

Abstract
Helical bundles which bind heme and porphyrin cofactors have been popular targets for cofactor-containing de novo protein design. By analyzing a highly non-redundant subset of the protein databank, we have determined a rotamer distribution for helical histidines bound to heme cofactors. Analysis of the entire non-redundant database for helical sequence preferences near the ligand histidine demonstrated little preference for amino acid side chain identity, size, or charge. Analysis of the database subdivided by ligand histidine rotamer, however, reveals strong preferences in each case, and computational modeling illuminates the structural basis for some of these findings. The majority of the rotamer distribution matches that predicted by molecular simulation of a single porphyrin-bound histidine residue placed in the center of an all-alanine helix, and the deviations explain two prominent features of natural heme protein binding sites: heme distortion in the case of the cytochromes C in the m166 histidine rotamer, and a highly prevalent glycine residue in the t73 histidine rotamer. These preferences permit derivation of helical consensus sequence templates which predict optimal side chain-cofactor packing interactions for each rotamer. These findings thus promise to guide future design endeavors not only in the creation of higher affinity heme and porphyrin binding sites, but in the direction of bound cofactor geometry.
Keywords: heme binding, rotamer, heme ruffle, bionanotechnology
Introduction
Designed proteins have great potential for a wide range of applications in medicine, sensing, ‘green’ industrial catalysis and energy production.1-3 An early direction in protein design was the creation of proteins which bind porphyrin and porphyrin-like cofactors.4-10 Designed proteins containing heme and chlorophyll cofactors have proven valuable in aiding the understanding of natural proteins: designed mimetic systems which recreate natural protein functions in a simplified context have enabled the separation and analysis of many of the underlying thermodynamics of cofactor-protein interactions.11-27 Several proteins which incorporate non-natural porphyrin and chlorin cofactors have been created as the initial steps in the creation of bionanoelectronic devices.28-35
For these reasons, improved methods for the design of high affinity heme and porphyrin binding sites are desirable. Heme binding is a molecular recognition problem that has many solutions. Metal-ligand coordination accounts for a large part of the binding affinity of hemes to proteins.3 Heme-protein packing interactions, however, also play a role. One common method used in protein engineering is that of ‘consensus design36-39: the amino acid sequences of a large number of related proteins are aligned and the most common amino acids at each position are chosen to engineer a new protein with improved properties such as thermostability or protease resistance. This method has the drawback that it requires a large number of related sequences, all of very similar folds, to be effective. Perhaps for this reason, it has not often been successfully applied to the design of cofactor-binding proteins, with the notable exception of the zinc finger family of divalent cation-binding proteins.36,40,41
Another method which has had some success, particularly in the design of mono- and dimetallic cofactor biding sites, is termed ‘retrostructural analysis42: the protein data bank (PDB) is surveyed for structural commonalities among the subset of proteins which bind the desired cofactor, and these simple structural features are then used as a consensus framework to implant cofactor binding sites into designed proteins.43-45 In contrast to consensus design, this method requires high resolution structures of a number of dissimilar folds in order to derive a minimal structural representation of the cofactor binding site. In the case of the diiron proteins, the analysis was performed on six different diiron proteins which are functionally and structurally unrelated.43 Each had, at the cofactor binding site, an arrangement of four helices each of which donates one chelating glutamate side chain into the center of the bundle, forming a distorted tetrahedral binding site. There was no other sequence or structural conservation in this limited set of proteins, but this minimal framework was sufficient to computationally design a number of dimetallic four helix bundle proteins with reactivities similar to their larger natural counterparts.44,46-48
Our goal is to derive a set of rules for binding porphyrin and porphyrin-like cofactors. Other groups have focused on aspects of these packing interactions using consensus methods: Huang et al. compiled the side chain torsion angles of histidine-bound hemes, identifying the major heme-bound histidine rotamer.21 Zaric et al looked at the relative orientation of the histidine and heme, finding that packing interactions with the histidine backbone and electrostatic interactions between the histidine NδH group and the heme propionate dominates.49 Paoli and coworkers surveyed the PDB for the frequency of amino acids involved in packing interactions with b-type heme cofactors, finding that the leucine, valine and phenylalanine side chains are most common in packing interactions with the heme face while leucine, alanine and phenylalanine are the most common heme edge packing residues.50 Benson and coworkers noted that the majority of packing interactions originated from amino acids in helical secondary structures11 and further noted that every heme protein structure in the PDB contained at least one aromatic side chain that engaged in a packing interaction with the heme cofactor.14
There are currently over 2000 structures of heme containing proteins in the PDB.3 This wealth of heme protein structures has allowed us to take a hybrid approach: starting with a retrostructural analysis of a nonredundent alpha helical subset of this database, we then analyze the sequence preferences within each structural subfamily. This has allowed us to isolate the amino acid identities that are forced upon the protein by heme cofactor packing interactions for each heme-bound histidine rotamer. Correspondingly, use of these packing interactions promises to allow the direction of bound porphyrin geometry. Finally, as is common in protein design endeavors, our analysis has explained several aspects of natural protein heme binding sites.
Materials and Methods
Defining a helical heme protein database
Dataset construction started with the March 2006 PDBSelect, a database of 3080 proteins derived from the entire PDB using a 25% sequence identity filter and a 3Ǻ resolution cutoff.51 Statistics were improved by adding selected nonhomologous multiheme structures. In house software, available by request from the authors, was used to identify α-helical histidines using the backbone torsion angles of residues from i-4 to i+4 thereby covering one full turn in each direction from the central histidine. If six or more of these nine residues had backbone torsion angles within helical boundaries (-82°<Φ<-42°, -60°<Ψ<-21°) the histidine was taken to be α-helical. Although these strict criteria will likely not identify every histidine on a helix, they identify histidines in the center of non-perturbed helices that consist of at least two full turns, resembling the intended target of most heme- and porphyrin-binding helical bundle designs. There were 6781 helical histidines in this database, and 215 of these were identified as heme ligands on the basis of the proximity of their imidazole ε nitrogen to a heme iron.
This database of histidines was further filtered by chain using the PISCES web server, which uses structure-based sequence alignments to identify more distant evolutionarily relationships.52 Using a 75% chain homology cutoff resulted in a database of 61 unrelated helical histidines ligated to hemes. PISCES uses a sorting algorithm that, at each step in the culling process, retains the chain with the highest resolution structure. The final database (see Supplementary Table 1) has an average resolution of 1.97 Ǻ and a resolution cutoff of 3.00 Ǻ. This database, while small, is equivalent in size to the original backbone-dependent rotamer library,53 the findings of which were not significantly changed in later studies using larger datasets.54 It represents a set of proteins unrelated in sequence or tertiary structure, allowing the determination of side chain identities forced on the helix by heme packing interaction geometry as opposed to evolution.
Molecular modeling
An idealized 17-residue alpha helix composed of alanine residues with the exception of the central histidine, was generated using Swiss PDB Viewer.55 A flat ferric 2,3,7,8,12,13,17,18-octamethyl porphyrin was generated and minimized using Jaguar (Shrödinger Inc.). This porphyrin was attached to the ligand residue using the histidine-porphyrin distance and relative planar orientations described as optimal by Zaric et al.49 Van der Waals energy surfaces were calculated as follows: histidine side chain χ1 and χ2 angles were incremented with a step size of 1°. After each rotation step the side chains and termini of the alpha helix were deleted and reoptimized. After this an energy minimization was performed keeping every atom but the alpha helix side chains, the termini and the heme core methyl groups fixed. Van der Waals energies were then calculated using CHARMM(22).56 Two surfaces were created by fixing the pseudo-torsion angle α, between the iron-porphyrin ring α carbon vector and the histidine N(ε)-C(ε), fixed at 0° and 45°. No significant difference was found between them. To examine the effects of shorter helices in the t-86 and m80 rotamers, energy surfaces were recalculated using idealized alanine helices which ended two residues before and two residues after the histidine. Neither alteration affected the energy landscape near that rotamer.
To calculate the structural consequences of a high-propensity glycine residue at i+4, energy surfaces were similarly generated using an idealized alpha helix containing a glycine residue at position 13. To calculate the structural consequences of heme ruffling, a ruffled heme taken from the 0.91 Ǻ resolution structure of the D. vulgaris cytochrome C (1J0P) was used. The propionates and vinyl substituents were replaced with methyl groups, leaving the central distorted 2,3,7,8,12,13,17,18-octamethyl porphyrin. The van der Waals energy surface was calculated as before, using the idealized 17-residue alanine helix. To recreate the shortened van der Waals packing interaction made possible by covalent bond formation, α-chloroglycine residues were introduced at positions i-1 and i-4 to the central histidine.
Results
Heme-ligand histidine rotamer distribution
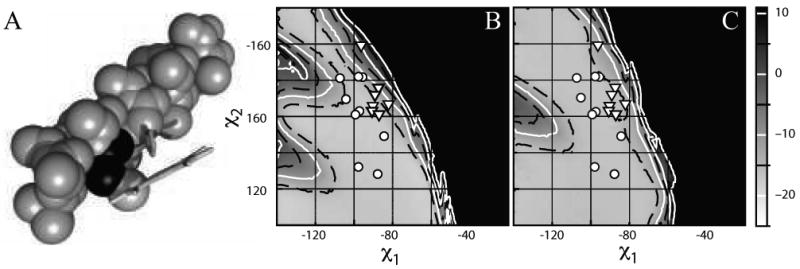
The torsional distribution of the heme ligand histidines in the nonredundant helical dataset are depicted in Figure 1A. The distribution clusters into four rotamers, two less than the distribution observed in the dataset of 6781 nonredundant helical non-ligand histidines (see Table 1) and two less than that observed by Lovell et al in their enumeration of backbone dependent non-ligand histidine side chain rotamers.57 Each of the four ligand histidine rotamers corresponds to a rotamer in the non-ligand distribution, leaving the t-177 and the m-64 non-ligand rotamers lacking heme-ligand congeners.
Figure 1.
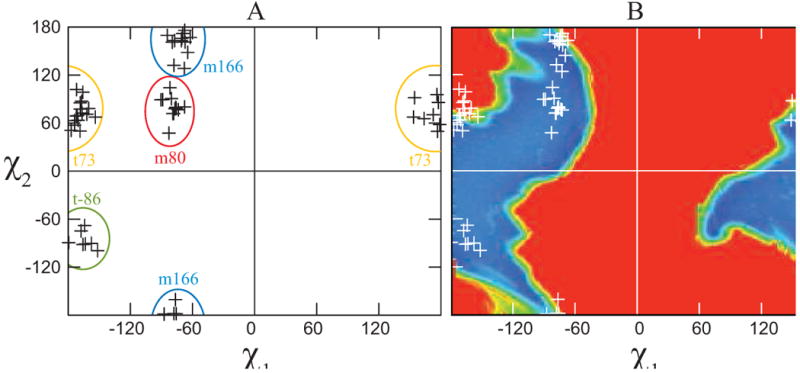
The rotameric preferences of a helical heme-bound histidine. (A) Distribution of the side chain torsion angles in the nonredundant dataset of helical, heme-bound histidines. Rotamers are separated by colored ovals. (B) Van der Waals energy landscape of a histidine-bound porphyrin in the center of a 17-residue alanine helix. A model complex, consisting of an idealized alanine helix with a central histidine ligated via the Nε atom to a porphyrin core consisting of a ferric porphyrin ring with methyl groups at each beta position was used to estimate the van der Waals energy landscape as a function of the histidine chi1 and chi2 using CHARMM22. For comparative purposes, the data from part A are superimposed on this landscape in white.
Table 1.
Rotamer distribution of helical histidines, with and without bound hemes.
| Heme ligand His | All helical His | Lovell et al.a | |||||||
|---|---|---|---|---|---|---|---|---|---|
| rotamer | # (%) | χ1 (S.D.) | χ2 (S.D.) | rotamer | # (%) | χ1 (S.D.) | χ2 (S.D.) | rotamer | # (%) |
| m80 | 12 (20%) | -79 (6) | 80 (14) | m98 | 869 (13%) | -73 (11) | 98 (26) | m80 | 17 (14%) |
| m166 | 16 (27%) | -73 (7) | 166 (18) | m172 | 591 (9%) | -69 (7) | 172 (14) | m170 | 11 (9%) |
| t73 | 24 (41%) | -175 (13) | 73 (15) | t73 | 1859 (28%) | 179 (10) | 73 (17) | t60 | 30 (24%) |
| t-86 | 7 (12%) | -164 (9) | -86 (11) | t-94 | 1114 (17%) | -179 (10) | -94 (19) | t-80 | 21 (17%) |
| t-177 | 364 (6%) | -170 (9) | -177 (20) | t-160 | 6 (5%) | ||||
| m-64 | 1554 (24%) | -71 (10) | -64 (29) | m-70 | 32 (26%) | ||||
Rotamer names obtained by Lovell are derived using all types of secondary structure, however the number and percent displayed are exclusively derived from helices.
