

Abstract
Background
Heart failure (HF) is a debilitating condition resulting in severe disability and death. In a subset of cases, clustered as Idiopathic Dilated Cardiomyopathy (iDCM), the origin of HF is unknown. In the brain of patients with dementia, proteinaceous aggregates and abnormal oligomeric assemblies of β-amyloid impair cell function and lead to cell death.
Methods and Results
We have similarly characterized fibrillar and oligomeric assemblies in the hearts of iDCM patients pointing to abnormal protein aggregation as a determinant of iDCM. We also showed that oligomers alter myocyte Ca2+ homeostasis. Additionally, we have identified two new sequence variants in the presenilin-1 (PSEN1) gene promoter leading to reduced gene and protein expression. We also show that presenilin-1 co-immunoprecipitates with SERCA2a.
Conclusions
Based on these findings we propose that two mechanisms may link protein aggregation and cardiac function: oligomer-induced changes on Ca2+ handling and a direct effect of PSEN1 sequence variants on EC-coupling protein function.
Keywords: Cardiomyopathy, Protein Folding, Calcium, Presenilin, Alzheimer
INTRODUCTION
In 1906 Alois Alzheimer discovered in the brains of a patient suffering from early age onset of dementia, aggregates of a proteinaceus material, later identified to be composed of amyloid fibers1. Alzheimer disease (AD) is associated with several genetic defects and the abnormal accumulation of the amyloid-β protein (Aβ) and tau in the form of extracellular (senile plaques) and intracellular (neurofibrillar tangles) aggregates respectively2. In the heart, amyloid degeneration leading to DCM has been limited to three conditions: 1) L-chain amyloidosis secondary to multiple myeloma; 2) transthyretin (TTR) cardiomyopathy3; and 3) desmin cardiomyopathy4,5. In a smaller number of cases of non-ischemic origin, the etiology of HF is infective, toxic or genetically determined. When one of these causative events cannot be recognized, the myocardial disease is classified as idiopathic (iDCM).
Genetically DCM has been found in only 35% of HF cases indicating that the cause of this disease remains largely unknown6. Mutations in genes encoding sarcomeric, cytoskeletal and nuclear proteins as well as proteins involved in the regulation of Ca2+ homeostasis have been described7,8. Allelic and locus heterogeneities occurs in DCM10 and these alterations, together with environmental factors, can disclose an otherwise silent genetic background as occurs in neurodegenerative diseases. Recently, two mutations in the familial early-onset AD-associated presenilin genes (PSEN1 D333G and PSEN2 S130L), 2, have been found in 0.9% of tested DCM families (3/325)9. The presenilins (PS1 and PS2) are highly conserved polytopic membrane proteins that are required for γ-secretase activity, an enzyme responsible for proteolytic processing of the amyloid precursor protein (APP) to generate As and a variety of membrane-associated proteins including the Notch receptor10. In the brain, the presenilins also regulate Ca2+ in the endoplasmic reticulum and modulate Ryanodine receptor (RyR) function by cleavage of Sorcin11. Both PSEN1 and PSEN2 are expressed in the heart and are essential for cardiac morphogenesis12,13.
Because of the role that presenilins play in protein processing and Ca2+ homeostasis, the possibility was advanced that mutations in PSEN1 and PSEN2 may account for the cardiac phenotype found in a subset of patients affected by iDCM. These mutations may result in alterations of Ca2+ homeostasis and/or buildup of misfolded substrates, which alter myocyte mechanical behavior. Here we address whether the accumulation of protein aggregates and genetic alterations in the PSEN and oligomeric fragments alter cardiomyocyte mechanics, Ca2+ homeostasis and ultimately ventricular performance.
METHODS
Patients
Myocardial tissue was collected from biopsy and explanted hearts from patients with iDCM, amyloid cardiomyopathies, or from donor non-failing hearts. All tissue from donor or transplant patients was collected in cold and oxygenated Wisconsin cardioplegic solution as soon as the heart was explanted from the patient. Non-failing hearts were available from National Disease Research Interchange (NDRI) supported by the National Institutes of Health. Tissues received as non-failing hearts can be found not suitable for transplantation for several reasons e.g., lack of identification of a suitable recipient, blood transfusion while in the emergency room, age of donor, need for resuscitation. All donations were with family approval. Donor hearts ruled out from transplantation were used if no macroscopic, laboratory or instrumental signs of cardiac diseases were present. The AD patients and non-AD affected family members were from The NIMH Genetics Initiative Alzheimer’s Disease Study Sample. Subjects were identified according to a standardized protocol applying the criteria of the National Institute of Neurological and Communicative Diseases and Stroke/Alzheimer’s Disease and Related Disorders Association (NINCDS/ADRDA) for the diagnosis of AD.
Sample collection
The hearts were dissected by regions and the tissue was frozen in liquid nitrogen and stored at −80°C. From the anterior wall a branch of the left anterior descending artery was cannulated and the tissue perfused with PLP (4% paraformaldehyde-lysine-sodium metaperiodate in 0.1M NaPO4). A sampleof the left ventricular anterior wall of the hearts was also collected in 2.4% glutarhaldehyde for electron microscopy (EM).
Structural staining
The frozen tissue was cut in 8μm slices. Samples were stained in a blinded fashion by the clinical pathology laboratory for protein aggregates using modified Congo-red14 or Thioflavin-S. The Congo-red sections were observed in random sequence under direct light as well as under polarized light. Polarized light microscopy could not, however, differentiate positive intracellular staining due to the natural anisotropy of the myosin filaments. Thioflavin-S slides were imaged using confocal microscopy (Leica TCS).
