n-hexane by mixing in a Vortex mixer. Phase separation was assisted by centrifugation, and the hexane phase was discarded. The MeOH phase was evaporated under nitrogen, resolubilized in 2 mL of 25% methanol, and cleaned up on LRC-C18 SPE columns. The columns were conditioned with 10 mL 100% MeOH, followed by LC grade water. The 25% MeOH phase containing the oyster extract was loaded onto the column, followed by 5 mL 25% MeOH as a rinse. The PbTx residues were eluted with 5 mL 100% MeOH into a glass test tube, evaporated under nitrogen, and resolubilized in 0.25 mL 100% MeOH by sonication and mixing in a Vortex mixer. The solution was then centrifuged, the supernatant was transferred to a small glass vial, and the sample was stored at −20°C until used.
MeOH (80%) extracts were prepared according to ref. 12. A 2 g portion of oyster homogenate was weighed into a 50 mL polypropylene centrifuge tube. Nine mL 80% MeOH was added, and the sample was mixed in a Vortex mixer for 30 s. The mixture was then heated at 60°C for 20 min in a dry heating block, then cooled for 5 min in an ice bath and centrifuged at 3000 μ g for 10 min. The supernatant was transferred to a clean tube, the pellet reextracted, and the supernatants combined. The combined extract was diluted to 20 mL with LC grade water and mixed. An 8 mL aliquot was transferred to a third tube, 10 mL n-hexane was added, and the tube was shaken vigorously and centrifuged for 10 min at 3000 × g. Five mL of the MeOH layer was transferred to a fourth tube, 15 mL water was added, and the tube was mixed gently. This extract solution was applied to a Strata-X SPE column that had been preconditioned with 9 mL each of 100% MeOH followed by 25% MeOH and fitted with a 15 mL reservoir. The column was washed with 4.5 mL 25% MeOH and eluted with 4.5 mL 100% MeOH. The eluate was diluted to 5 mL (0.1 g oyster tissue equiv/mL) for analysis.
Aqueous homogenates were prepared by homogenization of oysters in a blender with no added liquid. Aliquots (1 g) were transferred to 50 mL conical polypropylene centrifuge tubes, and PBS was added to bring the volume to 40 mL. Each sample was homogenized for 3 min at medium speed with a tissue homogenizer, and the solution was analyzed without further treatment.
Description of the Assay
The assay is formatted as a competitive displacement assay and performed in 96-well microtiter plates using BioVeris™ ECL technology. Biotinylated anti-PbTx antibodies are bound to streptavidin paramagnetic beads through the very stable avidin/biotin linkage. During the incubation phase, these antibodies bind the ruthenium-labeled brevetoxin (Ru-PbTx-3) from solution. During analysis, the sample enters the analyzer flow cell, where the beads are concentrated on a magnet and a continuous flow of buffer washes away unbound material. Then a voltage is applied, and light is emitted by the bound ruthenium label. In the presence of an unknown positive sample, unlabelled brevetoxin or brevetoxin metabolites compete with the Ru-PbTx-3 for antibody binding, reducing the ECL signal in a concentration-dependent manner. The M-SERIES® M8 analyzer can read 8 wells simultaneously and an entire 96-well plate in 15 min.
Assay Procedure
A fresh working solution of Ru-PbTx-3 (1 μL label stock in 50 mL assay buffer) was prepared daily. Toxin standards or unknown samples in matrix (40 μL) were added to 360 μL Ru-PbTx-3 working solution. This solution was divided into triplicate wells (100 μL/well) of 96-well microtiter plates, followed by 100 μL/well antibody/bead preparation (1 μL/mL in assay buffer). The plates were incubated 75–90 min at room temperature with shaking and then read.