In order to examine the origins of this restricted conformational space, a van der Waals energy surface was calculated using an idealized alanine helix with a central histidine ligand. This histidine was attached to an octamethyl porphyrin and the van der Waals energy calculated as a function of the histidine side chain conformation using CHARM22. Figure 1B explains the missing rotamers in the heme-ligand dataset: steric clash between the porphyrin macrocycle and the helix preclude the histidine occupying these rotamers in a helical context. Two of the rotamers, m80 and t-86, fit squarely in the center of minima in the van der Waals surface. Two others, however, lie at marginal regions of the energy surface: t73 and m166. These deviations explain two common features of natural helical heme protein binding sites, and will be discussed in detail below.
The majority of designed heme proteins created to date pack the porphyrin cofactor between a pair of helices, each of which donate a single histidine ligand.3 Thus it is informative to examine the rotameric preferences of the histidine residues involved in bis-histidine ligation where both histidines are on alpha helices. As Table 2 demonstrates in such binding sites almost 3 out of 4 hemes are ligated to two histidines in the t73 rotamer. This leads to the conclusion that for helical bundle protein designs with bis-histidine porphyrin binding sites, the t73 rotamer is the most energetically favorable rotamer for both histidine ligands.
Table 2.
Rotamer pair distribution in bis-histidine ligated hemes in which both histidines reside on a helix.
| m80 | 0% | |||
| m166 | 14% (3) | 0% | ||
| t73 | 9% (2) | 0% | 73% (16) | |
| t-86 | 0% | 5% (1) | 0% | 0% |
| m80 | m166 | t73 | t-86 | |
Helix-porphyrin contact frequencies
The observation of discrete rotameric preferences for helical heme-bound histidines leads to the prediction that each rotamer places the cofactor in a position to make contact with a different subset of helix residues. To examine this quantitatively, each residue in each heme-bound helix in the database was examined to determine whether it was in van der Waals contact with the cofactor. In order to derive results applicable to all porphyrin cofactors, contacts were only counted with the octamethyl porphyrin macrocycle core, not heme propionate groups or hydroxyfarnasyl tails. Positions in which the helical rotameric population was less than five were discarded as being not statistically significant. Supplementary Figure 1 depicts the helical residue counts by position for each rotamer in the database.
As Figure 2 shows, the frequencies with which different helical residues contact the porphyrin ring varies significantly with the χ1 angle: the m rotamers make contacts principally at the i-4 and i-1 positions of the helix on which they are ligated, while the t rotamers make contacts at i+4 and i+7. It is interesting to note that, for the t rotamer, there are a significant number of contacts another full turn of the helix from the heme ligand. Especially in t73 rotamer, the positions i-7, i+7 and i+8 all make contacts with the heme 25% of the time or more.
Figure 2.
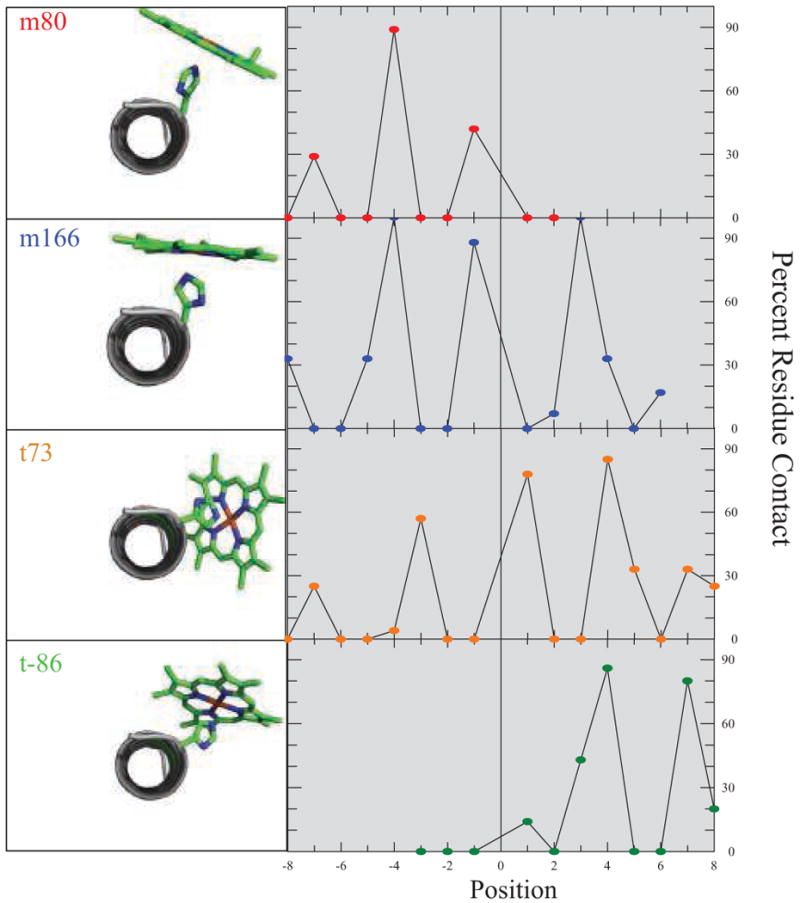
Porphyrin-helix contact frequencies are rotamer dependent. On the left of each panel is a C-terminal view of an identically oriented idealized alanine helix with a central Nε porphyrin-bound histidine side chain fixed at the mean χ1 and χ2 torsion angles for each rotamer. On the right is the positional contact frequency for that rotamer, determined using a 4.1 Å van der Waals contact cutoff. Only helical residues from each PDB file were used and contacts were catalogued only with the porphyrin macrocycle. Residue counts less than five are not depicted due to statistical uncertainty.
Rotamer dependence of helical residue frequencies and propensities
There is some debate as to whether residue frequencies or residue propensities are better indicators of the importance of a particular amino acid.58 Propensities, which are defined as the ratio of the frequency of a particular residue in a certain context to the frequency with which that residue appears in proteins as a whole or within a particular element of secondary structure, have proven valuable in discerning the importance of residues that are normally unlikely to appear.59 For example, as glycine residues energetically disfavor helix propagation,60 studies which use residue propensities to examine glycine populations in helices have uncovered the utility of glycine in helix capping motifs even when they are infrequently present in these motifs.59,61 However, propensities are not effective at revealing the importance of interactions involving residues which normally appear at a high frequency. For example Magliery et al. showed that leucine-leucine packing interactions within the core of the helical ROP-like proteins are critical to stability and function, but are not uncovered using propensity scales due to the high frequency with which leucine is normally present in helices.58 For this reason, we have elected to utilize both frequencies and propensities in our examination of the interactions between helices and bound hemes. Residue frequencies in the heme-ligand helix database are depicted in Table 3A-F. Propensities, determined using the ratio of the frequencies in Tables 3 to the helical residue frequencies present in the entire 6781-member nonredundant helical database, are enumerated in Supplementary Table 2A-F.
Table 3.
Position-Specific Amino Acid Frequencies for Helices with Hemes Bound to Histidines
| A. All Helices | |||||||||||||||||
| ALA | 13.8% | 5.4% | 9.5% | 17.4% | 4.0% | 10.9% | 14.3% | 19.0% | 0% | 14.8% | 10.5% | 13.3% | 20.0% | 5.7% | 3.2% | 0.0% | 8.3% |
| ARG | 0.0% | 5.4% | 4.8% | 8.7% | 4.0% | 10.9% | 5.4% | 1.7% | 0% | 1.6% | 7.0% | 6.7% | 0.0% | 2.9% | 12.9% | 4.2% | 8.3% |
| ASN | 0.0% | 2.7% | 4.8% | 2.2% | 0.0% | 3.6% | 1.8% | 1.7% | 0% | 1.6% | 7.0% | 2.2% | 2.5% | 0.0% | 3.2% | 8.3% | 0.0% |
| ASP | 0.0% | 0.0% | 0.0% | 6.5% | 2.0% | 0.0% | 3.6% | 0.0% | 0% | 4.9% | 7.0% | 2.2% | 0.0% | 0.0% | 0.0% | 0.0% | 4.2% |
| CYS | 0.0% | 0.0% | 0.0% | 0.0% | 18.0% | 0.0% | 0.0% | 15.5% | 0% | 4.9% | 0.0% | 6.7% | 0.0% | 0.0% | 12.9% | 0.0% | 0.0% |
| GLU | 0.0% | 0.0% | 9.5% | 0.0% | 2.0% | 1.8% | 5.4% | 0.0% | 0% | 4.9% | 7.0% | 4.4% | 2.5% | 8.6% | 3.2% | 0.0% | 4.2% |
| GLN | 0.0% | 0.0% | 7.1% | 4.3% | 0.0% | 1.8% | 1.8% | 1.7% | 0% | 4.9% | 3.5% | 2.2% | 0.0% | 5.7% | 0.0% | 8.3% | 0.0% |
| GLY | 10.3% | 13.5% | 0.0% | 4.3% | 4.0% | 0.0% | 7.1% | 1.7% | 0% | 4.9% | 8.8% | 6.7% | 22.5% | 8.6% | 6.5% | 0.0% | 0.0% |
| HIS | 0.0% | 8.1% | 0.0% | 4.3% | 0.0% | 5.5% | 1.8% | 0.0% | 100% | 0.0% | 7.0% | 2.2% | 0.0% | 5.7% | 3.2% | 20.8% | 0.0% |
| ILE | 13.8% | 16.2% | 2.4% | 6.5% | 14.0% | 7.3% | 7.1% | 8.6% | 0% | 3.3% | 1.8% | 4.4% | 12.5% | 22.9% | 19.4% | 12.5% | 4.2% |
| LEU | 6.9% | 13.5% | 9.5% | 10.9% | 12.0% | 9.1% | 7.1% | 15.5% | 0% | 11.5% | 3.5% | 13.3% | 10.0% | 8.6% | 0.0% | 8.3% | 4.2% |
| LYS | 3.4% | 2.7% | 14.3% | 4.3% | 6.0% | 7.3% | 3.6% | 6.9% | 0% | 4.9% | 7.0% | 2.2% | 0.0% | 5.7% | 0.0% | 0.0% | 4.2% |
| MET | 6.9% | 2.7% | 4.8% | 4.3% | 6.0% | 5.5% | 3.6% | 6.9% | 0% | 4.9% | 3.5% | 2.2% | 7.5% | 5.7% | 6.5% | 0.0% | 16.7% |
| PHE | 3.4% | 8.1% | 4.8% | 6.5% | 6.0% | 10.9% | 1.8% | 6.9% | 0% | 6.6% | 5.3% | 4.4% | 5.0% | 8.6% | 6.5% | 16.7% | 29.2% |
| PRO | 0.0% | 2.7% | 0.0% | 0.0% | 2.0% | 0.0% | 3.6% | 0.0% | 0% | 3.3% | 0.0% | 0.0% | 5.0% | 0.0% | 0.0% | 0.0% | 0.0% |
| SER | 3.4% | 0.0% | 2.4% | 2.2% | 6.0% | 5.5% | 10.7% | 3.4% | 0% | 3.3% | 0.0% | 2.2% | 0.0% | 8.6% | 6.5% | 0.0% | 0.0% |
| THR | 17.2% | 0.0% | 9.5% | 6.5% | 2.0% | 1.8% | 5.4% | 0.0% | 0% | 6.6% | 7.0% | 2.2% | 0.0% | 2.9% | 6.5% | 0.0% | 4.2% |
| TRP | 0.0% | 2.7% | 4.8% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0% | 1.6% | 5.3% | 2.2% | 0.0% | 0.0% | 6.5% | 4.2% | 0.0% |
| TYR | 10.3% | 5.4% | 7.1% | 4.3% | 6.0% | 1.8% | 7.1% | 8.6% | 0% | 4.9% | 1.8% | 8.9% | 0.0% | 0.0% | 3.2% | 8.3% | 0.0% |
| VAL | 10.3% | 10.8% | 4.8% | 6.5% | 6.0% | 16.4% | 8.9% | 1.7% | 0% | 6.6% | 7.0% | 11.1% | 12.5% | 0.0% | 0.0% | 8.3% | 12.5% |
| P-8 | P-7 | P-6 | P-5 | P-4 | P-3 | P-2 | P-1 | P0 | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | |
| Total | 29 | 37 | 42 | 46 | 50 | 55 | 56 | 58 | 60 | 61 | 57 | 45 | 40 | 35 | 31 | 24 | 24 |
| B. t73 | |||||||||||||||||
| ALA | 0.0% | 5.0% | 5.0% | 9.5% | 8.7% | 4.3% | 13.0% | 17.4% | 0% | 21.7% | 9.1% | 9.1% | 15.0% | 5.6% | 6.3% | 0.0% | 8.3% |
| ARG | 0.0% | 0.0% | 5.0% | 14.3% | 0.0% | 17.4% | 4.3% | 0.0% | 0% | 4.3% | 0.0% | 4.5% | 0.0% | 0.0% | 12.5% | 0.0% | 0.0% |
| ASN | 0.0% | 0.0% | 5.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 13.6% | 4.5% | 5.0% | 0.0% | 0.0% | 0.0% | 0.0% |