Immunoblotting, Immunohistochemistry, EM, Immunogold EM, PCR and sequencing, Luciferase activity
Standard techniques were applied for these methodologies and detailed in the supplemental material. Specific aspect for the present study included the use of anti-oligomer (A11) antibody that recognizes a generic, sequence independent epitope for prefibrillar amyloid oligomers at immunohistochemistry and EM. Anti-PS1 N-terminal and anti-Aβ antibody were used for immunoblotting. The entire coding and 5′-promoter regions of PSEN1 and PSEN2 gene were amplified from DNA for sequencing. Four pGL2 constructs were established containing PSEN1–92C, -92delC and PSEN1 -21G and -21A, for determining the transcriptional activity by luciferase reporter assay. The average transcriptional activities of PSEN1 -92delC variant vs. -92C WT allele, and -21G variant vs. -21A WT allele from four repeated experiments were compared by Wilcoxon Two-Sample Test. A p value <0.05 was taken to indicate statistical significance.
Preparation of Homogeneous Populations of Amyloid Peptide Monomers, Oligomers, and Fibrils
Soluble oligomers were prepared following the protocol kindly provided by Dr Charles Glabe. Aβ42 stock solutions (2 mM) were obtained by dissolving the lyophilized peptide (from 1:1 H20:acetonitrile) in hexafluoroisopropanol (HIFP) and sonicated. The resulting solution was added to double-distilled water in a siliconized Eppendorf tube. The solution was gently mixed and stirred and the HIFP evaporated. Aliquots were taken at 6–12-h intervals for the first 12-hrs then every 12-hrs. Oligomers were obtained after 4 days. Oligomer formation was monitored by A11 dot-blots. Annular protofibrils were obtained from the spherical oligomer preparation followed by the addition of 5% hexane. Once fibril formation was complete, the solution was centrifuged and the fibril pellet was washed three times with double-distilled water and then resuspended in phosphate buffered saline. The morphology was verified by negative stain electron microscopy.
Myocytes Oligomer Toxicity Measurements
Adult myocytes were isolated from C57/Bl6 mice and measurements performed as previously described15. Isolated cardiomyocytes were placed in a flow chamber on the stage of an inverted microscope, superfused with Tyrode solution and electrically stimulated with a biphasic pulse (5Hz, 50% above threshold). After obtaining a stable contraction and Ca2+ transient, 0.5 or 5 μg of oligomer solution was applied on a cell by rapid release (ALA scientific). In control cells, the buffer without oligomers was applied. Contraction amplitude and rates of contraction and relaxation were recorded online using a video-edge-detection system and data acquisition software (IonOptix Corp). The fluorescent Ca2+ indicator Fura-2 (Molecular Probes®) was used to measure intracellular Ca2+, as described previously16.
RESULTS
Detection of Protein Aggregates
To establish whether protein aggregates are present in iDCM, ventricular samples obtained from explanted failing (n=23) and donor non-failing (n=12) hearts were stained for amyloid and amyloid-like structures. Samples from patients with cardiac amyloidosis (n=6) were included as controls for the assessment of misfolded proteins. Amyloid specific positive staining were detected in 17 of the 23 iDCM hearts (Fig 1A, 1B) and in 2 donor hearts. In the latter case, patients were 75 years of age, the oldest in the group.
Figure 1.
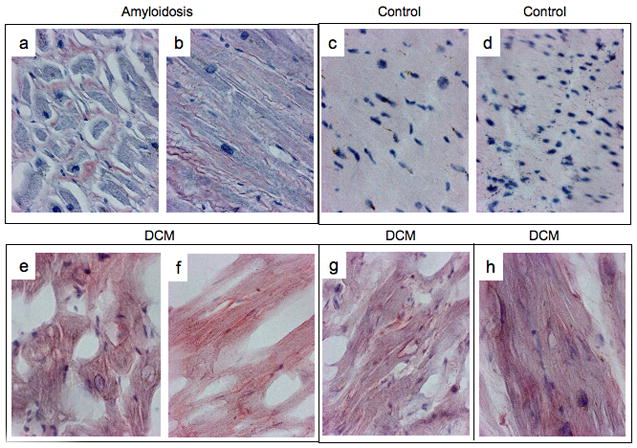



A) Congo-red staining of a case of senile amyloid (a, b) 2 non failing control (c, d) and 3 cases of iDCM (e–h)
B) Confocal microscopy of the Thioflavin-S staining of iDCM (a, b), control (c, d) and AL-amyloid cardiomyopathy (e, f). Panels a, d, e shows the sections under direct light, panels b, d, f show the sections under fluorescence light.
The AL-amyloid cardiomyopathy shows the distribution of the staining in the interstitial space whereas in the iDCM the staining occupies also the cytosol of the myocytes.
C) Electron micrographs of super-thin sections from AL-amyloid cardiomyopathy (a, b), iDCM left ventricle (c, d). Higher magnification of the fibrillar aggregates from AL-amyloidosis (b) and iDCM (d). The insert in panel d shows collagen fibers from the same heart for comparison. D) Electron micrographs of myocardial diagnostic biopsies from iDCM patients showing extracellular aggregates (plaques) (panel a–c, Panel b shows a higher magnification of panel a) and intracellular fibrils (tangles) (panel d–g). Panels e and g are higher magnifications of panels d and f respectively.
To strengthen these observations and provide evidence that protein occurs in iDCM, fibrillar deposits were identified by EM within the myocyte cytoplasm and in the extracellular compartment (Fig 1C). The distribution of the protein aggregates resembled, respectively, tangles and plaques commonly found in AD17. To exclude the possibility that myocardial aging and end-stage HF were implicated in the accumulation of degradation products, biopsy samples collected from patients with early stage iDCM (n=5) were studied by EM. The mean age of our patient sample was 52 (age range 31–65) in the transplant population and 45 (age range 24–65) in the biopsy group (Table 1a). Tangle- and plaque-like structures were found (Fig 1C–D), strongly suggesting that fibrillar deposits may constitute an integral component of the onset and progression of the pathology of iDCM.