Assay Calibration and Performance Checks
The assay was verified by spiking native brevetoxin directly into 80% methanol extracts of Gulf Coast oysters (Crassostrea virginica). This was done so that accuracy of the assay could be evaluated separately from the efficiency of the extraction technique. Control (nontoxic) oyster extract was spiked with the brevetoxin PbTx-3 to give final concentrations in the assay wells of 0.1, 0.5, and 5 ng/mL. Each plate contained a standard curve consisting of 10 points encompassing the range of 0.0015–33 ng/mL. In addition, 2 suitability standards of 0.05 and 10 ng/mL were added. These were spiked standards that acted as positive controls and defined the applicability of the assay; if these standards were not measured accurately to within ±25%, the assay was discarded and repeated. In addition, these standards were used as conservative estimates of the upper and lower limits of quantitation (LOQ). Finally, each of the 3 spiked “unknowns” described above was measured 4 times/assay plate, with each measurement being the mean of 3 triplicate wells. Each result was evaluated against the ±25% of the accuracy criteria, and the percent passing was noted. However, failures were not eliminated from the analysis. Two analysts performed 5 assays each over the course of 8 days.
After verification with 80% methanol extracts, the assay was then applied to spiked acetone extracts as well as human serum and human urine to assess performance in these matrixes.
Data Analysis
Curves were plotted and data were analyzed using GraphPad Prizm (Version 3.03; GraphPad Software, Inc., San Diego, CA). Curves were fitted to a sigmoidal dose response with a variable slope.
Results and Discussion
Standard Curves
A typical standard curve is shown in Figure 2. The mean slope and midpoint (IC50) of the 10 curves (2 analysts × 5 assays each) were reproducible and did not vary appreciably between analysts (0.97 ± 0.02 and 0.23 ± 0.01 ng/mL, respectively, for analyst No. 1 and 0.98 ± 0.02 and 0.25 ± 0.01 ng/mL for analyst No. 2). The working range of the assay, as defined by the suitability standards, was 0.05–10 ng/mL. Recoveries for the suitability standards were 92 ± 7.5% (lower limit) and 91 ± 7.5% (upper limit).
Figure 2.
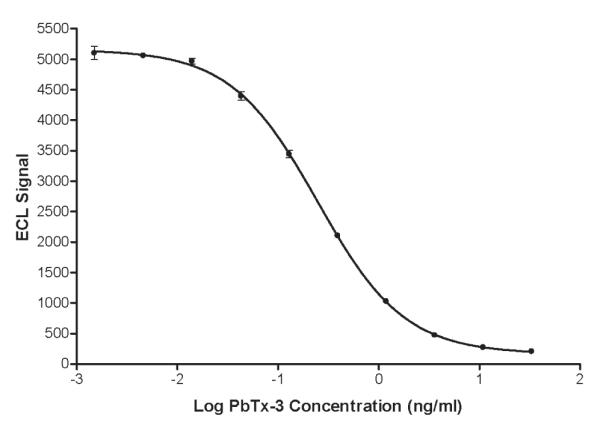
Standard curve for brevetoxin ECL assay. See Experimental section for methodology. Error bars span the range of triplicate determinations.
Accuracy and Repeatability
The combined results of all assays are shown in Table 1. The 0.1, 0.5, and 5.0 ng/mL standards were measured at 0.09, 0.52, and 4.6 ng/mL, respectively. The percentage of samples passing the acceptance criteria was 90% at 0.1 ng/mL, 97.5% at 0.5 ng/mL, and 100% at 5.0 ng/mL. Precision was also good, with relative standard deviation (RSD) ranging from 9.2 to 13.8%. Similar results were obtained when the data were broken down to evaluate the repeatability within the data sets of each analyst (data not shown). Analyst No. 1 had one assay failure, whereas analyst No. 2 had none. Measured concentrations and RSDs were very similar.
Table 1.
Combined assay repeatability results from 2 analysts performing 10 assays over 8 days
| PbTx-3 concn, ng/mL | Resulta | Pass, % | RSD, % |
|---|---|---|---|
| 0.1 | 0.09 (±0.01) | 90 | 13.8 |
| 0.5 | 0.52 (±0.05) | 97.5 | 9.2 |
| 5.0 | 4.6 (±0.45) | 100 | 10.0 |
Each concentration was determined 4 times in each assay (n = 40).