| ASP | 0.0% | 0.0% | 0.0% | 4.8% | 0.0% | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 0.0% | 4.5% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% |
| CYS | 0.0% | 0.0% | 0.0% | 0.0% | 4.3% | 0.0% | 0.0% | 0.0% | 0% | 4.3% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% |
| GLU | 0.0% | 0.0% | 5.0% | 0.0% | 4.3% | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% |
| GLN | 0.0% | 0.0% | 10.0% | 4.8% | 0.0% | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 16.7% | 0.0% |
| GLY | 14.3% | 15.0% | 0.0% | 0.0% | 8.7% | 0.0% | 0.0% | 0.0% | 0% | 8.7% | 4.5% | 9.1% | 45.0% | 0.0% | 6.3% | 0.0% | 0.0% |
| HIS | 0.0% | 0.0% | 0.0% | 4.8% | 0.0% | 0.0% | 0.0% | 0.0% | 100% | 0.0% | 13.6% | 0.0% | 0.0% | 11.1% | 0.0% | 16.7% | 0.0% |
| ILE | 28.6% | 25.0% | 5.0% | 14.3% | 17.4% | 8.7% | 13.0% | 17.4% | 0% | 0.0% | 4.5% | 4.5% | 5.0% | 33.3% | 31.3% | 25.0% | 8.3% |
| LEU | 7.1% | 15.0% | 10.0% | 9.5% | 4.3% | 21.7% | 13.0% | 26.1% | 0% | 13.0% | 9.1% | 13.6% | 10.0% | 11.1% | 0.0% | 16.7% | 8.3% |
| LYS | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 4.3% | 0% | 0.0% | 0.0% | 4.5% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% |
| MET | 7.1% | 5.0% | 5.0% | 9.5% | 8.7% | 8.7% | 8.7% | 17.4% | 0% | 8.7% | 4.5% | 4.5% | 5.0% | 11.1% | 6.3% | 0.0% | 16.7% |
| PHE | 7.1% | 10.0% | 10.0% | 14.3% | 8.7% | 17.4% | 4.3% | 4.3% | 0% | 13.0% | 9.1% | 9.1% | 0.0% | 16.7% | 12.5% | 16.7% | 41.7% |
| PRO | 0.0% | 5.0% | 0.0% | 0.0% | 4.3% | 0.0% | 0.0% | 0.0% | 0% | 4.3% | 0.0% | 0.0% | 10.0% | 0.0% | 0.0% | 0.0% | 0.0% |
| SER | 0.0% | 0.0% | 0.0% | 0.0% | 8.7% | 0.0% | 8.7% | 0.0% | 0% | 0.0% | 0.0% | 4.5% | 0.0% | 11.1% | 0.0% | 0.0% | 0.0% |
| THR | 21.4% | 0.0% | 10.0% | 4.8% | 0.0% | 0.0% | 13.0% | 0.0% | 0% | 4.3% | 0.0% | 0.0% | 0.0% | 0.0% | 6.3% | 0.0% | 0.0% |
| TRP | 0.0% | 0.0% | 10.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 13.6% | 4.5% | 0.0% | 0.0% | 12.5% | 0.0% | 0.0% |
| TYR | 7.1% | 10.0% | 10.0% | 4.8% | 13.0% | 4.3% | 13.0% | 8.7% | 0% | 4.3% | 4.5% | 9.1% | 0.0% | 0.0% | 6.3% | 0.0% | 0.0% |
| VAL | 7.1% | 10.0% | 10.0% | 4.8% | 8.7% | 17.4% | 8.7% | 4.3% | 0% | 13.0% | 13.6% | 13.6% | 5.0% | 0.0% | 0.0% | 8.3% | 16.7% |
| P-8 | P-7 | P-6 | P-5 | P-4 | P-3 | P-2 | P-1 | P0 | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | |
| Total | 14 | 20 | 20 | 21 | 23 | 23 | 23 | 23 | 24 | 23 | 22 | 22 | 20 | 18 | 16 | 12 | 12 |
| C.t73 bis-histidine | |||||||||||||||||
| ALA | 0.0% | 6.7% | 6.7% | 13.3% | 6.7% | 6.7% | 20.0% | 6.7% | 0% | 26.7% | 13.3% | 13.3% | 14.3% | 0.0% | 8.3% | 0.0% | 10.0% |
| ARG | 0.0% | 0.0% | 6.7% | 20.0% | 0.0% | 26.7% | 0.0% | 0.0% | 0% | 6.7% | 0.0% | 6.7% | 0.0% | 0.0% | 16.7% | 0.0% | 0.0% |
| ASN | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 13.3% | 6.7% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% |
| ASP | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% |
| CYS | 0.0% | 0.0% | 0.0% | 0.0% | 6.7% | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% |
| GLU | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% |
| GLN | 0.0% | 0.0% | 13.3% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 20.0% | 0.0% |
| GLY | 16.7% | 20.0% | 0.0% | 0.0% | 6.7% | 0.0% | 0.0% | 0.0% | 0% | 6.7% | 6.7% | 13.3% | 57.1% | 0.0% | 8.3% | 0.0% | 0.0% |
| HIS | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 100% | 0.0% | 6.7% | 0.0% | 0.0% | 14.3% | 0.0% | 20.0% | 0.0% |
| ILE | 16.7% | 33.3% | 0.0% | 20.0% | 20.0% | 6.7% | 13.3% | 20.0% | 0% | 0.0% | 6.7% | 6.7% | 0.0% | 35.7% | 33.3% | 30.0% | 10.0% |
| LEU | 8.3% | 13.3% | 13.3% | 6.7% | 6.7% | 26.7% | 13.3% | 20.0% | 0% | 13.3% | 13.3% | 6.7% | 14.3% | 14.3% | 0.0% | 10.0% | 10.0% |
| LYS | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 0.0% | 6.7% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% |
| MET | 8.3% | 0.0% | 6.7% | 13.3% | 6.7% | 6.7% | 13.3% | 26.7% | 0% | 6.7% | 6.7% | 0.0% | 0.0% | 7.1% | 8.3% | 0.0% | 20.0% |
| PHE | 8.3% | 13.3% | 13.3% | 13.3% | 13.3% | 13.3% | 6.7% | 6.7% | 0% | 6.7% | 6.7% | 13.3% | 0.0% | 14.3% | 8.3% | 20.0% | 30.0% |
| PRO | 0.0% | 6.7% | 0.0% | 0.0% | 6.7% | 0.0% | 0.0% | 0.0% | 0% | 6.7% | 0.0% | 0.0% | 14.3% | 0.0% | 0.0% | 0.0% | 0.0% |
| SER | 0.0% | 0.0% | 0.0% | 0.0% | 13.3% | 0.0% | 13.3% | 0.0% | 0% | 0.0% | 0.0% | 6.7% | 0.0% | 14.3% | 0.0% | 0.0% | 0.0% |
| THR | 25.0% | 0.0% | 13.3% | 6.7% | 0.0% | 0.0% | 0.0% | 0.0% | 0% | 6.7% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% |
| TRP | 0.0% | 0.0% | 13.3% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 20.0% | 6.7% | 0.0% | 0.0% | 8.3% | 0.0% | 0.0% |
| TYR | 8.3% | 0.0% | 6.7% | 6.7% | 6.7% | 6.7% | 20.0% | 13.3% | 0% | 6.7% | 0.0% | 0.0% | 0.0% | 0.0% | 8.3% | 0.0% | 0.0% |
| VAL | 8.3% | 6.7% | 6.7% | 0.0% | 6.7% | 6.7% | 0.0% | 6.7% | 0% | 13.3% | 6.7% | 13.3% | 0.0% | 0.0% | 0.0% | 0.0% | 20.0% |
| P-8 | P-7 | P-6 | P-5 | P-4 | P-3 | P-2 | P-1 | P0 | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | |
| Total | 12 | 15 | 15 | 15 | 15 | 15 | 15 | 15 | 16 | 15 | 15 | 15 | 14 | 14 | 12 | 10 | 10 |
| D.t-86 | |||||||||||||||||
| ALA | 0.0% | 16.7% | 50.0% | 0% | 14.3% | 0.0% | 28.6% | 0.0% | 0.0% | 0.0% | 0.0% | 20.0% | |||||
| ARG | 20.0% | 33.3% | 0.0% | 0% | 0.0% | 14.3% | 14.3% | 0.0% | 20.0% | 40.0% | 0.0% | 20.0% | |||||
| ASN | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 20.0% | 0.0% | |||||
| ASP | 0.0% | 0.0% | 0.0% | 0% | 14.3% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | |||||
| CYS | 0.0% | 0.0% | 16.7% | 0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 20.0% | 0.0% | 0.0% | |||||
| GLU | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 14.3% | 0.0% | 14.3% | 0.0% | 0.0% | 0.0% | 0.0% | |||||
| GLN | 20.0% | 0.0% | 0.0% | 0% | 14.3% | 0.0% | 0.0% | 0.0% | 20.0% | 0.0% | 0.0% | 0.0% | |||||
| GLY | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 28.6% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | |||||
| HIS | 0.0% | 0.0% | 0.0% | 100% | 0.0% | 0.0% | 14.3% | 0.0% | 0.0% | 20.0% | 0.0% | 0.0% | |||||
| ILE | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 0.0% | 0.0% | 28.6% | 40.0% | 0.0% | 0.0% | 0.0% | |||||
| LEU | 0.0% | 0.0% | 16.7% | 0% | 0.0% | 0.0% | 0.0% | 14.3% | 20.0% | 0.0% | 0.0% | 0.0% | |||||
| LYS | 40.0% | 16.7% | 0.0% | 0% | 0.0% | 14.3% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | |||||
| MET | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 14.3% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 20.0% | |||||
| PHE | 20.0% | 0.0% | 0.0% | 0% | 0.0% | 0.0% | 0.0% | 14.3% | 0.0% | 0.0% | 20.0% | 40.0% | |||||
| PRO | 0.0% | 16.7% | 0.0% | 0% | 14.3% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | |||||
| SER | 0.0% | 16.7% | 16.7% | 0% | 14.3% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | |||||
| THR | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 14.3% | 0.0% | 0.0% | 0.0% | 20.0% | 0.0% | 0.0% | |||||
| TRP | 0.0% | 0.0% | 0.0% | 0% | 14.3% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | |||||
| TYR | 0.0% | 0.0% | 0.0% | 0% | 14.3% | 0.0% | 28.6% | 0.0% | 0.0% | 0.0% | 40.0% | 0.0% | |||||
| VAL | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 0.0% | 14.3% | 28.6% | 0.0% | 0.0% | 20.0% | 0.0% | |||||
| P-3 | P-2 | P-1 | P0 | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | ||||||
| Total | 5 | 6 | 6 | 7 | 7 | 7 | 7 | 7 | 5 | 5 | 5 | 5 | |||||
| E.m166 | |||||||||||||||||
| ALA | 33.3% | 11.1% | 20.0% | 16.7% | 0.0% | 21.4% | 28.6% | 18.8% | 0% | 12.5% | 6.7% | 10.0% | 44.4% | 0.0% | 0.0% | ||
| ARG | 0.0% | 11.1% | 10.0% | 8.3% | 7.1% | 7.1% | 0.0% | 0.0% | 0% | 0.0% | 13.3% | 10.0% | 0.0% | 0.0% | 0.0% | ||
| ASN | 0.0% | 0.0% | 10.0% | 8.3% | 0.0% | 7.1% | 0.0% | 0.0% | 0% | 6.3% | 6.7% | 0.0% | 0.0% | 0.0% | 16.7% | ||
| ASP | 0.0% | 0.0% | 0.0% | 0.0% | 7.1% | 0.0% | 0.0% | 0.0% | 0% | 6.3% | 13.3% | 0.0% | 0.0% | 0.0% | 0.0% | ||
| CYS | 0.0% | 0.0% | 0.0% | 0.0% | 50.0% | 0.0% | 0.0% | 43.8% | 0% | 6.3% | 0.0% | 30.0% | 0.0% | 0.0% | 50.0% | ||
| GLU | 0.0% | 0.0% | 10.0% | 0.0% | 0.0% | 7.1% | 7.1% | 0.0% | 0% | 12.5% | 20.0% | 0.0% | 0.0% | 37.5% | 0.0% | ||
| GLN | 0.0% | 0.0% | 10.0% | 8.3% | 0.0% | 0.0% | 0.0% | 0.0% | 0% | 12.5% | 13.3% | 0.0% | 0.0% | 0.0% | 0.0% | ||
| GLY | 0.0% | 22.2% | 0.0% | 0.0% | 0.0% | 0.0% | 28.6% | 0.0% | 0% | 6.3% | 0.0% | 0.0% | 0.0% | 25.0% | 0.0% | ||
| HIS | 0.0% | 33.3% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 100% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | ||
| ILE | 0.0% | 0.0% | 0.0% | 0.0% | 14.3% | 14.3% | 7.1% | 6.3% | 0% | 0.0% | 0.0% | 0.0% | 22.2% | 0.0% | 0.0% | ||
| LEU | 11.1% | 0.0% | 0.0% | 25.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0% | 12.5% | 0.0% | 30.0% | 0.0% | 0.0% | 0.0% | ||
| LYS | 11.1% | 11.1% | 20.0% | 8.3% | 14.3% | 14.3% | 0.0% | 12.5% | 0% | 6.3% | 20.0% | 0.0% | 0.0% | 25.0% | 0.0% | ||