Table 1.
iDCM Patient’s sample
Patient’s population clinical data.
| CongoRed/Genetically tested | Age (years) | Ethnicity/Gender | Heart weight | EF (%) | Associated diseases | Medications |
|---|---|---|---|---|---|---|
| EXPLANTED | ||||||
| 1/16 | 53 | CF | 340 | 19 | Breast K, | D, DG, BB, ACEI, AA, AC |
| 2 | 60 | CM | 710 | 18 | HTN, gout, HL, HYPOT | D, BB, DG, LL, NT |
| 3 | 43 | CF | 320 | 15 | Anoxia brain inj | D, BB |
| 4 | 63 | CF | 330 | 10 | Anemia, AF, MVR, IDDM, HTN | D, ASA, AB |
| 5/7 | 56 | CM | 480 | 15 | NIDDM, PHT, AF | D |
| 6/8 | 57 | CM | 620 | 20 | AF, DM, HTN, HL | D, PEI, NT, LL, AIIRB, VD |
| 7/9 | 49 | CF | 420 | 20 | NIDDM, COPD, CRI | D, ACEI, NT, DG, INO |
| 8/10 | 31 | AAF | 376 | 10 | Asthma | D, BB, ACEI |
| 9/11 | 65 | HM | NR | 15 | COPD | D, DG, ACEI, AC |
| 10/12 | 48 | CM | 420 | 23 | Asthma, NIDDM, ETOH | NA |
| 11/13 | 54 | CM | 830 | 12 | HTN | D, DG, AIIRB, AC |
| 12/14 | 45 | HM | 340 | 19 | ETOH, AF | D, CA, AB, |
| 13/15 | 44 | CM | 490 | 27 | POST TX | D, ACEI, NT |
| 14/17 | 65 | CF | 570 | 10 | COPD, AF, ETOH, HYPOT | D, ACEI, DG, AA, LL |
| 15/18 | 47 | CM | 470 | 15 | HTN, PHTN, HL | D, ACEI, NT, DG |
| 16/19 | 33 | CF | 590 | 23 | HTN, PHTN, ETOH | D, ACEI, DG |
| 17/20 | 58 | CM | 535 | 22 | HTCM, Gout, AF, CRI | D, DG, BB, ASA, AA, AC |
| 18/21 | 55 | CF | 250 | 22 | DM, ASYHMA, AF, PHTN, HTN, | D, DG, ACEI, AIIRB |
| 19/22 | 65 | CM | 520 | 41 | AFl, ASD, HL, CRI | D, BB, ASA, LL, AA |
| 20/23 | 28 | CM | 880 | 9 | HTN, NIDDM, AF, CRI | D, BB, ACEI |
| 21 | 65 | CM | 600 | AF, HTN | D, DG, BB, ACEI | |
| 22 | 43 | CF | 320 | 15 | NONE | D, BB, CA, AB |
| 23 | 59 | CM | 450 | 18 | HTN | D, ACEI |
| BIOPSIES | ||||||
| 1 | 57 | CM | NA | 33 | HTN | D, ACEI, NT, LL |
| 2 | 65 | CM | NA | 30 | HTN | D, ACEI, DG, BB |
| 3 | 50 | CM | NA | 17 | NONE | ACEI, BB, NT |
| 4 | 31 | CF | NA | 16 | MS | AE, IMAO |
| 5 | 65 | CM | NA | 50 | D, DG, TNT, BB | |
| DONORS | ||||||
| 1 | 56 | CM | 630 | 76 | NA | NA |
| 2 | 75 | CM | 391 | UN | NA | NA |
| 3 | 42 | CM | 320 | UN | NA | NA |
| 4 | 67 | CM | 445 | UN | NA | NA |
| 5 | 60 | CM | NA | 60 | NA | NA |
| 6 | 63 | CM | NA | 70 | NA | NA |
| 7 | 39 | HF | 378 | UN | NA | NA |
| 8 | 33 | CM | 450 | UN | NA | NA |
| 9 | 56 | CM | 400 | UN | NA | NA |
| 10 | 58 | CF | 353 | UN | NA | NA |
| 11 | 73 | CF | 420 | 60 | NA | NA |
| 12 | 35 | CM | 363 | 55 | NA | NA |
| 13 | 75 | CF | 402 | UN | NA | NA |
| 14 | 59 | CM | 551 | UN | NA | NA |
Ethnicity/Gender abbreviations: CF=Caucasian Female, CM=Caucasian Male, AAF=African-American Female, HM=Hispanic Male, HF=Hispanic Female
Associated diseases abbreviations: HTN=hypertension, HL=hyperlipidemia, AF=Atrial fibrillation, MVR=Mitral valve regurgitation, IDDM=insulin dependent diabetes mellitus, NIIDM=non insulin dependent diabetes mellitus, PHTN=pulmonary hypertension, COPD=chronic obstructive pulmonary disease, CRI=chronic renal insufficiency, ETOH=alcoholism, AFl=atrial flutter, ASD=atrial septal defect, MS=multiple sclerosis
Medications abbreviations: D= diuretics, DG=digitalis, BB=beta-blocker, ACEI=ACE inhibitors, AA=antiarrhythmics, AC=anticoagulants, LL=lipid lowering, NT=Nitrates, ASA=aspirin, AB=alpha blocker, PEI=phosphodiesterase inhibitors, INO=inotropes, VD=vitamin D, AIIRB=Angiotensin II receptor blocker, IMAO=inhibitors monoaminoossidase
UN=unavailable, NA=non applicable
Since in AD patients Aβ accumulates in the brain, tissue sections from all iDCM and controls were also tested for the levels of Aβ in the myocardium. We did not detect differences in the level of Aβ in iDCM compared to controls at immunohistochemistry (Fig 2).
