

Abstract
We describe the synthesis, structure and DNA incorporation of a class of novel aromatic C-deoxynucleosides in which benzenes and larger polycyclic aromatics serve as DNA base analogs. Novel approaches have been developed for glycosidic bond formation and for epimenzation of the anomeric substitutents to β-configuration, and we describe some of the properties of such compounds in DNA.
1. Introduction
Natural C-nucleosides and their synthetic analogs constitute an important class of organic compounds having significant therapeutic promise as antiviral, antitumor and anticancer agents.1 The discovery and synthesis of several naturally occurring ribofuranosyl C-nucleosides, such as pseudouridine, showdomycin, formycin, and pyrazomycin have appeared in the literature.2–5 Many of these compounds are active as antibiotics and antitumor agents. The remarkable biological activity of such C-nucleosides has prompted organic chemists to design and attempt the preparation of nonnatural C-nucleosides for the study of structure-activity relationships. Thus a large number of C-nucleosides containing substituted and unsubstituted heteroaromatics, such as pyrroles,6 furans and thiophenes,7 pyridines,8 quinolines,9 imidazoles,10 pyrazoles,11 and thiazoles12 have been synthesized. Many of them also have been found to possess a diverse range of biological activity. The interest in such nonnatural C-nucleosides has been further enhanced as a result of the possibility of their application in the understanding and creation of novel nucleic acid structures.13 in the study of RNA-protein interactions,14 and as spectroscopic probes for the elucidation of DNA dynamics.15
As part of our current research in the study of macromolecular recognition of RNA and DNA, we initiated a program involving the use of nonpolar C-nucleosides as specific biophysical probes of noncovalent interactions in double and triple helical structures of nucleic acids.16 In this account we describe our approaches to the synthesis of several nonnatural aromatic C-nucleosides, their structures, their incorporation by solid-phase synthesis into designed DNA strands, and some of their physicochemical properties.
2. Nonpolar isosteres of natural nucleosides
Noncovalent interactions play a crucial role in the structure-function relationship of biomolecules. To study noncovalent interactions involving DNA and RNA, one common approach has been the use of modifed analogs of the natural DNA nucleosides, in which hydrogen bonding groups are deleted or sterically blocked. We have pointed out,16a however, that many of these modifications not only block hydrogen bonding potential but also alter the steric size and shape of the base, which can undoubtedly affect noncovalent interactions as well. To address this problem we proposed the design of nonpolar isosteres of the four natural nucleobases. These compounds utilize substituted benzenes as pyrimidine analogs and indoles or benzimidazoles as purine analogs (Fig. 1). These compounds are the closest possible mimics of size and shape for the natural compounds, but lack any significant hydrogen bonding potential.16a,17
Figure 1.

Structures of nonpolar isosteres for the four natural DNA bases.
The earliest work on this project was aimed toward the synthesis and study of two thymidine analogs (1.2) and an analog of 2-aminodeoxyadenosine (3).16a The synthesis of the latter was straightforward: the glycosidic bond was formed by displacement of an α-chlorodeoxyribose derivative with the sodium salt of the dimethylindole. Because the synthesis of such indole and benzimidazole nucleosides is straightforward and well precedented in the literature, we will not focus on them further in this account. By contrast, C-nucleosides of deoxyribose are not as well known in the literature, and synthesis of the glycosidic bond to such aromatic hydrocarbons had few precedents when this work began (see below).

3. Simple aromatic C-nucleosides
In addition to nucleosides of benzenes substituted to mimic the steric arrangement of pyrimidine bases, we have also focused our attention on simpler C-nucleosides derived from unsubstituted aromatics, such as benzene, naphthalene, phenanthrene and pyrene for the study of the origin of base stacking interactions in nucleic acids and as potential reporter groups.18 Their structures (4–8) are shown below. The smallest members of the series (4,5) were previously described:19 however, the remaining compounds in the series (6–8) were all unknown, as were good methods for glycosidic bond formation in such compounds.

4. Previous methods used for C-nucleosides derived from benzenes
The development of novel methods for the synthesis of C-glycosides via C-C bond formation at the anomeric carbon has gained great interest in recent years.1 Methods involving nucleophilic attack by carbon nucleophiles on the electrophilic anomeric center are among the most explored and efficient strategies for the formation of such C-glycosides. However, procedures which could be applied to the synthesis of C-deoxynucleosides containing fully carbocyclic aromatics are scarce in the literature. Millican and coworkers19a achieved the synthesis of 1.2-dideoxy-1-phenyl-β-D-ribofuranose (4) from protected hemiacetal (9) via Grignard addition, followed by acid-catalyzed ring closure, fractional crystallization and base-induced deprotection in 20% overall yield (Fig. 2A). In a study of ribosyl analogs of chloramphenicol, a protein synthesis inhibitor, Klein and coworkers20 prepared β-D-ribofuranosylbenzene (11) from 2.3.5-tri-O-benzoyl-D-ribosyl chloride (10) by reaction with diphenylcadmium, followed by removal of the hydroxyl protecting groups in 20% overall yield (Fig.2B). The major product of this reaction was a sugar ketal which arose from attack of the metallophenyl species on the C-2' ester carbonyl. Matsumoto and coworkers21 accomplished the synthesis of β-D-ribofuranosyl derivative (13) of 2-naphthol by C-glycosylation of 2-naphthol with the furanosyl fluoride (12) in the presence of a catalytic amount of Cp2HfCl2-AgClO4 (Fig.2C). The α:β-anomeric ratio for this reaction was found to be 1:9. Unfortunately, while efficient, this last method can be applied only to phenol and naphthol derivatives.
Figure 2.