| MET | 0.0% | 0.0% | 0.0% | 0.0% | 7.1% | 7.1% | 0.0% | 0.0% | 0% | 6.3% | 0.0% | 0.0% | 22.2% | 0.0% | 16.7% | ||
| PHE | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | ||
| PRO | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | ||
| SER | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 21.4% | 0.0% | 0% | 0.0% | 0.0% | 0.0% | 0.0% | 12.5% | 16.7% | ||
| THR | 11.1% | 0.0% | 20.0% | 8.3% | 0.0% | 7.1% | 0.0% | 0.0% | 0% | 12.5% | 6.7% | 10.0% | 0.0% | 0.0% | 0.0% | ||
| TRP | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | ||
| TYR | 22.2% | 0.0% | 0.0% | 8.3% | 0.0% | 0.0% | 0.0% | 18.8% | 0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | ||
| VAL | 11.1% | 11.1% | 0.0% | 8.3% | 0.0% | 14.3% | 7.1% | 0.0% | 0% | 0.0% | 0.0% | 10.0% | 11.1% | 0.0% | 0.0% | ||
| P-8 | P-7 | P-6 | P-5 | P-4 | P-3 | P-2 | P-1 | P0 | P1 | P2 | P3 | P4 | P5 | P6 | |||
| Total | 9 | 9 | 10 | 12 | 14 | 14 | 14 | 16 | 16 | 16 | 15 | 10 | 9 | 8 | 6 | ||
| F.m80 | |||||||||||||||||
| ALA | 16.7% | 0.0% | 11.1% | 22.2% | 0.0% | 16.7% | 0.0% | 8.3% | 0% | 8.3% | 20.0% | ||||||
| ARG | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 8.3% | 0% | 0.0% | 0.0% | ||||||
| ASN | 0.0% | 14.3% | 0.0% | 0.0% | 0.0% | 8.3% | 8.3% | 8.3% | 0% | 0.0% | 0.0% | ||||||
| ASP | 0.0% | 0.0% | 0.0% | 11.1% | 0.0% | 0.0% | 16.7% | 0.0% | 0% | 8.3% | 10.0% | ||||||
| CYS | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0% | 8.3% | 0.0% | ||||||
| GLU | 0.0% | 0.0% | 11.1% | 0.0% | 0.0% | 0.0% | 8.3% | 0.0% | 0% | 8.3% | 0.0% | ||||||
| GLN | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 8.3% | 8.3% | 0% | 0.0% | 0.0% | ||||||
| GLY | 16.7% | 0.0% | 0.0% | 11.1% | 0.0% | 0.0% | 0.0% | 8.3% | 0% | 0.0% | 20.0% | ||||||
| HIS | 0.0% | 0.0% | 0.0% | 11.1% | 0.0% | 25.0% | 8.3% | 0.0% | 100% | 0.0% | 10.0% | ||||||
| ILE | 0.0% | 14.3% | 0.0% | 0.0% | 11.1% | 0.0% | 0.0% | 0.0% | 0% | 16.7% | 0.0% | ||||||
| LEU | 0.0% | 28.6% | 22.2% | 0.0% | 55.6% | 0.0% | 8.3% | 16.7% | 0% | 8.3% | 0.0% | ||||||
| LYS | 0.0% | 0.0% | 33.3% | 11.1% | 0.0% | 0.0% | 8.3% | 8.3% | 0% | 8.3% | 0.0% | ||||||
| MET | 16.7% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 0.0% | ||||||
| PHE | 0.0% | 14.3% | 0.0% | 0.0% | 11.1% | 8.3% | 0.0% | 25.0% | 0% | 8.3% | 10.0% | ||||||
| PRO | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 8.3% | 0.0% | 0% | 0.0% | 0.0% | ||||||
| SER | 16.7% | 0.0% | 11.1% | 11.1% | 11.1% | 25.0% | 0.0% | 8.3% | 0% | 0.0% | 0.0% | ||||||
| THR | 16.7% | 0.0% | 0.0% | 11.1% | 0.0% | 0.0% | 0.0% | 0.0% | 0% | 8.3% | 20.0% | ||||||
| TRP | 0.0% | 14.3% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0% | 0.0% | 0.0% | ||||||
| TYR | 0.0% | 0.0% | 11.1% | 0.0% | 0.0% | 0.0% | 8.3% | 0.0% | 0% | 8.3% | 0.0% | ||||||
| VAL | 16.7% | 14.3% | 0.0% | 11.1% | 11.1% | 16.7% | 16.7% | 0.0% | 0% | 8.3% | 10.0% | ||||||
| P-8 | P-7 | P-6 | P-5 | P-4 | P-3 | P-2 | P-1 | P0 | P1 | P2 | |||||||
| Total | 6 | 7 | 9 | 9 | 9 | 12 | 12 | 12 | 12 | 12 | 10 | ||||||
Table 3A depicts the helical residue frequencies determined for every protein in the database as a function of the sequential distance from the histidine ligand. The only residue with a frequency greater than 25% is a phenylalanine residue at position i+8. This residue makes a t-stacking interaction with the porphyrin edge. This interaction has been examined in detail by Benson and coworkers, who determined using mutational analysis on the model cytochrome PSM-1 that this interaction stabilizes heme-bound helices.14 With this exception, there are no residues present at high enough frequency that they could be used to create a consensus sequence for heme-helix binding, and no significant variations in average volume or charge (data not shown).
This situation changes substantially when the database is re-examined as a function of the heme-ligand histidine rotamer. As Tables 3B-3F show, for each rotamer there are a number of helix positions with high residue preferences, and these preferences are significantly different for each rotamer. Furthermore, the positions of these preferences correlate strongly with the helical positions which make frequent contact with the cofactor in each rotamer.
The t73 rotamer
The t73 rotamer is the most populous, and by extrapolation the lowest energy,62 conformation for a helical heme-bound histidine. As mentioned above, this rotamer also forms the great majority of the rotamers found in dihelix bis-histidine heme binding sites. As such, this rotamer is the lowest energy target for designed bis-histidine porphyrin binding sites. Table 3B depicts the position-dependent helical residue frequencies near the helical heme ligand histidines in the t73 rotamer and Table 3C depicts these frequencies for the subset of these helices which take part in bis-histidine heme binding where both histidines reside on a helix. Likewise, Supplementary Tables 2B and 2C present the helical residue propensities near the ligand histidine for this rotamer alone and in a dihelix bis-histidine ligation motif.
As Table 1B shows, there are two highly preferred residues, both at positions which are in frequent contact with the cofactor: First, there is a high preference within this rotamer for a phenylalanine residue at the i+8 position. This is the t-stacking interaction with the cofactor that was discussed above, but at a much higher frequency. In fact, the appearance of this packing interaction in the entire database is almost entirely encompassed within the population of the t73 rotamer. As will be discussed below, the high frequency of appearance of this packing interaction suggests that it may aid in porphyrin cofactor binding, and should be included in future helical porphyrin binding designs which use this rotamer. Second, glycine is present at the i+4 position 45% of the time, corresponding to a residue propensity of 15 given the low frequency with which glycine is normally present in helices.59 This positional preference increases to 57% (19-fold propensity) within the subset of the dihelical bis-histidine heme ligands in the t73 rotamer.
Figure 3A depicts a molecular model created using a 17-residue idealized alanine helix with a central, porphyrin-bound histidine fixed at -175° and 73°: the mean χ1 and χ2 torsion angles, respectively, for the t73 rotamer. This model demonstrates that this histidine conformation causes a steric clash between the porphyrin cofactor and the side chain methyl group of the alanine at position i+4 from the histidine. The consequence of this clash is that many of the χ1, χ2 torsion angle pairs in t73 subset of the database fall outside of the van der Waals energy surface of Figure 1B. Glycine at this position relieves this clash. Figure 3B depicts an expansion of this energy surface with the histidine torsion angle pairs derived from each protein the database overlaid in white using symbols which designate the identity of the amino acid side chain at position i+4. The conformations which fall outside of the energy surface each have a glycine at this position. As Figure 3C demonstrates, recalculation of the energy surface using an idealized alanine helix with a glycine at position i+4 expands the allowable conformational space to include all but one of the histidine t73 rotamers. The sole exception, E. coli succinate dehydrogenase, has a non-helical glycine residue at the i+4 position.63 This steric clash is the result of the tilt angle that the porphyrin macrocycle makes with the helix. Even within different rotamer populations, this angle varies, and the nonideality of many natural helices makes exceptions such as this possible even when tilt angles are large. The tilt angles which each porphyrin makes with respect to its associated helix are enumerated in Supplementary Table 1.
Figure 3.
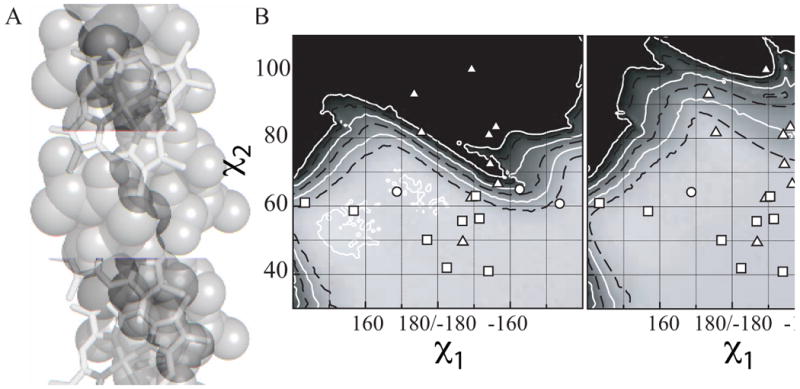
A glycine prevents steric clash in the t73 rotamer. (A) Space-filling model of a 17-residue idealized alanine helix with a central, porphyrin-bound histidine fixed at the mean χ1 and χ2 torsion angles for the t73 rotamer. The alanine four residues C-terminal from the histidine, which clashes with the porphyrin cofactor, is shaded in dark grey. (B) Expansion of the Van der Walls energy surface near the t73 rotamer. The χ1 and χ2 angles of each heme-bound histidine in the database are overlaid in white as a function of the identity of the residue four positions C-terminal to the to the histidine: ((x025B3)) Glycine, ((x025CB)) Alanine, ((x025A1)) All other residues. (C) Van der Waals energy surface calculated using an idealized 17-residue alanine helix identical to that used in part B but with a glycine four residues C-terminal to the central histidine. Histidine rotamers from the database are overlaid in white as in part B.