Figure 2.

Immunohistochemical analysis for Aβ of samples from non-failing donor and iDCM (representative) patients showing no difference in the Aβ expression in patients carrying PSEN mutations.
Oligomeric Assemblies and their Function
In the brain of AD patients, fibrillar deposits of β-amyloid represent the end product of misfolded peptides. Smaller oligomeric assemblies have been identified as soluble toxic aggregates that impair neural function17,18. This observation raised the possibility that a similar mechanism may be operative in the failing heart providing an unanticipated etiology for at least a subset of patients affected by iDCM. Therefore, a structural antibody, specific for the recognition of oligomeric structure, first described in the brain of subjects with AD17, was employed to detect oligomer fragments in iDCM hearts. We examined 6 cases of iDCM and 8 controls. Positive immunostaining was observed in 5 cases (Fig 3A) and in 37.5% of controls. A larger number of age and harvest time matched samples are required to assess the relative abundance of oligomers in the diseased hearts. Additionally, anti-oligo immuno-gold EM was utilized to confirm the presence of oligomeric peptide forms in biopsy samples of individuals in the initial phases of iDCM (Fig 3B). Importantly, similar pathology was found in the core of myocardium removed for LVAD implantation in a patient with end-stage iDCM.
Figure 3.

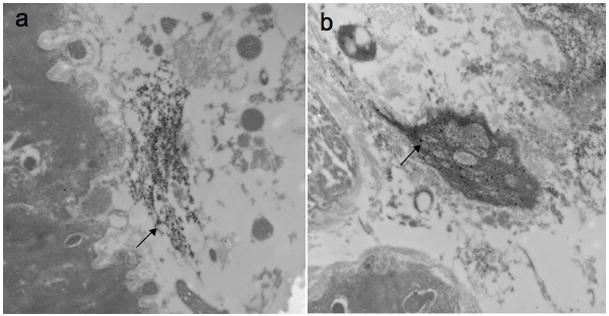
A) Immunohistochemical analysis of a sample from iDCM patient (panel a and b) vs. a non-failing donor (panel c and d). Oligomeric fibrils in red with anti-oligo antibodies (panel a and c) or aggregates with Thioflavin-S (panel b and d). The insert indicates absence of secondary antibody. DAPI stains nuclei in blue. B) Immuno-gold electron micrographs of myocardial diagnostic biopsies from 2 iDCM patients showing the presence of oligomeric fibrils in the myocardium.
However, these results leave unanswered the question whether formation of abnormal oligomers in the heart depresses myocyte mechanics and thereby ventricular function. Since the toxicity of oligomers is linked to the structure rather than the polypeptidic sequence of the fragments19, oligomers were generated from commercial lyophilized Aβ peptide (Fig 4a) to evaluate their effects on cardiomyocyte performance. Thus, freshly isolated adult mouse cardiomyocytes were exposed to oligomers (0.5–5 μg/ml) while Ca2+ transients and cell shortening were measured. Oligomers induced a sudden increase in peak systolic Ca2+ transient together with an enhanced velocity of Ca2+ release (Fig 4b).
Figure 4.

a) dot-blot of the preparation of oligomers showing the formation of oligomers after 4 days and subsequent hexane exposure of the oligomeric preparation forming fibrils (EM). b) Acute increase in contractility and Ca2+ transients in a representative tracing. This particular cell died after 80 sec of exposure. c) Quantitative changes of systolic and diastolic Ca2+ and velocity of Ca2+ release and reuptake.
Genetic Analysis
The coding region and promoters of PSEN1 and PSEN2 were sequenced in 20 patients with iDCM. In this report we describe four variants in PSEN1 and PSEN2 genes (Table 2). Two novel PSEN1 5′ promoter region variants, -92delC (UCSC Genome Chr 14 Database 72672839) upstream of alternative transcription initial exon 1A20 and -21G>A (UCSC Genome Chr 14 Database 72673257) upstream of alternative transcription initial exon 1B, were recognized in patients 21 and 4, respectively (allele frequency 0.025) (Fig 5A, upper panel). These variants were absent in our 14 controls. Patient 4 also possessed a previously reported PSEN1 missense mutation E318G21 (allele frequency 0.025). Additionally, a known PSEN2 missense mutation R62H22 (UCSC Genome Chr 1 Database 225138072) was identified in patients 1 and 17 (allele frequency 0.05). The 4 patients who possessed PSEN1 promoter region variants or PSEN2 missense variants were Caucasian females.
Table 2.
Sequence variants frequency detected by DNA sequencing analyses of PSEN1 gene in iDCM patients
| Gene | Variant | UCSC Genome Sequence Database (36.1) | SNP ID If available | Minor Allele frequency | Genotype frequency (variant/20 DCM patients) | |
|---|---|---|---|---|---|---|
| PSEN1 | -92delC | chr14:72672839 | 0.025 | CC 19/20 (95%) | C-1/20 (5%) | |
| PSEN1 | -21G>A | chr14: 72673257 | 0.025 | GG 19/20 (95%) | GA 1/20 (5%) | |
| PSEN1 | A953G/E318G | chr14:72742931 | rs17125721 | 0.025 | AA 19/20 (95%) | AG 1/20 (5%) |
| PSEN2 | G185A/R62H | chr1: 225138072 | 0.05 | GG 18/20 (90%) | GA 2/20 (10%) | |
Figure 5.