A–C Previous methods for glycosidic bond formation with benzene and naphthalene derivatives.
5. New approach to glycosidic bond formation
In our studies directed toward to the synthesis of nucleosides 1 and 2, we decided to investigate the reaction of organometallics with the α-Chlorosubstituted deoxyribose derivative 14, since the latter was readily available from 2-deoxyribose in good yields.22 Thus, when the Grignards derived from 5-bromo-2,4-difluorotoluene and 5-bromo-1,2,4-trimethylbenzene were reacted with chlorosugar 14 in THF at room temperature, the corresponding 3,5-di-O-protected α-C-nucleosides 15 and 16 were obtained in modest 23% and 22% yields (Fig.3).16a The low yields appeared to be due primarily to α,β-elimination of the chloride under the basic reaction conditions, and to a smaller extent, to the reaction of the Grignard reagents with the protecting group esters. The predominant formation of the α-isomers represents substitution with retention of configuration, and may be the result of a four-center transition state 17 at C-1 of the chlorosugar as suggested by Hurd and Miles23 in related pyranose systems, or by a dissociative mechanism with predominant alpha-face attack. Attempts to improve the yields of these coupling reactions with larger ratios or Grignard reagents to the chlorosugar were unsuccessful.
Figure 3.

Grignard-mediated synthesis leading to α-anomers of nucleosides 1 and 2.

6. Improvements with organocadmium and organozinc derivatives
In a study aimed at improving the yield of such coupling reactions we then considered less basic diarylcadmium, diarylzinc, and diarylmercury reagents.24 We found that both the cadmium and zinc reagents undergo facile reaction with the chlorosugar 14 to afford C-nucleosides 18 and 19 in good combined yields (61–83%) (Fig.4 and Table 1) on ~100–500 mg scales. Typically, finely ground, dry CdCl2 or ZnCl2 (1.2 equiv) is added to a preformed solution of aryl Grignard reagent (2.4 equiv) in 2 mL of anhydrous THF under dry nitrogen atmosphere. After heating the mixture under gentle reflux for 2–3 h. a solution of 14 (1.0 equiv) in THF is introduced at room temperature and the reaction is completed by heating the mixture for a further 2 h. Products are isolated by aqueous work-up and chromatographic separation.
Figure 4.

Arylcadmium-mediated glycosidic couplings giving C-deoxynucleosides.
Table 1.
Reactions of Chlorosugar 14 with various diarylcadmium species
| Entry | Ar | Yielda: | α-isomer 18 | β-isomer 19 | 20 | α/β |
|---|---|---|---|---|---|---|
| 1 | 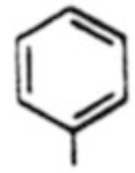 |
18a, 55% | 19a, 14% | -b | 3.9 | |
| 2 | 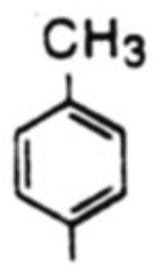 |
18b, 58% | 19b, 20% | -b | 2.9 | |
| 3 | 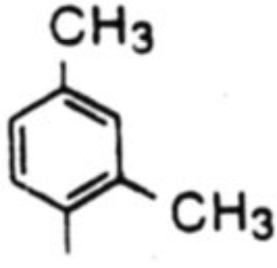 |
18c, 65% | 19c, 15% | -b | 4.3 | |
| 4 | 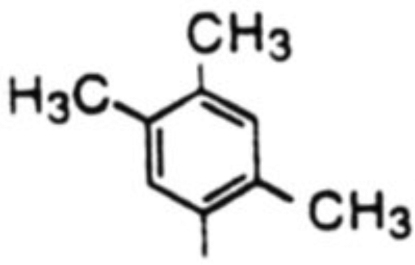 |
18d, 68% | 19d, 9% | -b | 7.6 | |
| 5 | 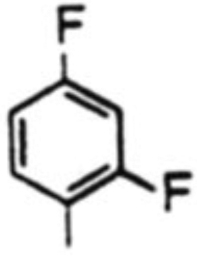 |
18e,c 9% | 19e, -b | 42% | - | |
| 18e,d 48% | 19e, 13% | 25% | 3.7 | |||
| 6 | 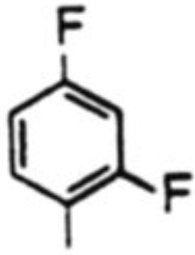 |
18f, 69% | 19f, 14% | -b | 4.9 | |
| 7 | 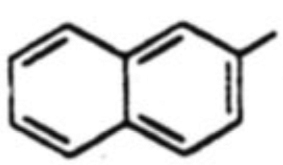 |
18g, 54% | 19g, 18% | -b | 3.0 |
Isolated yields
No product isolated
THF, reflux.
40 °C
As can be seen in Table 1, yields of the α-isomers 18a–d. 18f and 18g were consistently good to moderate (54–69%: entries 1–4 and 6, 7). The β-isomers 19a–d, 19f and 19g were obtained in 9–20% yields. The diastereoselectivities (α/β) were in the range of 2.9 : 1 to 7.6 : 1. In the case of entry 5C, reaction of the Grignard derived from l-bromo-2,4-difluorobenzene under the above conditions provided the α-isomer 18e in only 9% yield, the major product of this reaction being furfuryl p-toluate (20). However, when the reaction was performed at 40 °C, there was an increase in yield (61%, combined) of the C-nucleosides 18e and 19e, and the formation of 20 was significantly lowered (Entry 5d). As a control experiment, the reactivity of the chlorosugar 14 alone was examined and it was found that when heated under reflux in THF for 6 h, it underwent smooth dehydrochlorination followed by aromatization to furnish furfuryl p-toluate (20) in 70% isolated yield. It was thus concluded that the metallo-difluorobenzene species in Entry 5C is either less reactive or underwent a competing reaction resulting in its loss at THF/reflux, so that the chlorosugar 14 predominantly followed a heat-induced elimination reaction to provide 20 in 42% yield. Among various solvents, we found that ethereal solvents are well suited; THF was preferred because of better solubilities of the reactants in it.
Examination of several metallophenyl species24 indicated that while cadmium and zinc reagents worked equally well, the organomagnesium reagent provided only a moderate yield of 18 and 19. Lithium and mercury reagents do not produce the desired C-nucleosides; in these cases, only furfuryl p-toluate (20) was isolated.
7. Facile method for epimerization of α-C-deoxynucleosides to β-anomers
Improving the yields of this crucial C-C bond forming reaction from 20–25% to 61–83% was a major achievement for us. Although for some of our purposes the α-anomers of such aryl nucleosides are useful, more often it is the natural β-configuration that is desired. We therefore sought to develop an efficient method for the isomerization of the α-nucleosides to the β-configuration. To achieve this we took advantage of the benzylic nature of the C-1 center of the tetrahydrofuran ring. Acid catalyzed ring opening would generate a benzylic carbocation which might undergo ring closure by attack of the C-4 hydroxyl group under appropriate conditions to provide the thermodynamically more stable β-isomers. We indeed found that when heated with benzenesulfonic acid and a trace amount of water in refluxing xylene for ~2h, the α-isomers were smoothly converted to the corresponding β-isomers in good yields.18 A plausible mechanism for this acid-catalyzed isomerization is shown in Fig. 5.
Figure 5.