The planar tilt of the porphyrin with respect to the helix causes a counterbalancing increase in side chain volume on the other side of the helix from the small side chain at i+4 (see Supplementary Figure 2). The side chain volume at i-3 when there is a glycine residue at i+4 averages 108 ± 12 Å3. This is 28 Å3 larger than the average helical side chain volume in the entire database, 80 Å3. When the side chain at position i+4 is not a glycine, the i-3 side chain volume averages 100 ± 34 Å3, still 20 Å3 larger than the helical side chain average. This is a result of the porphyrin tilt requiring a larger side chain at i-3 to be able to pack against the cofactor face when it is further removed from the helix.
Besides the i+4 glycine residue and the i+8 phenylalanine residue, there are a number of other high frequency and/or high propensity side chain identities in the helices of the t73 rotamer, most of which occur at positions with high cofactor contact frequencies: isoleucine at i-7, leucine at i-3 and i-1, alanine at i+1, and isoleucine at i+5 and i+7. Table 4A depicts our predicted optimal helical sequences for each rotamer.
Table 4.
Rotamer-dependent helical consensus sequence templates derived from the database and comparison to previous designed protein sequences.
| A. Optimal sequences by rotamer (Contact residues highlighted in grey) | ||
|---|---|---|
| Ligand Histidine Rotamer | Sequence | |
| t73 | I XXX XXX XXH XXH XX XX I I
|
|
| m166 |
XX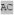 XA XA HXX HXX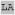
|
|
| t-86 | AHXX XX XX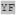
|
|
| m80 |
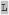 KXSXHXX KXSXHXX |
|
| B. The t73 sequence template compared to previous dihelical bis-histidine designs | ||
| Protein | Sequence | Reference |
| optimal t73 | IXXXXXHXXI
|
this work |
| HP-1 | EIWKQHEEALKKFEEALKQFEELKKL | (22) |
| ME1 | ILLASYGHRRLRKK | (9) |
| D2-heme | REKHRALAEQVYATGQMLKN | (8) |
| H2α17-FF | AEAFFKAHAEFFAKAA | (26) |
| Δ7-His | EIWKLHEEFIKLFEERIKKL | (25) |
| 6H7H | DKWKQHHQEFKQFLKELKQKLEEIA | (27) |
| AP1 | DPLVVAASIIGILHFIEWILDRGGNGEIFKQ | (18) |
| PAsc | QQKALQTAKEFQQAMQKHKQY | (34) |
| MOP-1 | NALELHEKALKQLEELLKKL | (35) |
| C. The m166 sequence template compared to the previous C-type cytochrome design | ||
| Protein | Sequence | Reference |
| optimal m166 |
XXXAHXX
|
this work |
| cHH | ELLKLHEELLKKFEECLKLHEERLKKL | (10) |
The m166 rotamer
The m166 rotamer, the second most populous rotamer, is composed of a mixture of covalently bound c-type cytochromes and noncovalently bound b-type cytochromes. The c-type cytochrome heme cofactors are most commonly covalently attached to the helix by the formation of thioether bonds between cysteine residues at positions i-1 and i-4 of the helix and the C(α) of the vinyl substituents of the heme cofactor using a consensus sequence called the CXXCH motif.64 Thioether bond formation is catalyzed in vivo by a complex biogenesis system which can vary extensively from organism to organism, although it has been shown that ferrous heme in a correctly positioned CXXCH binding site can spontaneously form thioether attachments provided the cysteines are not in a disulfide bond.65,66 Proteins with c-type cytochromes have diverse folds and functions, most famously in ferrying of electrons between the cytochrome bc1 and the cytochrome oxidase by the monoheme cytochrome c of the mitochondrial respiratory chain. There are a variety of advantages granted by covalent heme attachment.67,68 Every c-type cytochrome in the database is in this rotamer, and each has two thioether bonds embedded in a CXXCH motif.
The classic cytochrome C fold incorporates this motif into a short helix which terminates at the final cysteine residue, resulting in the ligand histidine being the first nonhelical residue.69 Our dataset selection screened out helices which do not extend one full turn on either side of the or in which the heme ligand histidine is not itself helical. Thus, our dataset does not contain a single incidence of this fold. Our data concentrates, rather, on heme binding sites which are incorporated on extended helices amenable to incorporation into helical bundle proteins.
This rotamer is also located at a marginal area of the van der Waals energy surface. Figure 4A details a 17-residue idealized alanine helix with a central, porphyrin-bound histidine fixed at -73° and 166°: the mean χ1 and χ2 torsion angles, respectively, for the m166 rotamer. This model predicts a steric clash between the alanine side chain methyl group at position i-4, one of the two cysteines which are covalently bound to the porphyrin cofactor, and the porphyrin ring: the C(γ) atom in the case of the c-cytochromes, as the thioether linkage enforces this particular cofactor orientation. Figure 4B presents the local expansion of a van der Waals energy surface calculated using α-chloroglycine residues at positions i-1 and i-4 to the central histidine. This substitution was utilized because it best recreates the ideal packing distance between the heme vinyl C(α)s and the cysteine thiolates in the covalent bond which links the helix and cofactor by effectively removing the hydrogen atoms that are present in an alanine methyl group but not present in a covalent bond. The figure is an expansion of the m166 region with the histidine torsion angle pairs derived from each protein the database overlaid in white using symbols which designate the attachment type. The c-type cytochromes are predominantly in marginal, high energy regions of the energy surface while the b-type cytochromes predominantly occur in low energy, nonmarginal areas.
Figure 4.
Heme ruffling is a consequence of steric clash between the heme C(γ) and the helix i-4 side chain. (A) Space-filling model of a 17-residue idealized alanine helix with a central, flat porphyrin-bound histidine fixed at the mean χ1 and χ2 torsion angles for the m166 rotamer. The alanine 4 residues N-terminal from the histidine, which clashes with the porphyrin C(γ) atom, is shaded in dark grey. (B) Expansion of the Van der Walls energy surface near the m166 rotamer. The χ1 and χ2 angles of each heme-bound histidine in the database are overlaid in white as a function of the connectivity of the heme: ((x025CB)) b-type, noncovalently bound hemes, ((x025BD)) c-type, covalently bound hemes. (C) Van der Waals energy surface calculated using the ruffled porphyrin derived from the 0.91 Ǻ resolution structure of the D. vulgaris cytochrome C (1J0P) connected as above to the idealized 17-residue alanine helix used in part B. Histidine rotamers from the database are overlaid in white as in part B.
In contrast to the i+4 position in the t73 rotamer, this clash cannot be avoided by reducing the volume of the side chain in the c-type cytochromes. Instead, the clash is alleviated by the deformation of the heme macrocycle. Figure 4C presents the recalculated energy surface created using the same idealized helix with a “ruffled” heme taken from the 0.91 Ǻ resolution structure of the D. vulgaris cytochrome C(3) (1J0P), itself a member of the helical heme protein database.70 Heme ruffling has been identified as a conserved cofactor conformation in cytochromes c and has been implicated in modulating reduction potentials and ligand affinities.71-73 Our data show that, at least in the case of c-type cytochromes bound in the center of extended helices, this ruffling is a consequence of the steric clash between the planar porphyrin macrocycle and the i-4 cysteine residue in helical CXXCH motifs. It is interesting to note that one of the two cytochromes b that have histidine torsion angles in the unfavorable region of the van der Waals energy surface depicted in Figure 4B near the cytochromes c, the high molecular mass cytochrome HmcA from Desulfovibrio vulgaris Hildenborough, itself has a significantly deformed heme cofactor.74
Besides the two cysteines at positions i-1 and i-4 on the helix (which are only present in the c-type cytochromes) there are other residues with high frequencies and or propensities in this rotamer. One preference is for a small residue at position i-2: 78.6% of the residues are glycine, alanine or serine (see Supplementary Figure 2 for the side average chain volume of each helical position in each rotamer). This includes every c-type cytochrome. This position faces away from the cofactor and the small size of the residues precludes them from making significant supporting contacts with the i-1 or i-4 cysteines.
Additionally, there are two more cysteine pairs present with high frequency in the database at positions i+3 and i+6. These represent the beginning of a CXXCH motif C-terminal to ligand histidine in the m166 rotamer which is itself not part of a CXXCH motif. The first binding site in each of these cases is also a bis-histidine c-type cytochrome binding site in which the covalently attached heme binds to cysteines on the portion of the protein which carries the other histidine ligand. Multiheme attachment sites such as this are found in multiheme proteins such as the cytochromes c′.75 This second site begins one full heptad away from the first, resulting in a second covalently attached heme on almost the same face of the helix one heptad, or 10.5 Å C-terminal to the first heme attachment site. Further residue preferences in this rotamer include alanine at position i-8, histidine at position i-7, leucine at i+3, alanine at i+4 and glutamic acid at i+5. These residue preferences are substantially different than those determined for the t73 rotamer (see Table 4A).
The t-86 and m80 rotamers
The position-dependent helical residue frequencies near the helical heme ligand histidines in the t-86 and m80 rotamer are depicted in Figures 3E and 3F respectively. Despite the fact that the remaining two rotamers in the database, t-86 and m80, are each in broad minima of the van der Waals energy surface, both are present at comparatively low frequencies. It is interesting to note that these are the only rotamers have mean torsion angles significantly different than the histidines without hemes in the same rotamer (see Table 1). This suggests that they represent higher energy conformations, and therefore poor choices for heme-binding protein design. In contrast to the t73 and m166 rotamers, both are present in low enough populations that the frequency and/or propensity data derived from them are not as statistically significant. However, each of these rotamers again displays distinct preferences for residue identities, most commonly at positions which are in frequent contact with the porphyrin macrocycle: the t-86 rotamer correlates with an alanine at i-1, a glycine at i+2, a tyrosine at i+3, and an isoleucine at i+4. Residue preferences for the m80 rotamer include leucine at i-7, lysine at i-6, leucine at i-4, serine at i-3 and phenylalanine at i-1. There are no residue preferences at positions C-terminal to the histidine in the m80 rotamer due to the fact that the helical population declines rapidly two residues after the ligand histidine (see Supplementary Figure 1).
Heme propionate orientational specificity
Heme cofactors have two propionate groups which are energetically highly unfavorable to bury in the hydrophobic core of a soluble protein in the absence of an engineered charge compensation or ‘salt-bridge’ interaction.76 Therefore it is important to determine whether the rotamer-based consensus sequences enforce a particular propionate orientation when binding heme cofactors: in other words, are the derived packing interactions specific enough that they not only enforce a particular porphyrin planar geometry, but also the direction of the porphyrin substituents? Figure 5A depicts the definition of the relative propionate and heme vector geometries – starting with the helix aligned upright, pointing at the observer, the azimuthal angle, θ1, is formed between the vector normal to the helix vector which passes through the histidine C(α) and the iron-heme CHA vector. The zenith angle, θ2, is measured between helical vector and the iron-heme CHA vector oriented such that both are in the same plane. As Figure 5B indicates, the t73 rotameric heme propionate orientations are relatively disperse while the t-86, m166 and m80 rotameric hemes clearly cluster.
Figure 5.
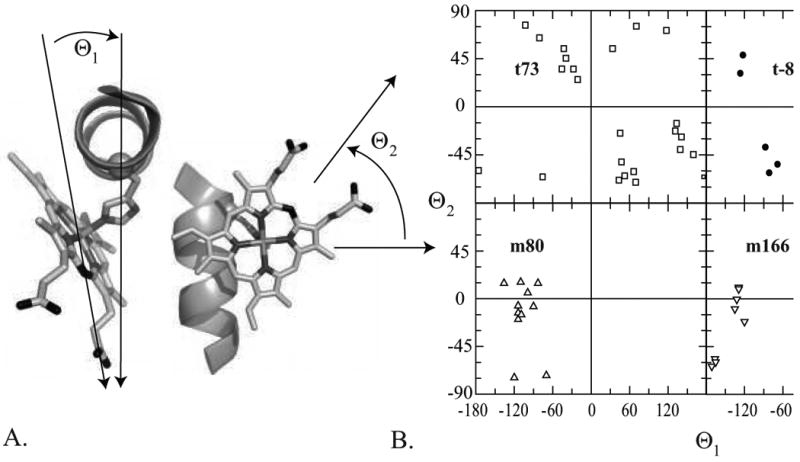
t73 rotamers have non-specific heme propionate orientations in the dataset while the other rotamers cluster at specific orientations. (A) Starting with the helix aligned upright with the N-terminus pointing at the observer, the azimuthal angle θ1 is formed between the vector normal to the helix vector which passes through the histidine C(α) and the iron-heme CHA vector. The zenith angle θ2 is measured between helical vector and the iron-heme CHA vector oriented such that both are same plane. The heme-helix model depicted has been placed at the mean θ1 and θ2 angles for the c-type cytochromes in the m166 rotamer. (B) The distribution of θ1 and θ2 angles of the hemin-containing proteins in the dataset subdivided by rotamer. In the m166 dataset, b-type hemes are depicted using open triangles and c-type hemes are depicted using solid triangles.