Measurements of intermediate precision are shown in Table 2. Variability between days and between analysts was generally less than that between assays, suggesting that interassay variability dominated the precision of the assay.
Table 2.
Measurements of intermediate precision expressed as RSD, %
| 0.1 ng/mLa | 0.5 ng/mLa | 5.0 ng/mLa | |
|---|---|---|---|
| Between analysts (n = 2) | 2.3% | 7.5% | 3.6% |
| Between assays (n = 10) | 12.7% | 8.8% | 8.3% |
| Between days (n = 8) | 11.9% | 7.3% | 9.0% |
Concentrations are those of PbTx-3 in the spiked “unknown” samples.
Assay Performance in Other Matrixes
Although the assay was verified in the 80% methanol extraction procedure developed in New Zealand, it was important to determine whether the assay would perform equally well in the 100% acetone extract typically used in the United States for metabolite characterization. We also wanted to know if the assay would perform satisfactorily in clinical matrixes that might support pharmacokinetic studies as well as medical confirmation of NSP. Thus, we compared samples of 80% methanol extract spiked with PbTx-3 to identical concentrations of PbTx-3 spiked into 100% acetone extract and 1:100, 1:20, and 1:2 dilutions of human urine and human serum. These results are shown in Figure 3. The acetone extract and the 1:100 and 1:20 dilutions of human serum and human urine resulted in values within 97–105% of the control, suggesting that the assay performed equally in these matrixes. The 1:2 dilutions of serum and urine showed slight matrix effects (22 and 10%, respectively). These effects were probably due to matrix components that slowed the binding process and can be eliminated by increasing the incubation time to 2 h(data not shown).
Figure 3.
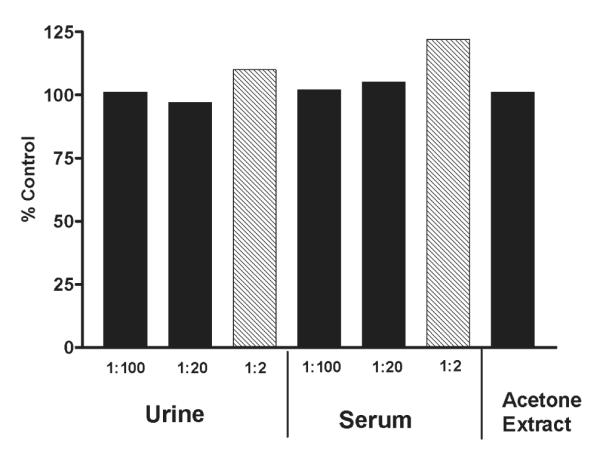
Assay performance in other matrixes. PbTx-3 (1.2 ng/mL, final concentration) was spiked into a 100% acetone extract of oysters, human urine, human serum, and a control (80% methanol) extract. Each extract was then analyzed in triplicate. The 1:2 dilutions of both serum and urine (shaded bars) indicated a small amount (22 and 10%, respectively) of matrix interference. All other samples were 97–105% of the control value.
Evaluation of Toxic Shellfish from a Naturally Occurring Red Tide Event
Verification of the assay with naturally occurring toxic shellfish is problematic due to the lack of validated assays with which to compare results. At present, the only validated assay for brevetoxins in shellfish is the mouse bioassay, which measures total combined toxicity of all parent and metabolite compounds in an ethyl ether extract after intraperitoneal injection. It measures pharmacological activity at the receptor site, as opposed to the structural components of the molecule recognized by specific antibodies. Thus, metabolites that share critical antigenic structural components but differ in potency will be measured differently in the 2 assays.