Proposed mechanism for acid-catalyzed isomerization of α- to β-C-deoxynucleosides.
This isomenzation appears quite general for this class of C-aryl deoxynucleosides. In all cases the major product formed is the desired β-anomer, and the ratios of β- to α-isomers after reaction range from 2.5:1 to 4:1 (see Table 2). Over longer reaction times some decomposition is evident, and thus yields are lowered somewhat in some cases. We found that separation of the isomers was occasionally difficult by column chromatography, and we commonly submitted the re-isolated α-isomer and mixed fractions to the isomenzation conditions again.18
Table 2.
Yields and anomeric ratios for acid-catalyzed isomerization
| compounda | aryl substitutent | beta isomer yieldb | beta / alpha ratio |
|---|---|---|---|
| 8 | 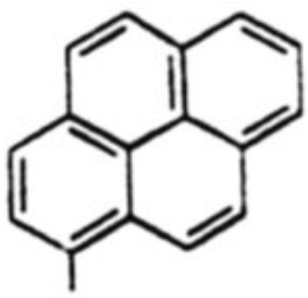 |
38 % | 3:1 |
| 7 | 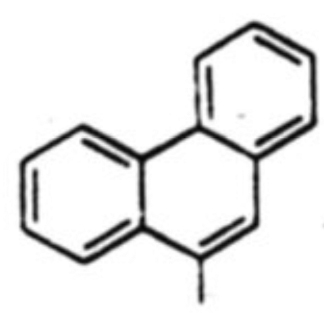 |
28 % | 3:1 |
| 5 | 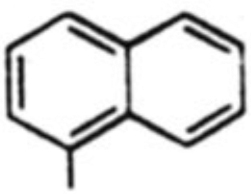 |
37 % | 2.5:1 |
| 6 | 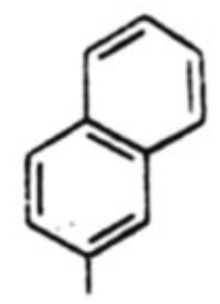 |
41 % | 2.5:1 |
| 2 | 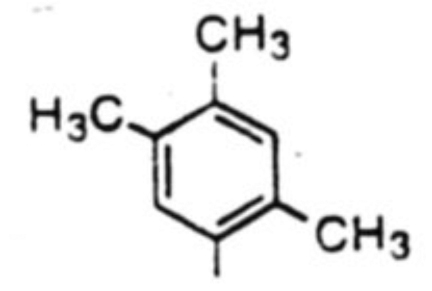 |
54 % | 4:1 |
| 1 | 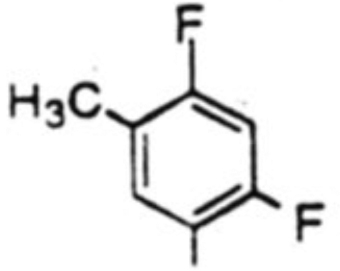 |
46 % | 3:1 |
Reactions were carned out on the bis-p-toluoyl ester derivatives
Isolated yields
8. Structural characterization
Structural characterization of α- and β-isomers was carried out by means of 1H NMR and NOE studies. In N-nucleoside compounds, the diagnostic tool routinely used for the determination of α/β anomeric configuration is based on the apparent multiplicity of the signal corresponding to the proton on C-1'.25 According to this rule, an α-isomer should feature an evenly spaced doublet of doublets for the 1'-H (J1'H-2'Hα - J1'H-2'Hβ), and a β-isomer a pseudo-triplet (J1'H-2'Hα = J1'H-2'Hβ). In C-nucleosides however, this rule does not always hold true.26 Quite the reverse: we have observed that a pseudo-triplet with J = 6.4–6.6 Hz for 1'-H was characteristic of the α-isomers, whereas for the β-isomers the same proton appeared as a doublet of doublets with J = 10.7–10.9 and 5.0 Hz (Table 3). Chemical shift values and splitting patterns of the 2'-protons were also quite diagnostic. In the case of the α-isomers, 2'-Hα (m) appeared upfield relative to 2'-Hβ (m). This trend was reversed in the case of the β-isomers; moreover, a doublet of doublets with J = 13.8–14.0 and 4.9–5.0 Hz was observed for the 2'-Hα. The 3'-H for the β-isomers appeared as a doublet with J = 5.8–6.0 Hz, whereas in α-isomers it appeared as a multiples. The 4'-H and 5',5"-H resonances appeared as two sets of complex multiplets in both α- and β-isomers (not shown); but their chemical shift values were generally reversed in these two classes of isomers.
Table 3.
Chemical Shift, Multiplicity and J values for α and β-C-Nucleosides
| Compound | 1'-H | 2'-Hα | 2'-Hβ | 3'-H |
|---|---|---|---|---|
| 18a | 5.4 t | 2.44-2.3 m | 3.02-2.92 m | 5.69-5.61 m |
| J = 6.6 | ||||
| 18b | 5.35 t | 2.38-2.28 m | 3.0-2.9 m | 5.67-5.59 m |
| J = 6.6 | ||||
| 18c | 5.5 t | 2.28-2.15 m | 3.03-2.96 m | 5.68-5.62 m |
| J = 6.6 | ||||
| 18d | 5.49 t | 2.3-2.16 m | 3.02-2.94 m | 5.66-5.61 m |
| J = 6.6 | ||||
| 18e | obscured | 2.38-2.27 m | 3.04-2.95 m | 5.68-5.58 m |
| 18f | 5.32 t | 2.36-2.26 m | 2.98-2.89 m | 5.63-5.59 m |
| J = 6.4 | ||||
| 18g | 5.59 t | 2.48-2.4 m | 3.1-3.0 m | 5.74-5.68 m |
| J = 6.6 | ||||
| 19a | 5.28 dd | 2.57 dd | 2.38-2.21 m | 5.63 d |
| J = 10.9, 5.0 | 13.8, 4.9 | 5.8 | ||
| 19b | 5.25 dd | 2.54 dd | 2.32-2.2 m | 5.63 d |
| J = 10.9. 5.0 | 13.8, 5.0 | 6.0 | ||
| 19c | 5.43 dd | 2.55 dd | 2.27-2.25 m | 5.63 d |
| J = 10.9, 5.0 | 13.8, 5.0 | 6.0 | ||
| 19d | 5.41 dd | 2.6-2.51 m | 2.3-2.15 m | 5.64 d |
| J = 10.9, 5.0 | 6.0 | |||
| 19e | 5.47 dd | 2.66 dd | 2.23-2.16 m | 5.63 d |
| J = 10.7, 5.0 | 14.0, 4.9 | 5.8 | ||
| 19f | 5.23 dd | 2.52 dd | 2.38-2.2 m | 5.63 d |
| J = 10.7, 5.0 | 14.0, 5.0 | 6.0 | ||
| 19g | 5.46 dd | 2.63 dd | 2.4-2.3 m | 5.63 d |
| J = 10.7, 5.0 | 13.8, 5.0 | 6.0 | ||
Proton NOE studies were conducted after deprotection of the toluoyl ester groups. Results of the NOE studies are shown in Table 4 for a pair isomers of 1.2-dideoxyribofuranosylbenzene. Irradiation of 1'-H for the α-isomer gave rise to NOE on Ph-H (7.5%), 3'-H (2.2%) and on 2'-Hβ (3.8%). For the β-isomer, irradiation of 1'-H gave NOE on Ph-H (12.1%), 4'-H (3.9%) and 2'-Hα (4.7%). Most convincing was the the irradiation of 2'-Hβ for the α-isomer where large NOE's were observed on both 1'-H (11.3%) and 3'-H (11.5%), in addition to 31.0% NOE on the geminal 2'-Hα. Irradiation of 2'-Hα had nominal effect on 1'-H (1.1%) and 3'-H (1.6%), but it had modest effect on 4'-H (3.2%) which is on the same face of the furanose ring as 2'-Hα. In the case of the β-isomer, irradiation of 2'-Hβ had a large effect on 3'-H (15.3%), but only a small effect on 1'-H (1.7%): irradiation of 2'-Hα showed a small effect on 3'-H (2.7%) but a large effect on 1'-H (14.4%). Similar results were obtained in all other aryl-C-deoxynucleoside cases as well.18
Table 4.
Proton NOE data for aromatic C-nucleosides derived from benzenea
| C-Nucleoside | Irradiation | Observed NOE (%) |
|---|---|---|
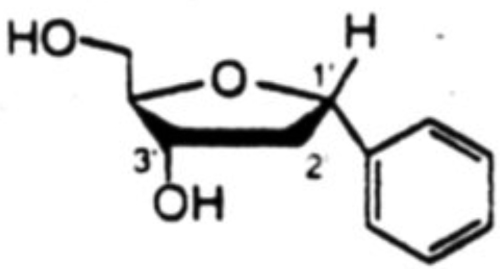 |
1'-H | Ph-H(7.5), 3'-H(2.2), 2'-Hβ(3.8) |
| 2'-Hβ | 1'-H(11.3), 3'-H(11.5), 2'-Hα(31.0) | |
| 2'-Hα | Ph-H(6.8), 1'-H(1.1), 3'-H(1.6), 4'-H(3.2), 2'-Hβ(24.7) | |
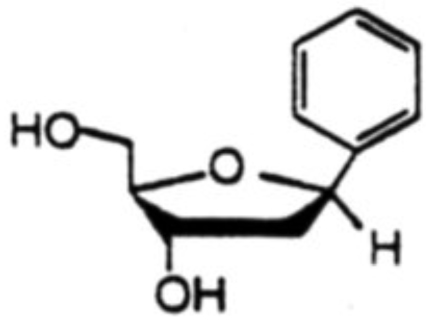 |
1'-H | Ph-H(12.1), 4'-H(3.9), 2'-Hα(4.7) |
| 2'-Hα | 1'-H(14.4), 3'-H(2.7), 2'-Hb(19.0) | |
| 2'-Hβ | Ph-H(6.9), 1'-H(1.7), 3'-H(15.3), 2'-Hα(28.5) |
Measured in degassed methanol-d4 at ambient temperature
9. Ring conformations of α- and β-anomers
The distinct differences in the 1H NMR splitting patterns of individual proton sets of α- and β-C-nucleosides discussed above are indicative of their existence in different preferred conformations. Because of the conformational flexibility of five membered rings in solution, deduction of stereochemical relationships between vicinal hydrogens from observed 3JH.H coupling constants is not straightforward. It has been argued that theoretically, a furanose ring can assume twenty different envelope and twist conformations.27 From X-ray diffraction studies28–30 of nucleosides and nucleotides however, it has become apparent that the five membered furanose ring in N-nucleosides occurs in two ranges of conformations, usually referred to as C2'-endo-C3'-exo (2T3 or S-type) and C3'-endo-C2'-exo (3T2 or N-type) forms. In the present C-nucleosides the situation is further complicated by the α/β anomery of the C-1' center.
X-ray crystallographic studies carried out on a relevant deoxynucleoside. 3,5-di-O-benzoyl-1,2-dideoxy-1-phenyl-β-D-ribo-furanose,30 show a 2T3 (S-type) conformation with C2'-endo-C3'-exo pucker of the ribose sugar ring. By analogy to these findings, it is possible that all aryl β-C-nucleosides in the present work (including the phenyl deoxynucleoside 4) have similar 2T3 conformations, since the coupling constants (and also structures and electronic distributions) are all very similar (Table 3).31 The coupling constants for the α-anomer series, however, indicate that they have conformations different from those of the β-series. We obtained the structure of the α-1-naphthyl nucleoside by x-ray crystallographic analysis (Fig. 6), and this sheds light on the issue. This structure has a different (although related) ring conformation than that of the β-benzene nucleoside described above. The naphthyl furanose ring has the C3'-exo (T3) conformation, which also falls into the S-type family, but which has different H1'-H2' dihedral angles than the 2T3 conformation, thus giving different splitting patterns. By analogy to this crystal structure, we propose that the entire α-anomeric series discussed herein falls into the C3'-exo conformational class. It should be noted that we observe small changes in coupling constants for toluoyl-protected and deprotected nucleosides, and so the comparison between the two crystal structures above is not completely analogous.
Figure 6.