For the t73 rotamers, there is no relationship between the helical packing interactions and the heme orientation beyond that of the porphyrin plane. This is true even when subsets of the dataset are examined, such as the dihelical bis-histidine dataset or the t73 data with a glycine residue at position i+4. This implies that for dihelical bis-histidine heme protein design endeavors using the t73 rotameric residue consensus sequence the propionate orientation can find an energy minimum, most likely that which projects the propionate side chains into solution, without it being specifically engineered during the protein design process.
The m rotamers behave in the opposite manner - both m166 and m80 cluster significantly. The c-type cytochromes in the m166 rotamer are fixed in orientation by their covalent attachment to the cysteines at i-1 and i-4. This results in tight clustering of the propionate orientations around θ1 and θ2 angles of 0° and +50° respectively. The heme-helix pair in Figure 5A depicts the propionate orientation of a heme-bound to a histidine in the m166 rotamer with the propionates projecting along the mean vector for the c-type cytochromes. The b-type cytochromes in the m166 rotamer cluster more loosely around θ1 and θ2 angles of +165° and -110°. The m80 rotameric heme propionate orientations likewise cluster around 0° and -110°. Some degree of the restriction in the propionate orientations in the b-type m rotamers derives from the fact that the rather steep angle with respect to the helix imposed by the m rotamer limits the possible propionate orientations via steric interactions with the helix, but this does not explain the full degree of orientational restriction. The t-86 rotamer distribution is somewhat clustered, but its population is too small to be statistically significant.
It is interesting to note that the majority b-type m166 rotameric heme propionates cluster at a substantially different orientation than the c-type m166 heme propionates. This is despite the fact that the rotameric helical residue frequencies do not differ markedly between the two subpopulations. It is possible that this change in orientation is a consequence of the heme ruffle induced in the porphyrin macrocycle as a consequence of the c-type covalent attachment. Support for this hypothesis is given by the fact that the lone b-type heme in the c-type heme propionate orientation cluster is the aforementioned substantially deformed b-type heme from the cytochrome HmcA of Desulfovibrio vulgaris Hildenborough.74
Discussion
We have, for the first time, derived and explained the rotameric distribution of heme bound histidine side chains. This has allowed us to isolate rotamer-dependent helical consensus sequence templates which predict optimal side chain-cofactor packing interactions for each rotamer. These sequence templates are intended to be used to implant high-affinity porphyrin binding into designed helical bundle proteins. Table 4A presents the helical consensus sequence for each rotamer. There are two types of binding sites that the data provides clear instructions for creating: dihelical bis-histidine porphyrin sites and covalent c-type cytochromes.
Implications for protein design – b-type helical bundle proteins
The dihelical bis-histidine binding site, bound via ligand histidines both in the t73 rotamer, is the structural target for the majority of designed helical bundle proteins. Table 4B compares the t73 helical template derived in this work with the helical sequences of several designed helical bundle heme- and porphyrin-binding proteins. Several contrasts are immediately apparent. First, it is clear that helix-heme packing extends to at least two full turns or one heptad in each direction from the bound cofactor. Most designed porphyrin binding proteins created to date terminate the helix after one turn in at least one direction. Extension of the helix in designed proteins to include both i-7 and i+8 residues should enhance cofactor affinity by expanding the packing interface to accommodate the entire hydrophobic porphyrin surface.
Second, the helical positions which make frequent contact with the porphyrin cofactor in this rotamer: i-7, i-3, i+1, i+4, i+5, i+7 and i+8, all show a strong preference for hydrophobic side chains – aliphatic hydrophobic residues in particular with the exception of the phenylalanine at i+8. Indeed, this preference for aliphatic hydrophobic residues at contact sites holds true for each rotamer in the database. These residues presumably stabilize cofactor binding via hydrophobic packing interactions with the porphyrin ring. With the exception of one of the designed proteins in Table 4B, the designs place a charged residue in at least one of these positions if not several. Appropriate selection of side chain hydrophobicity in residues which contact the porphyrin macrocycle promises to increase cofactor affinity by increasing van der Waals interactions and avoiding the possibility of burying charges near the hydrophobic porphyrin macrocycle. As the positions of contact vary by rotamer, this may offer a simple method for the design of proteins which bind porphyrin cofactors at a specific geometry.
Third, the appearance of a significant population of phenylalanine residues at i+8, each forming an identical t-stacking interaction with the porphyrin in sequentially and structurally unrelated heme proteins, points to its utility in stabilizing heme and porphyrin binding. This specific interaction has not appeared, to our knowledge, in any designed porphyrin-binding helical bundle proteins, and its prevalence argues for its inclusion in any future designs.
Finally, the prevalence for a glycine residue at i+4 leads to the conclusion that it also should be included in helical bundle protein design, and the optimal sequence template depicted in Table 4B contains it. However, glycine residues in the center of helices are significantly destabilizing.60 Given the appearance of other hydrophobic residues at this position, alanine may be a better choice. However, the negative correlation between side chain volume at i+4 and side chain volume at i-3 (see supplementary Figure 2) suggests that the identity of these two residues can be used to explicitly govern the tilt angle between the porphyrin ring plane and the helix to which it is bound.
The cofactor geometry enforced by the t73 rotamer restricts the possible placement of porphyrin ligands in between pairs of adjacent helices (see Figure 6). Cofactor binding using both a core and an interfacial residue is not possible given the histidine ligand rotameric constraints. The porphyrin cofactor must instead lie between matching side chains – either two core position amino acids or two interfacial residues. This has important implications for helical bundle design. First, as porphyrin ring solvent exposure has been shown to significantly modulate cofactor reduction potentials,77 the choice of core or interfacial binding modes could be utilized to direct the electron affinity of the bound cofactor.
Figure 6.

The optimal interhelical position of porphyrin ligands varies by histidine rotamer. The hydrophobic core is demarked in grey. Geometric constraints force the t73 rotameric ligands to be either at the core A and D positions or the interfacial E and G positions at parallel helical interfaces. At antiparallel helical interfaces, the ligand residues must both occupy either the A or the E position in the helical heptad. In the m166 rotamer, typified by the c-type cytochromes, the geometry of the covalently bound heme and its associated propionates necessitates that the CXXCH ligand histidine occupy an E position in the helical heptad, while the remaining ligand occupies a G position in parallel helix interfaces or an E position in antiparallel helix interfaces.
Second, the majority of helical bundle porphyrin binding proteins designed to date consist of multiple copies of the same helix designed to self-associate by hydrophobic sequestration.78 For single, bis-histidine binding sites implanted on one helical pair, this results in heme- and porphyrin-binding sites being located at an interface between parallel helices. As the ligand residues have to lie between either at core A and D positions or interfacial E and G positions in the heptad helical repeat (see Figure 6), it is energetically unfavorable to create this binding motif using two copies of the same peptide due to helical heptad constraints: if the same residue takes an E position on one peptide and a G position on another, the residue i+1 to it must simultaneously occupy a solvent exposed F position and a hydrophobic core A position respectively, resulting in either hydrophobic side chain exposure or polar side chain burial depending on the residue identity. The binding site must instead be composed of two different helices which are either covalently linked (i.e. in a single chain three helix or larger bundle), or designed to specifically form noncovalent heterodimers. Antiparallel helices have the advantage that the helical repeat position of each residue near the histidine ligand is retained in both peptides, but unless the ligand residues are exactly in the middle of the helix, two different helices must again be used or else they will be offset from each other due to the reversal of the helical orientation. This could be readily created using a single helix-loop-helix peptide or a similar arrangement implanted in a larger designed protein.
c-Type helical proteins
The m166 rotamer and its associated helical template are the design elements derived in this work for the creation of c-type cytochrome proteins. There have been several attempts, both successful and not, to modify naturally occurring b-type cytochromes by implanting the CXXCH motif near one of the histidine ligands.65,79-81 To our knowledge, however, there has been only one partially successful attempt at the de novo design of a c-type cytochrome.10
Unlike b-type cytochromes, the binding site for naturally occurring c-type cytochromes need not be particularly complementary to the bound cofactor because the heme is covalently attached to the protein by the host organism's biogenesis system before the protein is folded.64 As inspection of the heme depicted in Figure 5A shows, a given histidine rotamer propionate orientation still has two possible heme binding arrangements, best envisioned as the one shown in Figure 5A and a new one created by rotating the heme 180° around the Fe-CHA axis. These two orientations are called heme insertion isomers, mixtures of which have long been known to exist in some natural b-type cytochrome proteins.82 Biogenesis systems all attach both cysteines to both vinyl groups in a manner that enforces one insertion isomer: the 4-position vinyl is covalently attached to the i-1 cysteine and the 2-posiiton vinyl is connected to the i-4 cysteine. While every c-type cytochrome in the database has the heme cofactor in this orientation with respect to the helix, the b-type cytochromes in the m166 rotamer are mixed in insertion isomers, and the isomer does not correlate with the identity of any of the helical template residues. This means that the derived helical template for this rotamer may not direct the bound cofactor insertion isomer, even while it directs both the heme rotamer and the propionate orientation.
This, again, has important implications for c-type cytochrome protein design. c-Type cytochromes can spontaneously form under benign solution conditions in the absence of any biosynthetic apparatus provided the bound heme is reduced and properly oriented and the cysteines are not in a disulfide bond.66 However, early attempts to re-engineer natural b-type cytochromes using this method resulted in a mixture of products, likely the result of heme insertion isomers further complicated by incomplete and non-regiospecific thioether bond formation.65 The de novo design of a c-type hemoprotein which can spontaneously form will require the creation of a protein which holds the noncovalently bound heme in the correct rotamer and in the correct orientation, including both the heme insertion isomer and the propionate orientation.81 This is made difficult by the apparent large difference in heme geometry preference, especially propionate orientation, which is engendered by the porphyrin ruffling that is concomitant with thioether bond formation. Molecular modeling (data not shown) suggests that a better position for the two cysteines given the preferred geometry of the bound b-type hemes in the m166 rotamer may be at i+1 and i+4. An additional requirement is for a degree of binding site complementarity sufficient to allow the holoprotein to either attain or retain a native-like tertiary structure after covalent attachment. The m166 helical sequence template, which includes aliphatic hydrophobic residues at each of the helical contact positions other than the two cysteines, should provide such a framework.
An alternate method for the creation of a c-type cytochrome protein lies in the use of natural systems which can generate the two thioether bonds in vivo with absolute regiospecificity. A single plasmid containing the E. coli cytochrome c maturation apparatus has been recently become available.83 This biogenesis system has been exogenously added to bacteria which express re-engineered natural proteins, and shown to successfully generate c-type cytochromes in at least two cases.79,81 While heme attachment utilizing biological recognition instead of precise cofactor binding site fabrication is a promising solution, it currently only provides relatively low yields of holoprotein. It remains to be seen how broadly applicable this system and others of its kind will prove to be.
The cofactor binding geometry places significant constraints on the placement of a porphyrin bound to a histidine in the m166 rotamer, whether covalently attached or not. The severe tilt of the porphyrin ring with respect to the helix means it must occupy an interfacial E position residue in the helical repeat (see Figure 6). More than half of the histidine ligands in this rotamer in the database are part of a bis-histidine ligation motif. In the majority of these, however, the other histidine ligand is not itself in a helix. If the cofactor is to be bound at the interface between two parallel helices, the other ligand residue must occupy the G position of that helix. If the other ligand residue is to be a histidine, both the database and molecular modeling predicts that the second histidine ligand is best placed in the m80 rotamer. If the cofactor is to be bound between antiparallel helices, both ligands must occupy the E position on their respective helices. There is one bis-histidine example of this binding motif in the database, which has the other ligand histidine in the t-86 rotamer. This observation is further complimented by molecular models. As in the case of the dihelical bis-histidine t73 rotamer, this would be very difficult to accomplish using helical bundles composed of multiple copies of identical helices.