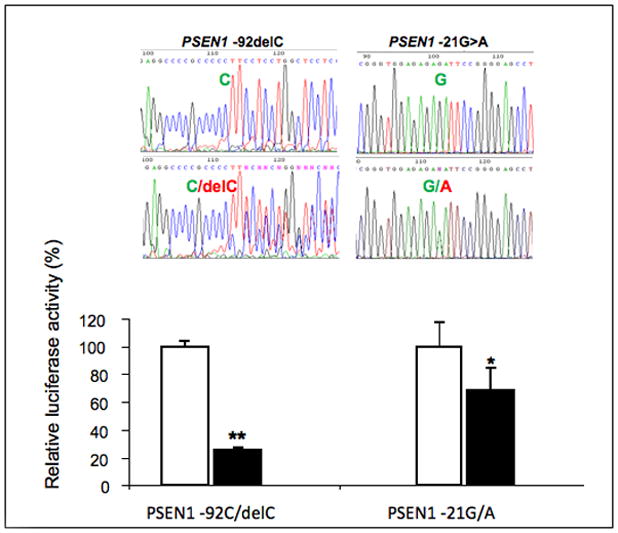


A) Presenilin sequencing electropherograms (upper panel). PSEN1–92delC and -21G>A detected in iDCM patients are shown (top panels are sequences from controls). Measurement of the transcriptional activity by luciferase reporter gene assay (lower panel). PSEN1 -92delC variant exhibited 25% of transcriptional activity over the basal expression of PSEN1 promoter with wild-type allele -92C by the measurement of relative luciferase activity. PSEN1 -21A variant reduced the transcriptional activity to 68% compared with the basal expression of PSEN1 promoter with wild-type allele -21G. *p=0.0029. B) Immunoblotting for PS1 in donor controls, iDCM (p=0.01 vs controls) and iDCM with the genetic variations in the PS promoter (indicated by asterisk). PS1 expression normalized by GAPDH in the patients carrying the genetic variations was 0.79±0.18 vs 0.93±25 in the iDCM patients without the genetic variants (the level in the non-failing population was 0.56±0.24) p=0.046 at Kruskal-Wallis test.
C) Co-immunoprecipitation of PS and SERCA2a. Panel a: PS detection after immunoprecitpitation with SERCA2a. Panel b: SERCA2a detection after immunoprecitpitation with PS1. Patients carrying the genetic variations are indicated by asterisk.
The functional relevance of the new genetic variations was tested at the transcriptional level by luciferase assay and at the protein level by SDS page. We cloned the PSEN1 5′ promoter regions spanning -92delC and -21G>A variants into a reporter vector and their transcriptional activity was measured. The constructs contained all known positive promoter elements that are required for PSEN1 expression, including two putative TATA boxes and two putative transcription initiation CAP sites upstream of exon 1A23. In comparison to the basal level of PSEN1 promoter wild-type -92C allele, PSEN1 -92delC variant reduced transcriptional activity by 75% (p=0.002, Fig 5A lower panel). Constructs for PSEN1 -21G or -21A included sequences from the end of exon 1A to the end of alternative transcription initial exon 1B. With respect to -21A wild-type allele, PSEN1 -21G variant reduced transcriptional activity by 32% (p=0.002, Fig 5A, lower panel). The construct containing PSEN1 wild-type -92C to alternative transcription initial exon 1A exhibited 5-fold higher transcriptional activity than the construct containing PSEN1 wild-type -21G to alternative transcription initial exon 1B. Consistently, these variants decreased the expression of PS1 protein in the myocardium (Fig 5B). Importantly, PS1 co-immunoprecipitated with the cardiac isoform of the SR Ca2+-ATPase pump SERCA2a (Fig 5C) suggesting that changes in PS1 may be accompanied by defects in SERCA2a function as shown to occur in the in vitro for Serca2b24.
Population frequency
By SNP analysis, PSEN1 -21 G>A was not detected in 265 AD patients (0/530chromosomes). PSEN1 -92delC was found in AD patients with an allele frequency 0.005 (8/1632 chromosomes). PSEN1 E318G was previously detected in AD patients with an allele frequency 0.02 (12/596 chromosomes)21,25 which is the same as the frequency we obtained by testing late-onset AD patients from NIMH samples. PSEN2 R62H missense mutation was detected in AD patients with an allele frequency 0.016 (5/320 chromosomes). The family members of identified AD patients who possessed PSEN1 -92delC, E318G and PSEN2 R62H were also analyzed for segregation by SNP genotyping. None of PSEN1–92delC, E318G or PSEN2 R62H was segregated with AD affection status in identified AD families.
DISCUSSION
The mechanisms responsible for the onset of iDCM and its evolution to overt HF and death are currently unknown. The documentation in the present study that fibrillar deposits with the formation of tangle- and plaque-like structures within the myocardium occur in the early and late stages of the disease point to protein misfolding as an important determinant of the pathogenesis of iDCM. Protein misfolding impairs cell function in neurodegenerative disorders and other chronic diseases suggesting that a similar effect may be operative in the human heart18,26,27. Although the negative impact of protein aggregates on the mechanical and Ca2+ handling properties of human myocytes was not tested, these deposits modified the functional behavior of normal adult mouse myocytes supporting the notion that similar consequences may occur on human myocytes.
Protein degradation characteristically occurs with myocardial aging. Senile systemic amyloidosis occurs in 25% of individuals 80 years of age and older28. Amyloid-like deposits are described in non-diseased individuals between the sixth and ninth decade of life29,30. In this report, aggregates were also markedly pronounced in relatively younger patients at early stages of HF, excluding age and disease stage as accounting for protein misfolding in our iDCM population. On the other hand, the presence of protein aggregates, common in advanced age, may suggest that iDCM can be an early senility of the heart, similarly to AD where the common senile dementia occurs at early age.