Structure of 1'-α-1-naphthyl-2-deoxy-D-ribose as determined by x-ray crystallography.
10. The anomeric effect and deoxyribose conformation
The anomeric effect of N-nucleosides has very strong influence on ribose geometry.32 This phenomenon evidently results from the interaction of the dipoles operating in the C1'-O and C1'-N bonds, which tend to become oriented in the opposite directions. The C1'-N bond in the most stable conformation of N-nucleosides tends to occupy a quasi-axial position, thus moving out of the average plane of the furanose ring. Such an orientation would place one of the two lone pair containing orbitals of the ring oxygen in an approximately trans-diaxial disposition with the C1'-N bond. This also permits a favorable interaction between the oxygen lone pair and the antibonding σ* orbital of the C1'-N bond. In C-nucleosides, however, this effect is subdued because of the absence of a strong dipole in the C1'-C bond. Therefore the predominant conformation in a C-nucleoside appears to be the one in which bulky substituents are staggered as much as possible, and assume quasi-equatorial positions on the most staggered carbon atoms. The substituents therefore, tend to move into the plane of the furanose ring. A dramatic example of the anomeric effect is seen in the comparison of the anomers of the natural N-nucleoside thymidine and its isosteric analog, the difluorotoluene nucleoside 1 (see Fig.7). We studied the H1'-H2' coupling constants for the two anomers of the isostere18 and compared them to literature values for α- and β-thymidine.33 In the literature it is observed that β-anomers of the natural deoxynucleosides give splitting patterns for H-1' which resemble a triplet, meaning the two coupling constants are similar. Interestingly, we observe that it is the opposite (α)-anomer of the isostere which appears as a triplet. Conversely, the splitting pattern of the α-anomer of thymidine resembles the β-anomer of the isostere. Thus, although the bases are almost perfectly isosteric, the C-1' nitrogen in the N-nucleoside causes a significantly different ring conformation than that for C-nucleoside 1.
Figure 7.