Another interesting component of the database analysis is the detection of sequential repeats of heme binding sites in this rotamer placed one heptad apart. This repeating heme attachment motif may prove useful in the creation of robust bioinspired molecular wires and chlorin-based antennae complexes that have been the subject of recent activity.84,85 Efforts in our lab to realize these and other heme- and porphyrin containing proteins using the principles derived here are currently underway.
Supplementary Material
Acknowledgments
C.N. is thankful for support from the NIH (MARC grant# T34 GM007639) and the Mellon Mays foundation. C.F. gratefully acknowledges support from the Alexander von Humboldt foundation.
References
- 1.Koder RL, Dutton PL. Intelligent design: the de novo engineering of proteins with specified functions. Dalton Transactions. 2006;25:3045–3051. doi: 10.1039/b514972j. [DOI] [PubMed] [Google Scholar]
- 2.Razeghifard R, Wallace BB, Pace RJ, Wydrzynski T. Creating functional artificial proteins. Current Protein & Peptide Science. 2007;8(1):3–18. doi: 10.2174/138920307779941479. [DOI] [PubMed] [Google Scholar]
- 3.Reedy CJ, Gibney BR. Heme protein assemblies. Chemical Reviews. 2004;104(2):617–649. doi: 10.1021/cr0206115. [DOI] [PubMed] [Google Scholar]
- 4.Choma CT, Lear JD, Nelson MJ, Dutton PL, Robertson DE, Degrado WF. Design of a Heme-Binding 4-Helix Bundle. Journal of the American Chemical Society. 1994;116(3):856–865. [Google Scholar]
- 5.Robertson DE, Farid RS, Moser CC, Urbauer JL, Mulholland SE, Pidikiti R, Lear JD, Wand AJ, Degrado WF, Dutton PL. Design and Synthesis of Multi-Heme Proteins. Nature. 1994;368(6470):425–431. doi: 10.1038/368425a0. [DOI] [PubMed] [Google Scholar]
- 6.Sharp RE, Diers JR, Bocian DF, Dutton PL. Differential binding of iron(III) and zinc(II) protoporphyrin IX to synthetic four-helix bundles. Journal of the American Chemical Society. 1998;120(28):7103–7104. [Google Scholar]
- 7.Gibney BR, Isogai Y, Rabanal F, Reddy KS, Grosset AM, Moser CC, Dutton PL. Self-assembly of heme A and heme B in a designed four-helix bundle: Implications for a cytochrome c oxidase maquette. Biochemistry. 2000;39(36):11041–11049. doi: 10.1021/bi000925r. [DOI] [PubMed] [Google Scholar]
- 8.Ghirlanda G, Osyczka A, Liu WX, Antolovich M, Smith KM, Dutton PL, Wand AJ, DeGrado WF. De novo design of a D-2-symmetrical protein that reproduces the diheme four-helix bundle in cytochrome bc(1) Journal of the American Chemical Society. 2004;126(26):8141–8147. doi: 10.1021/ja039935g. [DOI] [PubMed] [Google Scholar]
- 9.Cordova JM, Noack PL, Hilcove SA, Lear JD, Ghirlanda G. Design of a functional membrane protein by engineering a heme-binding site in glycophorin A. Journal Of The American Chemical Society. 2007;129(3):512–518. doi: 10.1021/ja057495i. [DOI] [PubMed] [Google Scholar]
- 10.Ishida M, Dohmae N, Shiro Y, Oku T, Iizuka T, Isogai Y. Design and synthesis of de novo cytochromes c. Biochemistry. 2004;43(30):9823–9833. doi: 10.1021/bi049546e. [DOI] [PubMed] [Google Scholar]
- 11.Cowley AB, Kennedy ML, Silchenko S, Lukat-Rodgers GS, Rodgers KR, Benson DR. Insight into heme protein redox potential control and functional aspects of six-coordinate ligand-sensing heme proteins from studies of synthetic heme peptides. Inorganic Chemistry. 2006;45(25):9985–10001. doi: 10.1021/ic052205k. [DOI] [PubMed] [Google Scholar]
- 12.Cowley AB, Benson DR. Weak-field anions displace the histidine ligand in a synthetic heme peptide but not in N-acetylmicroperoxidase-8: Possible role of heme geometry differences. Inorganic Chemistry. 2007;46(1):48–59. doi: 10.1021/ic060682c. [DOI] [PubMed] [Google Scholar]
- 13.Cowley AB, Lukat-Rodgers GS, Rodgers KR, Benson DR. A possible role for the covalent heme-protein linkage in cytochrome c revealed via comparison of N-acetylmicroperoxidase-8 and a synthetic, monohistidine-coordinated heme peptide. Biochemistry. 2004;43(6):1656–1666. doi: 10.1021/bi035531p. [DOI] [PubMed] [Google Scholar]
- 14.Liu DH, Williamson DA, Kennedy ML, Williams TD, Morton MM, Benson DR. Aromatic side chain-porphyrin interactions in designed hemoproteins. Journal Of The American Chemical Society. 1999;121(50):11798–11812. [Google Scholar]
- 15.Reddi AR, Reedy CJ, Mui S, Gibney BR. Thermodynamic investigation into the mechanisms of proton-coupled electron transfer events in heme protein maquettes. Biochemistry. 2007;46(1):291–305. doi: 10.1021/bi061607g. [DOI] [PubMed] [Google Scholar]
- 16.Gibney BR, Huang SS, Skalicky JJ, Fuentes EJ, Wand AJ, Dutton PL. Hydrophobic modulation of heme properties in heme protein maquettes. Biochemistry. 2001;40(35):10550–10561. doi: 10.1021/bi002806h. [DOI] [PubMed] [Google Scholar]
- 17.Shifman JM, Gibney BR, Sharp RE, Dutton PL. Heme redox potential control in de novo designed four-alpha- helix bundle proteins. Biochemistry. 2000;39(48):14813–14821. doi: 10.1021/bi000927b. [DOI] [PubMed] [Google Scholar]
- 18.Discher BM, Noy D, Strzalka J, Ye SX, Moser CC, Lear JD, Blasie JK, Dutton PL. Design of amphiphilic protein maquettes: Controlling assembly, membrane insertion, and cofactor interactions. Biochemistry. 2005;44(37):12329–12343. doi: 10.1021/bi050695m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noy D, Moser CC, Dutton PL. Bacteriochlorophyll Protein Maquettes. In: Grimm B, Porra W, Ruediger W, Scheer H, editors. Biochemistry and Biophysics of Chlorophylls. Kluwer; Dorderecht: 2003. In Press. [Google Scholar]
- 20.Shifman JM, Moser CC, Kalsbeck WA, Bocian DF, Dutton PL. Functionalized de novo designed proteins: Mechanism of proton coupling to oxidation/reduction in heme protein maquettes. Biochemistry. 1998;37(47):16815–16827. doi: 10.1021/bi9816857. [DOI] [PubMed] [Google Scholar]
- 21.Huang SS, Koder RL, Lewis M, Wand AJ, Dutton PL. The HP-1 maquette: From an apoprotein structure to a structured hemoprotein designed to promote redox-coupled proton exchange. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(15):5536–5541. doi: 10.1073/pnas.0306676101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koder RL, Valentine KG, Cerda JF, Noy D, Smith KM, Wand AJ, Dutton PL. Native-like structure in designed four helix bundles driven by buried polar interactions. Journal of the American Chemical Society. 2006;128(45):14450–14451. doi: 10.1021/ja064883r. [DOI] [PubMed] [Google Scholar]
- 23.Rosenblatt MM, Wang JY, Suslick KS. De novo designed cyclic-peptide heme complexes. 2003;100(23):13140–13145. doi: 10.1073/pnas.2231273100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reedy CJ, Kennedy ML, Gibney BR. Thermodynamic characterization of ferric and ferrous haem binding to a designed four-alpha-helix protein. Chemical Communications. 2003;(5):570–571. doi: 10.1039/b211488g. [DOI] [PubMed] [Google Scholar]
- 25.Sakamoto S, Obataya I, Ueno A, Mihara H. Effects of amino acids substitution of hydrophobic residues on haem-binding properties of designed two-alpha-helix peptides. Journal Of The Chemical Society-Perkin Transactions 2. 1999;(10):2059–2069. [Google Scholar]
- 26.Xu ZJ, Farid RS. Design, synthesis, and characterization of a novel hemoprotein. Protein Science. 2001;10(2):236–249. doi: 10.1110/ps.30801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noy D, Discher BM, Rubtsov IV, Hochstrasser RA, Dutton PL. Design of amphiphilic protein maquettes: Enhancing maquette functionality through binding of extremely hydrophobic cofactors to lipophilic domains. Biochemistry. 2005;44(37):12344–12354. doi: 10.1021/bi050696e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cochran FV, Wu SP, Wang W, Nanda V, Saven JG, Therien MJ, DeGrado WF. Computational de novo design and characterization of a four-helix bundle protein that selectively binds a nonbiological cofactor. Journal of the American Chemical Society. 2005;127(5):1346–1347. doi: 10.1021/ja044129a. [DOI] [PubMed] [Google Scholar]
- 29.Xu T, Wu SP, Miloradovic I, Therien MJ, Blasie JK. Incorporation of designed extended chromophores into amphiphilic 4-helix bundle peptides for nonlinear optical biomolecular materials. Nano Letters. 2006;6(11):2387–2394. doi: 10.1021/nl062091p. [DOI] [PubMed] [Google Scholar]
- 30.Strzalka J, Xu T, Tronin A, Wu SP, Miloradovic I, Kuzmenko I, Gog T, Therien MJ, Blasie JK. Structural studies of amphiphilic 4-helix bundle peptides incorporating designed extended chromophores for nonlinear optical biomolecular materials. Nano Letters. 2006;6(11):2395–2405. doi: 10.1021/nl062092h. [DOI] [PubMed] [Google Scholar]
- 31.Ye SX, Discher BM, Strzalka J, Xu T, Wu SP, Noy D, Kuzmenko I, Gog T, Therien MJ, Dutton PL, Blasie JK. Amphiphilic four-helix bundle peptides designed for light-induced electron transfer across a soft interface. Nano Letters. 2005;5(9):1658–1667. doi: 10.1021/nl050542k. [DOI] [PubMed] [Google Scholar]
- 32.Kovaric BC, Kokona B, Schwab AD, Twomey MA, de Paula JC, Fairman R. Self-assembly of peptide porphyrin complexes: Toward the development of smart biomaterials. Journal Of The American Chemical Society. 2006;128(13):4166–4167. doi: 10.1021/ja056357q. [DOI] [PubMed] [Google Scholar]
- 33.Dunetz JR, Sandstrom C, Young ER, Baker P, Van Name SA, Cathopolous T, Fairman R, de Paula JC, Akerfeldt KS. Self-assembling porphyrin-modified peptides. Organic Letters. 2005;7(13):2559–2561. doi: 10.1021/ol050644h. [DOI] [PubMed] [Google Scholar]
- 34.Bender GM, Lehmann A, Zou H, Cheng H, Fry HC, Engel D, Therien MJ, Blasie JK, Roder H, Saven JG, DeGrado WF. De novo design of a single-chain diphenylporphyrin metalloprotein. Journal Of The American Chemical Society. 2007;129(35):10732–10740. doi: 10.1021/ja071199j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fahnenschmidt M, Bittl R, Schlodder E, Haehnel W, Lubitz W. Characterization of de novo synthesized four-helix bundle proteins with metalloporphyrin cofactors. Physical Chemistry Chemical Physics. 2001;3(18):4082–4090. [Google Scholar]
- 36.Krizek BA, Amann BT, Kilfoil VJ, Merkle DL, Berg JM. A Consensus Zinc Finger Peptide - Design, High-Affinity Metal-Binding, a Ph-Dependent Structure, and a His to Cys Sequence Variant. Journal of the American Chemical Society. 1991;113(12):4518–4523. [Google Scholar]
- 37.Dai MH, Fisher HE, Temirov J, Kiss C, Phipps ME, Pavlik P, Werner JH, Bradbury ARM. The creation of a novel fluorescent protein by guided consensus engineering. 2007;20(2):69–79. doi: 10.1093/protein/gzl056. [DOI] [PubMed] [Google Scholar]
- 38.Forrer P, Binz HK, Stumpp MT, Pluckthun A. Consensus design of repeat proteins. 2004;5(2):183–189. doi: 10.1002/cbic.200300762. [DOI] [PubMed] [Google Scholar]