We then evaluated whether the aggregates simply represent a tombstone of discarded material or are they relate to the disease. Protein aggregates are composed of misfolded proteins at various maturation stages, from monomeric peptides to intermediate oligomers and complex fibrillar structures. Intermediate oligomers have been shown to be present in the brain of patients with AD and, in neurons, to be associated with cell toxicity31,32. Here we showed that intermediate oligomers are also present in the myocardium of patients with iDCM with a distribution similar to what is observed in the brain of patients with AD17. We also demonstrated for the first time that, on cardiomyocytes, oligomers promote an increase in systolic [Ca2+]that may lead to cell dysfunction and death33–35. An increase in cytosolic Ca2+ in the failing heart is coupled with Ca2+ depletion in the sarcoplasmic reticulum which, in turn, favors protein misfolding26,36,37. In the failing heart, Ca2+ dishomeostasis and contractile dysfunction are well known abnormalities and are characterized by changes in excitation-contraction coupling proteins, with SERCA2a playing a key role37.
Several proteins involved in the control of Ca2+ cycling have been shown to be substrates of presenilin/γ secretase11. PS has been recently shown to interact with SERCA2b attenuating its function when PS is downregulated24. Importantly, PS1 also co-immunoprecitpitate with SERCA2a in our iDCM cases suggesting that PS1 may also depress SERCA2a thereby contributing to the observed alterations in Ca2+ homeostasis and myocyte performance in HF. Sequencing of PSEN1 and PSEN2 led to the identification of 26 DNA variants including two novel PSEN1 gene regulatory region variants, -92delC and -21G>A, in our iDCM patient sample. These two previously unknown variants significantly reduced mRNA and protein expression of PS1 indicating loss of function of the genetic variants. Thus these variants likely influence Ca2+ homeostasis by altering SERCA2a function.
In our cases of sporadic iDCM, two PSEN missense mutations (PSEN1 D333G and PSEN2 R62H) previously described in familial DCM9 were detected in addition to the variants in the PSEN promoter. A PSEN1 missense polymorphism E318G was also found. This mutation causes amino acid changes from polar acidic Glutamic acid or Aspartic acid to non-polar neutral Glycine, similar to the PSEN1 D333G mutation. E318G and D333G are located 15 amino acids apart within a “hot spot” in a large hydrophilic cytoplasmic loop between transmembrane domains six and seven. This region contains critical functional domains which are processed by endoproteolysis38 and are essential for the interaction between PS1 and other proteins39. The PSEN2 R62H mutation identified in patients 1 and 17 is located in a “hot spot” of the NH2 terminus and has been associated with AD22 and breast cancer40. PSEN2 R62H enhances PS2 degradation, reduces PS2 stability and compromises Notch function40.
The two presenilin missense substitutions, E318G and R62H observed in iDCM are present in highly divergent regions of PS1 and PS2; PS1 and PS2 proteins share extensive sequence identity along their entire length with the exception of the NH2-terminal domain and the second half of the hydrophilic loop region. Thus, the E318G and R62H missense substitutions may play an important role in the pathogenesis of a subset of patients with iDCM by modulating different functions of the presenilin proteins. An important aspect is whether PSEN variants could affect APP processing in the heart and determine the accumulation of amyloid aggregates. Since the sequence variants we observed are loss of function any effect on APP processing would reduce the Aβ production. Aβ level was tested by immunohistochemistry in our samples and no difference in Aβ expression was present in iDCM compared to controls.
Four of the seven Caucasian female with iDCM possessed either a PSEN1 promoter region variant or a PSEN2 missense mutation. This gender dependent cardiac genotype is consistent with the higher prevalence of AD in women than men41. Estrogen deficiency typical in postmenopausal women may be implicated since cardiomyocytes and fibroblasts contain estrogen receptors and estrogens regulate the expression of specific cardiac genes42.
In conclusion, we have provided the first evidence that protein misfolding is implicated in the etiology and pathogenesis of iDCM in humans. The formation of oligomeric fragments has toxic consequences on cardiomyocytes, which are mediated by alterations in Ca2+ homeostasis. Additionally, missense mutations in the PSEN1 and PSEN2 genes alter presenilin expression and may alter its interaction with proteins involved in excitation-contraction coupling. Further studies in a larger cohort and in animal models will be needed to evaluate the role and mechanisms of these sequence variants and the respective protein expression in the familial and sporadic cases compared with the non-failing population. Furthermore cardiac function is needed to be assessed in AD patients and the AD unaffected family members.
The elucidation of these new pathogenetic mechanisms will facilitate the advancement of novel strategies for early diagnosis and treatment of iDCM.
Heart failure (HF) is a progressive and ultimately fatal disease, which represents a leading public health problem worldwide. Despite substantial advances in the clinical management of HF, the only current permanent therapeutic option is heart transplantation. However, the limited supply of functional organs for transplantation and the age threshold for the surgical intervention constrains the scope of this treatment modality. The majority of cases of HF are ischemic in origin. The second most frequent origin of non-ischemic HF is “idiopathic” dilated cardiomyopathy (iDCM) where no other causative events can be recognized. Recent studies highlight the recognition of the public health importance of a large number of diseases associated with defects in the ability of proteins to fold, leading to the accumulation of cytotoxic protein deposits. These group of diseases include amyloidosis and various neurodegenerative disorders such as Alzheimer’s Disease. Preliminary evidence suggests that iDCM may be included among these misfolding diseases. Also, mutations in the same genes causing Alzheimer’s Disease in a significant percentage of cases can also be at the origin of iDCM. Interestingly, a dual mechanism seems to mediate the effect of these genetic variations: changes in the control of calcium (Ca2+) handling proteins causing defect in the contractile function, and cell damages associated with the protein aggregation process. The incidence, prognosis, and therapeutic option for iDCM may, therefore, be greatly advanced by establishing a fundamental understanding of key factors leading to the disease.