H1' – H2' coupling constants for thymidine and its isostere (C-nucleoside 1). The splitting pattern for the N-nucleoside is found to be similar to that of the opposite anomer of the C-nucleoside. The anomeric effect causes an inverse relationship by influencing ring geometry.
11. Nucleosides derived from larger polycyclic aromatic hydrocarbons
The C-deoxynucleosides derived from polycyclic aromatic hydrocarbons naphthalene, phenanthrene, and pyrene (5 – 8 above) were synthesized using the same cadmium-mediated glycosidic coupling method described above.18 Isolated yields for the major isomers (the α-anomers) after coupling ranged from 31 – 52%. These were then epimerized to the desired β-configuration by our acid-catalyzed equilibration, as tabulated in Table 2. In the case of the pyrene nucleoside we also carried on the α-anomer since we felt it might have fluorescence properties as interesting as those of the β-anomer (see below).
We chose to synthesize these compounds chiefly because of our interest in aromatic stacking, especially as it applies to DNA. Some researchers have cited a hydrophobic contribution to DNA base stacking in aqueous solution, and so we felt that a series of simple hydrocarbons standing in as "bases" would serve as a test of how increasing hydrophobicity might effect the energetics of stacking. To study stacking we then carried these compounds on with the aim of incorporating them into synthetic DNA oligonucleotides.18
12. Incorporation into DNA
The 3.5-di-O-protected C-nucleosides 1–334 and 5–818 were deprotected by reaction with 0.1M NaOMe/MeOH at room temperature for 1 h (Fig.8). These products were converted to 5'-O-dimethoxytrityl derivatives in 60–75% isolated yields by reaction with 4,4'-dimethoxytrityl chloride and N,N-diisopropylethylamine in CH2Cl2 under gentle reflux for 3–4 h. For incorporation into DNA by solid-phase procedure, these DMT derivatives were transformed into 3'-O-N,N-diisopropyl-2-cyanoethyl phosphoramidites by standard literature procedures. Each phosphoramidite coupled with >95% efficiency as determined by the released trityl cation. High resolution NMR and reversed-phase HPLC were employed to characterize these synthetic DNA's.
Figure 8.