- 39.Main ERG, Xiong Y, Cocco MJ, D'Andrea L, Regan L. Design of stable alpha-helical arrays from an idealized TPR motif. 2003;11(5):497–508. doi: 10.1016/s0969-2126(03)00076-5. [DOI] [PubMed] [Google Scholar]
- 40.Krizek BA, Merkle DL, Berg JM. Ligand Variation and Metal-Ion Binding-Specificity in Zinc Finger Peptides. Inorganic Chemistry. 1993;32(6):937–940. [Google Scholar]
- 41.Krizek BA, Berg JM. Complexes of Zinc Finger Peptides with Ni-2+ and Fe-2+ Inorganic Chemistry. 1992;31(13):2984–2986. [Google Scholar]
- 42.Lombardi A, Summa CM, Geremia S, Randaccio L, Pavone V, DeGrado WF. Retrostructural analysis of metalloproteins: Application to the design of a minimal model for diiron proteins. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(12):6298–6305. doi: 10.1073/pnas.97.12.6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Summa CM, Lombardi A, Lewis M, DeGrado VF. Tertiary templates for the design of diiron proteins. Current Opinion in Structural Biology. 1999;9(4):500–508. doi: 10.1016/S0959-440X(99)80071-2. [DOI] [PubMed] [Google Scholar]
- 44.Marsh ENG, DeGrado WF. Noncovalent self-assembly of a heterotetrameric diiron protein. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(8):5150–5154. doi: 10.1073/pnas.052023199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nanda V, Rosenblatt MM, Osyczka A, Kono H, Getahun Z, Dutton PL, Saven JG, DeGrado WF. De novo design of a redox-active minimal rubredoxin mimic. 2005;127(16):5804–5805. doi: 10.1021/ja050553f. [DOI] [PubMed] [Google Scholar]
- 46.Summa CM, Rosenblatt MM, Hong JK, Lear JD, DeGrado WF. Computational de novo design, and characterization of an A(2)B(2) diiron protein. Journal of Molecular Biology. 2002;321(5):923–938. doi: 10.1016/s0022-2836(02)00589-2. [DOI] [PubMed] [Google Scholar]
- 47.Kaplan J, Degrado WF. De novo design of catalytic proteins. Proceedings of the National Academy of Sciences. 2004;101(32):11566–11570. doi: 10.1073/pnas.0404387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Calhoun JR, Nastri F, Maglio O, Pavone V, Lombardi A, DeGrado WF. Artificial diiron proteins: From structure to function. Biopolymers. 2005;80(2-3):264–278. doi: 10.1002/bip.20230. [DOI] [PubMed] [Google Scholar]
- 49.Zaric SD, Popovic DM, Knapp EW. Factors determining the orientation of axially coordinated imidazoles in heme proteins. Biochemistry. 2001;40(26):7914–7928. doi: 10.1021/bi010428q. [DOI] [PubMed] [Google Scholar]
- 50.Schneider S, Marles-Wright J, Sharp KH, Paoli M. Diversity and conservation of interactions for binding heme in b-type heme proteins. 2007;24(3):621–630. doi: 10.1039/b604186h. [DOI] [PubMed] [Google Scholar]
- 51.Hobohm U, Sander C. Enlarged Representative Set of Protein Structures. Protein Science. 1994;3(3):522–524. doi: 10.1002/pro.5560030317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang GL, Dunbrack RL. PISCES: a protein sequence culling server. Bioinformatics. 2003;19(12):1589–1591. doi: 10.1093/bioinformatics/btg224. [DOI] [PubMed] [Google Scholar]
- 53.MacGregor MJ, Islam SA, Sternberg MJE. Analysis of the relationship between side-chain conformation and secondary structure in proteins. J Mol Biol. 1987;198:295–310. doi: 10.1016/0022-2836(87)90314-7. [DOI] [PubMed] [Google Scholar]
- 54.Creamer TP. Side-chain conformational entropy in protein unfolded states. Proteins-Structure Function And Genetics. 2000;40(3):443–450. doi: 10.1002/1097-0134(20000815)40:3<443::aid-prot100>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 55.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. 1997;18(15):2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 56.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. Charmm - A Program For Macromolecular Energy, Minimization, And Dynamics Calculations. Journal Of Computational Chemistry. 1983;4(2):187–217. [Google Scholar]
- 57.Lovell SC, Word JM, Richardson JS, Richardson DC. The penultimate rotamer library. Proteins-Structure Function and Genetics. 2000;40(3):389–408. [PubMed] [Google Scholar]
- 58.Magliery TJ, Regan L. Beyond consensus: Statistical free energies reveal hidden interactions in the design of a TPR motif. 2004;343(3):731–745. doi: 10.1016/j.jmb.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 59.Engel DE, DeGrado WF. Amino acid propensities are position-dependent throughout the length of alpha-helices. Journal Of Molecular Biology. 2004;337(5):1195–1205. doi: 10.1016/j.jmb.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 60.Lyu PC, Liff MI, Marky LA, Kallenbach NR. Side-Chain Contributions To The Stability Of Alpha-Helical Structure In Peptides. Science. 1990;250(4981):669–673. doi: 10.1126/science.2237416. [DOI] [PubMed] [Google Scholar]
- 61.Aurora R, Rose GD. Helix capping. Protein Science. 1998;7(1):21–38. doi: 10.1002/pro.5560070103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pohl FM. Empirical Protein Energy Maps. Nature-New Biology. 1971;234(52):277–&. doi: 10.1038/newbio234277a0. [DOI] [PubMed] [Google Scholar]
- 63.Yankovskaya V, Horsefield R, Tornroth S, Luna-Chavez C, Miyoshi H, Leger C, Byrne B, Cecchini G, Iwata S. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science. 2003;299(5607):700–704. doi: 10.1126/science.1079605. [DOI] [PubMed] [Google Scholar]
- 64.Stevens JM, Daltrop O, Allen JWA, Ferguson SJ. C-Type Cytochrome Formation: Chemical and Biological Enigmas. Acc Chem Res. 2004;37:999–1007. doi: 10.1021/ar030266l. [DOI] [PubMed] [Google Scholar]
- 65.Barker PD, Nerou EP, Freund SMV, Fearnley IM. Conversion of Cytochrome b562 to C-type Cytochromes. Biochemistry. 1995;34:15191–15203. doi: 10.1021/bi00046a027. [DOI] [PubMed] [Google Scholar]
- 66.Daltrop O, Allen JWA, Willis AC, Ferguson SJ. In Vitro Formation of a C-type Cytochrome. Proc Natl Acad Sci U S A. 2002;277:35703–35711. doi: 10.1073/pnas.132259099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barker PD, Ferguson SJ. Still a puzzle: why is haem covalently attached in c-type cytochromes? Structure. 1999;7(12):R281–R290. doi: 10.1016/s0969-2126(00)88334-3. [DOI] [PubMed] [Google Scholar]
- 68.Allen JWA, Barker PD, Daltrop O, Stevens JM, Tomlinson EJ, Sinha N, Sambongi Y, Ferguson SJ. Why isn't ‘standard’ heme good enough for c-type and d(1)-type cytochromes? Dalton Transactions. 2005;(21):3410–3418. doi: 10.1039/b508139b. [DOI] [PubMed] [Google Scholar]
- 69.Bertini I, Cavallaro G, Rosato A. Cytochrome c: Occurrence and functions. Chemical Reviews. 2006;106(1):90–115. doi: 10.1021/cr050241v. [DOI] [PubMed] [Google Scholar]
- 70.Ozawa K, Takayama Y, Yasukawa F, Ohmura T, Cusanovich MA, Tomimoto Y, Ogata H, Higuchi Y, Akutsu H. Role of the aromatic ring of Tyr43 in tetraheme cytochrome c(3) from Desulfovibrio vulgaris Miyazaki F. Biophysical Journal. 2003;85(5):3367–3374. doi: 10.1016/S0006-3495(03)74756-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hobbs JD, Shelnutt JA. Conserved Nonplanar Heme Distortions In Cytochromes-C. Journal Of Protein Chemistry. 1995;14(1):19–25. doi: 10.1007/BF01902840. [DOI] [PubMed] [Google Scholar]
- 72.Shelnut JA, Song X, Ma J, Jia S, Jentzen W, Medforth CJ. Nonplanar Porphyrins and their Significance in Proteins. Chem Soc Rev. 1998;27:31–41. [Google Scholar]
- 73.Ma JG, Vanderkooi JM, Zhang J, Jia SL, Shelnutt JA. Resonance Raman investigation of nickel microperoxidase-11. Biochemistry. 1999;38(9):2787–2795. doi: 10.1021/bi982332a. [DOI] [PubMed] [Google Scholar]
- 74.Matias PM, Coelho AV, Valente FMA, Placido D, LeGall J, Xavier AV, Pereira IAC, Carrondo MA. Sulfate respiration in Desulfovibrio vulgaris Hildenborough - Structure of the 16-heme cytochrome c HmcA at 2.5-angstrom resolution and a view of its role in transmembrane electron transfer. Journal Of Biological Chemistry. 2002;277(49):47907–47916. doi: 10.1074/jbc.M207465200. [DOI] [PubMed] [Google Scholar]
- 75.Mowat CG, Chapman SK. Multi-heme cytochromes - new structures, new chemistry. Dalton Transactions. 2005;(21):3381–3389. doi: 10.1039/b505184c. [DOI] [PubMed] [Google Scholar]
- 76.Warshel A, Russell ST. Calculations Of Electrostatic Interactions In Biological-Systems And In Solutions. Quarterly Reviews Of Biophysics. 1984;17(3):283–422. doi: 10.1017/s0033583500005333. [DOI] [PubMed] [Google Scholar]
- 77.Stellwagen E. Heme Exposure As Determinate Of Oxidation-Reduction Potential Of Heme Proteins. Nature. 1978;275(5675):73–74. doi: 10.1038/275073a0. [DOI] [PubMed] [Google Scholar]
- 78.Ghosh D, Pecoraro VL. Probing metal-protein interactions using a de novo design approach. Current Opinion in Chemical Biology. 2005;9(2):97–103. doi: 10.1016/j.cbpa.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 79.Faraone-Mennella J, Tezcan FA, Gray HB, Winkler JR. Stability and folding kinetics of structurally characterized cytochrome c-b(562) Biochemistry. 2006;45(35):10504–10511. doi: 10.1021/bi060242x. [DOI] [PubMed] [Google Scholar]
- 80.Lin YW, Wang WH, Zhang Q, Lu HJ, Yang PY, Xie Y, Huang ZX, Wu HM. Converting cytochrome b(5) into cytochrome c-like protein. Chembiochem. 2005;6(8):1356–1359. doi: 10.1002/cbic.200500030. [DOI] [PubMed] [Google Scholar]
- 81.Allen JWA, Barker PD, Ferguson SJ. A cytochrome b(562) variant with a c-type cytochrome CXXCH heme-binding motif as a probe of the Escherichia coli cytochrome c maturation system. Journal Of Biological Chemistry. 2003;278(52):52075–52083. doi: 10.1074/jbc.M307196200. [DOI] [PubMed] [Google Scholar]
- 82.Lamar GN, Budd DL, Viscio DB, Smith KM, Langry KC. Proton Nuclear Magnetic-Resonance Characterization of Heme Disorder in Hemoproteins. Proceedings of the National Academy of Sciences of the United States of America. 1978;75(12):5755–5759. doi: 10.1073/pnas.75.12.5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Arslan E, Schulz H, Zufferey R, Kunzler P, Thony-Meyer L. Overproduction of the Bradyrhizobium japonicum c-type cytochrome subunits of the cbb(3) oxidase in Escherichia coli. Biochemical And Biophysical Research Communications. 1998;251(3):744–747. doi: 10.1006/bbrc.1998.9549. [DOI] [PubMed] [Google Scholar]
- 84.Baldwin AJ, Bader R, Christodoulou J, MacPhee CE, Dobson CM, Barker PD. Cytochrome display on amyloid fibrils. Journal Of The American Chemical Society. 2006;128(7):2162–2163. doi: 10.1021/ja0565673. [DOI] [PubMed] [Google Scholar]
- 85.Noy D, Dutton PL. Design of a minimal polypeptide unit for bacteriochlorophyll binding and self-assembly based on photosynthetic bacterial light-harvesting proteins. Biochemistry. 2006;45(7):2103–2113. doi: 10.1021/bi052175x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.