Supplementary Material
Acknowledgments
We wish to acknowledge Dr Anthony Rosenzweig for support and critical revision of the manuscript, Dr Anne Marie Lompre' for critical discussion, Dr Charles Glabe for kindly providing to us with the protocol for oligomers preparation, Michele Parkinson, Jason Divito, Donna M Romano and Libin Cui for technical assistance. Mary McKee for technical assistance with EM, Alexander Ivanov for assisting with oligomer preparation, Jan Kajstura and Konrad Urbanek for assistance with iummunohistochemistry.
FUNDING SOURCES
This work was supported by funds from NIH, (K08-HL69842-FdM, R01AG014713, R01MH60009-RET), Beth Israel Deaconess Medical Center Discretional funds (FdM), Cure Alzheimer’s fund (RET), the Neuroscience Education and Research Foundation (AL).
Footnotes
Conflicts of interest: None
References
- 1.Alzheimer A. A new disease of the cortex. Allg Z Psych. 1907;64:146–148. [Google Scholar]
- 2.Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 3.Kelly DP, Strauss AW. Inherited cardiomyopathies. N Engl J Med. 1994;330:913–919. doi: 10.1056/NEJM199403313301308. [DOI] [PubMed] [Google Scholar]
- 4.Bova MP, Yaron O, Huang Q, Ding L, Haley DA, Stewart PL, Horwitz J. Mutation R120G in alphaB-crystallin, which is linked to a desmin-related myopathy, results in an irregular structure and defective chaperone-like function. Proc Natl Acad Sci U S A. 1999;96:6137–6142. doi: 10.1073/pnas.96.11.6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li D, Tapscoft T, Gonzalez O, Burch PE, Quinones MA, Zoghbi WA, Hill R, Bachinski LL, Mann DL, Roberts R. Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation. 1999;100:461–464. doi: 10.1161/01.cir.100.5.461. [DOI] [PubMed] [Google Scholar]
- 6.Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, Jessup M, Konstam MA, Mancini DM, Michl K, Oates JA, Rahko PS, Silver MA, Stevenson LW, Yancy CW, Antman EM, Smith SC, Jr, Adams CD, Anderson JL, Faxon DP, Fuster V, Halperin JL, Hiratzka LF, Jacobs AK, Nishimura R, Ornato JP, Page RL, Riegel B. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation. 2005;112:e154–235. doi: 10.1161/CIRCULATIONAHA.105.167586. [DOI] [PubMed] [Google Scholar]
- 7.Schonberger J, Seidman CE. Many roads lead to a broken heart: the genetics of dilated cardiomyopathy. Am J Hum Genet. 2001;69:249–260. doi: 10.1086/321978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olson EN. A decade of discoveries in cardiac biology. Nat Med. 2004;10:467–474. doi: 10.1038/nm0504-467. [DOI] [PubMed] [Google Scholar]
- 9.Li D, Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Partain J, Nixon RR, Allen CN, Irwin RP, Jakobs PM, Litt M, Hershberger RE. Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am J Hum Genet. 2006;79:1030–1039. doi: 10.1086/509900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- 11.Pack-Chung E, Meyers MB, Pettingell WP, Moir RD, Brownawell AM, Cheng I, Tanzi RE, Kim TW. Presenilin 2 interacts with sorcin, a modulator of the ryanodine receptor. J Biol Chem. 2000;275:14440–14445. doi: 10.1074/jbc.m909882199. [DOI] [PubMed] [Google Scholar]
- 12.Donoviel DB, Hadjantonakis AK, Ikeda M, Zheng H, Hyslop PS, Bernstein A. Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 1999;13:2801–2810. doi: 10.1101/gad.13.21.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakajima M, Moriizumi E, Koseki H, Shirasawa T. Presenilin 1 is essential for cardiac morphogenesis. Dev Dyn. 2004;230:795–799. doi: 10.1002/dvdy.20098. [DOI] [PubMed] [Google Scholar]
- 14.Elghetany MT, Saleem A, Barr K. The congo red stain revisited. Ann Clin Lab Sci. 1989;19:190–195. [PubMed] [Google Scholar]
- 15.Lim CC, Apstein CS, Colucci WS, Liao R. Impaired cell shortening and relengthening with increased pacing frequency are intrinsic to the senescent mouse cardiomyocyte. J Mol Cell Cardiol. 2000;32:2075–2082. doi: 10.1006/jmcc.2000.1239. [DOI] [PubMed] [Google Scholar]
- 16.del Monte F, Harding SE, Schmidt U, Matsui T, Kang ZB, Dec GW, Gwathmey JK, Rosenzweig A, Hajjar RJ. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation. 1999;100:2308–2311. doi: 10.1161/01.cir.100.23.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 18.Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 19.Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem. 2005;280:17294–17300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- 20.Rogaev EI, Sherrington R, Wu C, Levesque G, Liang Y, Rogaeva EA, Ikeda M, Holman K, Lin C, Lukiw WJ, de Jong PJ, Fraser PE, Rommens JM, St George-Hyslop P. Analysis of the 5′ sequence, genomic structure, and alternative splicing of the presenilin-1 gene (PSEN1) associated with early onset Alzheimer disease. Genomics. 1997;40:415–424. doi: 10.1006/geno.1996.4523. [DOI] [PubMed] [Google Scholar]