Scheme for conversion of protected nucleosides to phosphoramidite derivatives for incoporation into DNA by automated solid-phase synthesis.
13. Properties in DNA
Since the successful development of synthetic approaches to these C-nucleosides, we have begun to utilize them in studies of noncovalent interactions in DNA. In an examination of the importance of hydrogen bonding to the pairing of bases in DNA, we inserted single-base andpairwise substitutions of compounds 1–3 in oligonucleotides.16b It was found that pairing with the natural bases was weak and nonselective, whereas the pairing of two nonpolar bases was significantly stronger. The results suggested that hydrogen bonding in natural DNA is probably more important for pairing selectivity than for stabilization. Interestingly, some of the data indicated that such nonpolar nucleosides might be efficient at base stacking.
A number of studies have identified hairpin-type structures in DNA and RNA that are specially stable: this has been attributed to cross-loop hydrogen bonding.34 To examine this effect we replaced natural nucleotides (thymidines) in a hairpin loop with non-H bonding isosteres (nucleosides 1 and 2) (see Fig. 9). Interestingly, we found13 that the nonnatural structures are even more stable than the natural ones, indicating that loops in folded nucleic acid structures can be stabilized without hydrogen bonding, and that base stacking may be more important in such folded structures. Further studies on stacking properties of these compounds are underway.35
Figure 9.

Stabilization of folded DNA hairpins by nonpolar nucleoside isosteres 1 and 2 relative to their natural analog thymidine (T). Conditions: pH 7.0. 10 mM NaCl.
Since the larger polycyclic aromatic hydrocarbons are commonly fluorescent, we examined phenanthrene and pyrene nucleosides 7 and 8 after being placed in DNA strands.18 The results showed (Figure 10) that both are indeed fluorescent, with the phenanthrene giving the more intense emission under these conditions. It seems possible that such simple fluorescent nucleosides may have use as reporter groups in DNA. We are currently examining this series of C-nucleosides for their influence on helix stability,35 for their interaction with proteins,36 and for their utility as biological probes in general.
Figure 10.