A competitive ELISA produced by the Center for Marine Science at the University of North Carolina at Wilmington (4) has shown great promise for detecting brevetoxins in shellfish homogenates as well as other matrixes. This assay is also antibody-based, and thus offers the best comparison to our assay. We tested and compared 3 samples of toxic oysters collected during a Gulf Coast red tide by testing them in both assays. Oyster samples were collected and extracted at the U.S. Food and Drug Administration, Dauphin Island, AL. Extracts of each sample were prepared in both 80% methanol and in 100% acetone. Extracts were shipped to the Florida Fish and Wildlife Conservation Commission, Fish and Wildlife Research Institute, St. Petersburg, FL, for testing in the brevetoxin ELISA, as well as to our laboratory for testing by the ECL assay. Results are shown in Table 3 and represented graphically in Figure 4. The correlation between the 2 assays was excellent (r2 = 0.997). The slope of the correlation line was 1.7. Given that these assays were performed on different days by different analysts in different laboratories, we believe the agreement to be quite good.
Table 3.
Comparison of 80% methanol, 100% acetone, and rapid aqueous extracts in the ECL assay performed in our laboratories and the brevetoxin ELISA performed by the Florida Fish and Wildlife Conservation Commission, Fish and Wildlife Research Institute
| Sample No.a | ECLb (n = 3) | ELISAb (n = 2) | |
|---|---|---|---|
| MeOH | 4231 | 0.5 (±0.03) | 0.6 (±0.01) |
| 0521 | 1.5 (±0.06) | 2.7 (±0.28) | |
| 0531 | 5.9 (±0.15) | 9.8 (±0.25) | |
| Acetone | 4231 | 0.3 (±0.06) | 0.5 (±0.04) |
| 0521 | 1.3 (±0.12) | 1.8 (±0.05) | |
| 0531 | 5.0 (±0.22) | 8.7 (±0.20) | |
| Aqueous | 4231 | 0.5 (±0.05) | NDc |
| 0521 | 2.0 (±0.1) | NDc | |
| 0531 | 9.6 (±0.9) | NDc |
Each sample was provided at 0.1 g eq/mL in the stated extract.
Results are expressed in μg/g shellfish (± standard deviation).
ND = Not detected.
Figure 4.
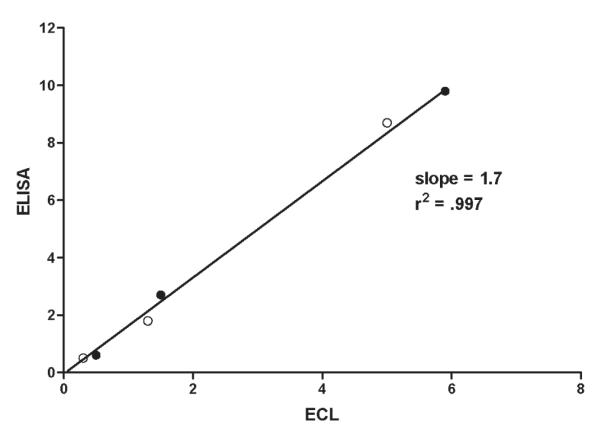
Correlation between ECL assay results and the ELISA assay results using the method of Naar et al. (ref. 4) on a common sample set. Open circles represent samples extracted with acetone and closed circles represent samples extracted with 80% methanol. See the text for method details.
Naar et al. (4) reported that their ELISA performed equally well on aqueous oyster homogenates and solvent extracts. This could be a distinct advantage, as it eliminates sample extraction and preparation time. We, therefore, compared our ECL assay to aqueous homogenates of the same positive oyster samples tested above. The results of that experiment confirmed the conclusions of Naar et al. (4). Our determinations were in good agreement with those of the ECL and ELISA (Table 3).
This suggests that screening of shellfish can proceed with only the much more rapid aqueous homogenation, a procedure that does not require a hexane wash step and SPE. However, more extensive comparisons are needed to confirm this. Positive samples requiring LC/MS confirmation will still likely require solvent extraction.