- 21.Sandbrink R, Zhang D, Schaeffer S, Masters CL, Bauer J, Forstl H, Beyreuther K. Missense mutations of the PS-1/S182 gene in German early-onset Alzheimer’s disease patients. Ann Neurol. 1996;40:265–266. doi: 10.1002/ana.410400225. [DOI] [PubMed] [Google Scholar]
- 22.Cruts M, van Duijn CM, Backhovens H, Van den Broeck M, Wehnert A, Serneels S, Sherrington R, Hutton M, Hardy J, St George-Hyslop PH, Hofman A, Van Broeckhoven C. Estimation of the genetic contribution of presenilin-1 and -2 mutations in a population-based study of presenile Alzheimer disease. Hum Mol Genet. 1998;7:43–51. doi: 10.1093/hmg/7.1.43. [DOI] [PubMed] [Google Scholar]
- 23.Pastorcic M, Das HK. Alternative initiation of transcription of the human presenilin 1 gene in SH-SY5Y and SK-N-SH cells. The role of Ets factors in the regulation of presenilin 1. Eur J Biochem. 2004;271:4485–4494. doi: 10.1111/j.1432-1033.2004.04453.x. [DOI] [PubMed] [Google Scholar]
- 24.Green KN, Demuro A, Akbari Y, Hitt BD, Smith IF, Parker I, LaFerla FM. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J Cell Biol. 2008;181:1107–1116. doi: 10.1083/jcb.200706171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Albani D, Roiter I, Artuso V, Batelli S, Prato F, Pesaresi M, Galimberti D, Scarpini E, Bruni A, Franceschi M, Piras MR, Confaloni A, Forloni G. Presenilin-1 mutation E318G and familial Alzheimer’s disease in the Italian population. Neurobiol Aging. 2007;28:1682–1688. doi: 10.1016/j.neurobiolaging.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 26.Gorza L, FdM Protein unfolding in cardiomyopathies. Heart Failure Clinic. 2005;1:237–250. doi: 10.1016/j.hfc.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 27.Gething MJ, Sambrook J. Protein folding in the cell. Nature. 1992;355(6355):33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- 28.Cornwell GG, 3rd, Murdoch WL, Kyle RA, Westermark P, Pitkanen P. Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am J Med. 1983;75:618–623. doi: 10.1016/0002-9343(83)90443-6. [DOI] [PubMed] [Google Scholar]
- 29.de Exel Nunes LM, Salge AK, de Oliveira FA, Teixeira Vde P, Dos Reis MA. Cerebral and cardiac amyloidosis in autopsied elderly individuals. Clinics. 2006;61:113–118. doi: 10.1590/s1807-59322006000200005. [DOI] [PubMed] [Google Scholar]
- 30.Steiner I, Hajkova P. Patterns of isolated atrial amyloid: a study of 100 hearts on autopsy. Cardiovasc Pathol. 2006;15:287–290. doi: 10.1016/j.carpath.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 31.Lansbury PT, Lashuel HA. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature. 2006;443:774–779. doi: 10.1038/nature05290. [DOI] [PubMed] [Google Scholar]
- 32.Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 33.del Monte F, Johnson CM, Stepanek AC, Doye AA, Gwathmey JK. Defects in calcium control. J Card Fail. 2002;8:S421–431. doi: 10.1054/jcaf.2002.129285. [DOI] [PubMed] [Google Scholar]
- 34.Trump BFBI, Toshihide S, Phelps PC, DeClaris N. Cell calcium, cell injury and cell death. Environmental health perspective. 1984;57:281–287. doi: 10.1289/ehp.8457281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chandrashekhr YNJ. Death Hath a Tousand Doors to let our life. Circulation Research. 2003;92:710–714. doi: 10.1161/01.RES.0000069364.24820.8B. [DOI] [PubMed] [Google Scholar]
- 36.Stevens FJ, Argon Y. Protein folding in the ER. Semin Cell Dev Biol. 1999;10:443–454. doi: 10.1006/scdb.1999.0315. [DOI] [PubMed] [Google Scholar]
- 37.Del Monte F, Hajjar RJ. Intracellular devastation in heart failure. Heart Fail Rev. 2008;13:151–162. doi: 10.1007/s10741-007-9071-9. [DOI] [PubMed] [Google Scholar]
- 38.Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, Hardy J, Levey AI, Gandy SE, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 39.Mah AL, Perry G, Smith MA, Monteiro MJ. Identification of ubiquilin, a novel presenilin interactor that increases presenilin protein accumulation. J Cell Biol. 2000;151:847–862. doi: 10.1083/jcb.151.4.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.To MD, Gokgoz N, Doyle TG, Donoviel DB, Knight JA, Hyslop PS, Bernstein A, Andrulis IL. Functional characterization of novel presenilin-2 variants identified in human breast cancers. Oncogene. 2006;25:3557–3564. doi: 10.1038/sj.onc.1209397. [DOI] [PubMed] [Google Scholar]
- 41.Barnes LL, Wilson RS, Bienias JL, Schneider JA, Evans DA, Bennett DA. Sex differences in the clinical manifestations of Alzheimer disease pathology. Arch Gen Psychiatry. 2005;62:685–691. doi: 10.1001/archpsyc.62.6.685. [DOI] [PubMed] [Google Scholar]
- 42.Grohe C, Kahlert S, Lobbert K, Stimpel M, Karas RH, Vetter H, Neyses L. Cardiac myocytes and fibroblasts contain functional estrogen receptors. FEBS Lett. 1997;416:107–112. doi: 10.1016/s0014-5793(97)01179-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.