Fluorescence emission spectra of DNA oligonucleotides (sequence 5'-XCGCGCG) containing nucleosides 8 (pyrene) and 7 (phenanthrene). Experimental methods are described in ref. 18.
Acknowledgements
We thank the National Institutes of Health (GM52956) and the Army Research Office for support. E.T.K. gratefully acknowledges awards from the Beckman Foundation. American Cyanamid, the Office of Naval Research, and the Army Research Office, a Dreyfus Teacher-Scholar Award and an Alfred P. Sloan Foundation fellowship.
Biography

Eric T. Kool was born in Evanston, Illinois in 1960. He received a National Science Foundation Predoctoral Fellowship to pursue graduate studies at Columbia University with Ronald Breslow, receiving his Ph.D. in 1988. He did postdoctoral work with Peter Dervan at the California Institute of Technology as an NIH Postdoctoral Fellow. In July 1990 Kool joined the faculty at the University of Rochester, and he was promoted to the rank of Associate Professor in 1995. His research interests in organic chemistry include the design of molecules which can recognize DNA and RNA sequence and structure, and of molecules which mimic biological structure and function.

Narayan C. Chaudhuri received his B.Sc. (Chemistry) and M.Sc. (Organic Chemistry) degrees from the University of Calcutta, India. He then obtained his Ph.D. in synthetic organic chemistry from the Indian Institute of Technology, Kanpur in 1989. After some industrial work, Chaudhuri joined the laboratory of Prof. Eric T. Kool as a postdoctoral research associate, where he worked (1992–95) on the solid-phase synthesis oligonucleotides, on the synthesis of novel structures for high-affinity recognition of DNA, and on the chemistry of C-nucleosides. Currently he is finishing another postdoctoral assignment at the University of Illinois at Chicago.