Conclusions
We describe here the development and initial suitability studies of a new competitive ECL-based immunoassay for the Type-2 brevetoxins in oyster extracts. The assay was verified by spiking known amounts of PbTx-3 into 80% methanol extracts of Gulf Coast oysters. Preliminary data suggest that acetone extracts, aqueous homogenates, and the clinical matrixes urine and serum can also be analyzed without significant matrix interferences. The assay offers the advantages of speed (≃2 h analysis time); simplicity (only 2 additions, one incubation period, and no wash steps before analysis); low LOQ (conservatively, 50 pg/mL = 1 ng/g tissue equivalents); and a stable, nonradioactive label. Due to the variety of brevetoxin metabolites present and the lack of certified reference standards for LC/MS confirmation, a true validation of brevetoxins in shellfish extracts is not possible at this time. However, when 3 naturally incurred samples of toxic oysters were extracted with either methanol or acetone, our data correlated well with another brevetoxin immunoassay currently in use in the United States.
We believe this assay offers the convenience, precision, and sensitivity to support regulatory testing of shellfish, pharmacokinetic studies in animals, and clinical laboratory research.
Footnotes
The views, opinions, and/or findings contained herein are those of the authors and should not be construed as an official Department of the Army position, policy, or decision unless so designated by other documentation.
References
- (1).Poli MA. In: Baden DG, editor. Proceedings of the Workshop Conference on Seafood Intoxications: Pan-American Implications of Natural Toxins in Seafood; Miami, FL: University of Miami; 1996. [Google Scholar]
- (2).Pierce RH. Toxicon. 1986;24:955–965. doi: 10.1016/0041-0101(86)90001-2. [DOI] [PubMed] [Google Scholar]
- (3).Poli MA, Hewetson JF. In: Tosteson TR, editor. Proceedings of the Third International Conference on Ciguatera; Quebec, Canada: Polyscience; 1992. pp. 115–127. [Google Scholar]
- (4).Naar J, Bourdelais A, Tomas C, Kubanek J, Whitney PL, Flewelling L, Steidinger K, Lancaster J, Baden DG. Environ. Health Perspect. 2002;110:179–185. doi: 10.1289/ehp.02110179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Garthwaite I, Ross K, Miles CO, Briggs LR, Towers NR. J. AOAC Int. 2001;84:1643–1648. [PubMed] [Google Scholar]
- (6).Van Dolah FM, Finley EL, Hanes BL, Doucette GJ, Moeller PD, Ramsdell JS. Nat. Toxins. 1994;2:189–196. doi: 10.1002/nt.2620020407. [DOI] [PubMed] [Google Scholar]
- (7).Poli MA, Musser SM, Dickey RW, Eilers PP, Hall S. Toxicon. 2000;38:981–993. doi: 10.1016/s0041-0101(99)00191-9. [DOI] [PubMed] [Google Scholar]
- (8).Wang Z, Plakas SM, El Said KR, Jester ELE, Granade HR, Dickey RW. Toxicon. 2004;43:455–465. doi: 10.1016/j.toxicon.2004.02.017. [DOI] [PubMed] [Google Scholar]
- (9).Ishida H, Nozawa A, Nukaya H, Tsuji K. Toxicon. 2004;43:779–789. doi: 10.1016/j.toxicon.2004.03.007. [DOI] [PubMed] [Google Scholar]
- (10).Melinek R, Rein KS, Schultz DR, Baden DG. Toxicon. 1994;32:883–890. doi: 10.1016/0041-0101(94)90367-0. [DOI] [PubMed] [Google Scholar]
- (11).Plakas SM, El Said KR, Jester ELE, Granade HR, Musser SM, Dickey RW. Toxicon. 2002;40:721–729. doi: 10.1016/s0041-0101(01)00267-7. [DOI] [PubMed] [Google Scholar]
- (12).McNabb P. Cawthron Report 899. Cawthron Institute; Nelson, New Zealand: 2004. [Google Scholar]