Rex X.-F. Ren was born in the Peoples Republic of China in 1964. He obtained his Bachelor’s degree in Chemistry from Wuhan University in 1985. He then moved to New York State to do graduate studies with Prof. E. Turos at SUNY-Buffalo, and he obtained his Ph.D. in 1994, with his thesis focusing on synthetic methods and on beta-lactam synthesis. His subsequent postdoctoral work with Prof. E. Kool at Rochester involved the development of synthetic methods for novel C-nucleosides. He is now doing postdoctoral studies with Prof. K. Nalanishi at Columbia University.
References
- 1.For reviews of C-glycoside synthesis, see: Hanessian S, Pernet AG. Adv. Carbo. Chem. Biochem. 1976;33:111–188. doi: 10.1016/s0065-2318(08)60281-4. Postema MHD. Tetrahedron. 1992;48:8545–8599.
- 2.(a) Lerch U, Burdon MG, Moffat JG. J. Org. Chem. 1971;36:1507. doi: 10.1021/jo00810a016. [DOI] [PubMed] [Google Scholar]; (b) Brown DM, Burdon MG, Slatcher RP. J. Chem. Soc., Section C. 1968:1051. [Google Scholar]; (c) Shapiro R, Chambers RW. J. Am. Chem. Soc. 1961;83:3920. [Google Scholar]
- 3.(a) Kalvoda L, Farkas J, Sorm F. Tetrahedron Lett. 1970:2297. doi: 10.1016/s0040-4039(01)98213-9. [DOI] [PubMed] [Google Scholar]; (b) Trummlitz G, Moffat JG. J. Org. Chem. 1973;38:1841. doi: 10.1021/jo00950a015. [DOI] [PubMed] [Google Scholar]; (c) Barton DHR, Ramesh M. J. Am. Chem. Soc. 1990;112:891. [Google Scholar]
- 4.Buchanan JG, Jumaah AO, Kerr G, Talekar RR, Wightman RH. J. Chem. Soc., Perkins Trans. 1991;1:1077. [Google Scholar]
- 5.Farkas J, Flegelova Z, Sorm F. Tetrahedron Lett. 1972:2279. [Google Scholar]
- 6.(a) Casiraghi G, Cornia M, Rassu G, Sante CD, Spanu P. Tetrahedron. 1992;48:5619. [Google Scholar]; (b) Patil SA, Otter BA, Klein RS. Tetrahedron Lett. 1994;35:5339. [Google Scholar]
- 7.(a) Yokoyama M, Tanabe T, Toyoshima A, Togo H. Synthesis. 1993:517. [Google Scholar]; (b) Yokoyama M, Toyoshima A, Akiba T, Togo H. Chem. Lett. 1994:265. [Google Scholar]; (c) Dondoni A, Marra A, Sherrmann MC. Tetrahedron Lett. 1993;34:7323. [Google Scholar]; (d) Yokoyama M, Akiba T, Togo H. Synthesis. 1995:638. [Google Scholar]
- 8.(a) Belmans M, Esmans E, Dommisse R, Lepoivre J, Alderweireldt F, Balzarini J, Clercq ED. Nucl. & Nucleotides. 1985;4:523. [Google Scholar]; (b) Mertes MP, Zielinki J, Pillar C. J. Med. Chem. 1967;10:320. doi: 10.1021/jm00315a007. [DOI] [PubMed] [Google Scholar]; (c) Togo H, Ishigami S, Fujii M, Ikuma T, Yokoyama M. J Chem. Soc. Perkin Trans. 1994;1:2931. [Google Scholar]
- 9.(a) Solomon MS, Hopkins PB. J Org. Chem. 1993;58:2232. [Google Scholar]; (b) Togo H, Aoki M, Kuramochi T, Yokoyama M. J. Chem. Soc. Perkin Trans. 1993;1:2417. [Google Scholar]
- 10.Bergstrom DE, Zhang P. Tetrahedron Lett. 1991;32:6485. [Google Scholar]
- 11.(a) Buchanan JG, Quijano ML, Wightman RH. J. Chem. Soc. Perkins Trans. 1992;1:1573. [Google Scholar]; (b) Zhang HC, Brakta M, Daves GD., Jr Nucleosides & Nucleotides. 1995;14:105. [Google Scholar]
- 12.(a) Du J, Qu F, Lee D, Newton MG, Chu CK. Tetrahedron Lett. 1995;36:8167. [Google Scholar]; (b) Dondoni A, Scherrmann MC. Tetrahedron Lett. 1993;34:7319. [Google Scholar]; (c) Dondoni A, Scherrmann MC. J. Org. Chem. 1994;59:6404. [Google Scholar]
- 13.Ren X-F, Schweitzer BA, Sheils CJ, Kool ET. Angew. Chem. 1996;35:743. doi: 10.1002/anie.199607431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puglisi JD, Chen L, Blanchard S, Frankel AD. Science. 1995;270:1200. doi: 10.1126/science.270.5239.1200. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 15.(a) Miller TR, Hopkins PB. Bioorg. & Med. Chem. Lett. 1994;4:981. [Google Scholar]; (b) Miller TR, Alley SC, Reese AW, Solomon MS, McCallister WV, Mailer C, Robinson BH, Hopkins PB. J. Am. Chem. Soc. 1995;117:9377. [Google Scholar]
- 16.(a) Schweitzer BA, Kool ET. J. Org. Chem. 1994;59:7238. doi: 10.1021/jo00103a013. ibid, 1995, 60, 8326. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schweitzer BA, Kool ET. J. Am. Chem. Soc. 1995;117:1863. doi: 10.1021/ja00112a001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schweitzer BA, Sheils CJ, Ren X-F, Chaudhuri NC, Kool ET. J. Biomol. Struct. Dyn. 1996;209 [Google Scholar]
- 18.Ren RX-F, Chaudhuri NC, Paris PL, Rumney S, IV, Kool ET. J. Am. Chem. Soc. 1996;118:7671. doi: 10.1021/ja9612763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Millican TA, Mock GA, Chauncey MA, Patel TP, Eaton MAW, Gunning J, Cutbush SD, Neidle S, Mann J. Nucleic Acids Res. 1984;12:7435. doi: 10.1093/nar/12.19.7435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klein RS, Kotick MP, Watanabe KA, Fox JJ. J. Org. Chem. 1971;36:4113. doi: 10.1021/jo00825a022. [DOI] [PubMed] [Google Scholar]
- 21.Matsumoto T, Katsuki M, Suzuki K. Tetrahedron Lett. 1988;29:6935. [Google Scholar]
- 22.Hoffer M. Chem. Ber. 1960;93:2777. [Google Scholar]
- 23.Hurd CD, Miles HT. J. Org. Chem. 1964;29:2976. [Google Scholar]
- 24.(a) Chaudhuri NC, Kool ET. Tetrahedron Lett. 1995;36:1795. [Google Scholar]; (b) Chaudhuri NC, Kool ET. Tetrahedron Lett. 1995;36:4910. [Google Scholar]
- 25.Robins MJ, Robins RK. J. Am. Chem. Soc. 1965;87:4934. doi: 10.1021/ja00949a042. [DOI] [PubMed] [Google Scholar]
- 26.(a) Srivastava PC, Robins PK, Takusagawa F, Berman HM. J. Het. Chem. 1981;18:1659. [Google Scholar]; (b) Hacksell U, Cheng JC-Y, Daves GD., Jr J. Carbohydr. Chem. 1986;5:287. [Google Scholar]
- 27.Smith M, Jardetzky CD. J. Mol. Spectroc. 1968;28:70. [Google Scholar]
- 28.(a) Altona C, Sundaralingam M. J. Am. Chem. Soc. 1972;94:8205. doi: 10.1021/ja00778a043. [DOI] [PubMed] [Google Scholar]; (b) Altona C, Sundaralingam M. J. Am. Chem. Soc. 1973;95:2333. doi: 10.1021/ja00788a038. [DOI] [PubMed] [Google Scholar]
- 29.Francois P, Sonveaux E, Touillaux R. Nucleosides & Nucleotides. 1990;9:379. [Google Scholar]
- 30.Ford KG, Neidle S, Eaton MAW, Millican TA, Mann J. Acta. Cryst. 1987;C43:1988. [Google Scholar]
- 31.Karplus M. J. Chem. Phys. 1959;30:11. [Google Scholar]
- 32.Angyal SJ. Angew. Chem. Int. Ed. Engl. 1969;8:157. [Google Scholar]
- 33.Cleve G, Hoyer G, Schulz G, Vorbruggen H. Chem. Ber. 1973;106:3062. [Google Scholar]
- 34.Senior M, Jones RA, Breslauer KJ. Proc. Natl. Acad. Sci. USA. 1988;85:6242. doi: 10.1073/pnas.85.17.6242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guckian KM, Schweitzer BA, Ren X-F, Sheils CJ, Paris PL, Kool ET. J. Am. Chem. Soc. 1996;118:8182. doi: 10.1021/ja961733f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moran S, Ren X-F, Sheils CJ, Kool ET. Nucleic Acids Res. 1996;24:2044. doi: 10.1093/nar/24.11.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]