

Abstract
Previous studies have established that abrogation of monoamine oxidase (MAO) A expression leads to a neurochemical, morphological, and behavioral specific phenotype with increased levels of serotonin (5-HT), norepinephrine, and dopamine, loss of barrel field structure in mouse somatosensory cortex, and an association with increased aggression in adults. Forebrain-specific MAO A transgenic mice were generated from MAO A knock-out (KO) mice by using the promoter of calcium-dependent kinase IIα (CaMKIIα). The presence of human MAO A transgene and its expression were verified by PCR of genomic DNA and reverse transcription-PCR of mRNA and Western blot, respectively. Significant MAO A catalytic activity, autoradiographic labeling of 5-HT, and immunocytochemistry of MAO A were found in the frontal cortex, striatum, and hippocampus but not in the cerebellum of the forebrain transgenic mice. Also, compared with MAO A KO mice, lower levels of 5-HT, norepinephrine, and DA and higher levels of MAO A metabolite 5-hydroxyin-doleacetic acid were found in the forebrain regions but not in the cerebellum of the transgenic mice. These results suggest that MAO A is specifically expressed in the forebrain regions of transgenic mice. This forebrain-specific differential expression resulted in abrogation of the aggressive phenotype. Furthermore, the disorganization of the somatosensory cortex barrel field structure associated with MAO A KO mice was restored and became morphologically similar to wild type. Thus, the lack of MAO A in the forebrain of MAO A KO mice may underlie their phenotypes.
This study shows that a forebrain-specific expression of monoamine oxidase (MAO)3 A is able to restore the disorganized somatosensory cortex barrel field structure and an aggressive behavior seen in MAO A KO mice. MAO A and B are outer membrane mitochondrial enzymes responsible for the metabolic degradation of biogenic amines in humans (1, 2). MAO A prefers serotonin (5-HT) and norepinephrine (NE) as substrates, whereas MAO B prefers phenylethylamine (PEA) and benzylamine (1-5). Dopamine (DA), tyramine, and tryptamine are common substrates for both forms. MAO A and B are encoded by different genes (6, 7) closely linked on the X chromosome (8). Both isoenzymes are widely present in most brain regions (9). MAO A gene deficiency in a Dutch family shows borderline mental retardation and impulsive aggression (10, 11). A promoter region was associated with high or low MAO A promoter activity (12). Individuals with low MAO A promoter activity who are maltreated in early childhood have increased risk for antisocial behavior as adults (13, 14). These results indicated that the interaction of the gene and the environment of early childhood predispose the adult behavior. Functional magnetic resonance imaging in healthy human volunteers shows that the low expression variant, associated with increased risk of violent behavior, correlated with pronounced limbic volume reductions, hyper-responsive amygdala during emotional arousal, and diminished reactivity of regulatory prefrontal regions as compared with the high expression allele (15, 16).
MAO A knock-out (KO) mice showed elevated brain levels of MAO A substrates 5-HT, DA, and NE in all brain regions (17, 18). In contrast to MAO A deficiency, only an increase in PEA level was found in the brain of MAO B-deficient mice and humans (19, 20). They do not show aggressive behavior. These studies suggest that the changes in brain monoamines such as 5-HT, NE, and DA may be related to aggression in MAO A/B double KO mice. Brain levels of 5-HT, NE, DA, and PEA all increased to a much greater degree than in either MAO A or B single KO mice (21). They show chase/escape and anxiety-like behavior different from MAO A or B single KO, suggesting that varying monoamine levels result in a unique behavioral phenotype.
MAO A KO mice show complete absence of barrels in the somatosensory cortex and aggressive behavior (17). Neonatal administration of the tryptophan hydroxylase inhibitor para-chlorophenylalanine reduced 5-HT and partly restored the capacity to form cortical barrels (17). Restoration of the barrel field can be accomplished before postnatal day 7 (22, 23). These studies suggest the neuronal developmental stage is critically important for the aggressive behavior in adulthood in both human and mice, prompting us to create a transgenic mouse that would restore the somatosensory cortex structure in the early developmental period and alleviate the aggressive behavior in the adult male mouse. To do this, we used the mouse calcium-calmodulin-dependent kinase IIα (CaMKIIα) promoter to drive expression of a human MAO A transgene in MAO A KO mice. CaMKIIα is an abundant protein specifically expressed in forebrain synapses from postnatal day 1 to adult life. This promoter has been well characterized with regard to specific forebrain expression, including neocortex, striatum, and hippocampus (24-27).
This is the first study showing that the forebrain of transgenic mice with the mouse CaMKIIα promoter/human MAO A construct in MAO A KO mice is able to modify the phenotypes observed in MAO A KO mice to a phenotype more like wild type. This study further confirms the role of MAO A itself in somatosensory cortex organization and aggressive behavior.
EXPERIMENTAL PROCEDURES
Construction of pCaMKIIα-MAO A cDNA Vector
The 20-kb human MAO A cDNA was excised by EcoRI digestion from a pECE vector. The insert was ligated into the EcoRV site of pNN265. This vector contained a 5′ intron and a 3′ intron plus a poly(A) signal from SV40 (28). The insert containing human MAO A cDNA was digested by NotI and was ligated into the unique NotI site of pMM403 that contains the 8.6-kb CaMKIIα promoter (24). The orientation of the insert was determined by digestion with HindIII. The linearized plasmid DNA (Fig. 1) was then microinjected into fertilized eggs from MAO A KO mice.
FIGURE 1.
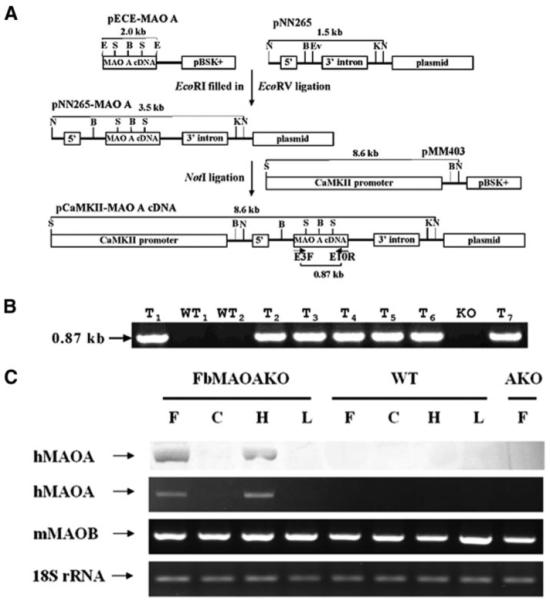
A, construction of the transgene, pCaMKII-MAOcDNA (see text for details). Abbreviations of the restriction sites are as follows: E, EcoRI; S, SacI, B, BamHI; K, KpnI; N, NotI; and Ev, EcoRV. B, identification of forebrain transgenic mice by PCR. The 0.87-kb PCR product band was only found in forebrain transgenic mice (n = 7) and not in wild type (n = 2) or MAO A KO mice. Abbreviations used are as follows: WT, wild type mice; T, forebrain transgenic mice; KO, MAO A knock-out mice. C, forebrain-specific expression of hMAO A protein and RNA by Western blot and RT-PCR. F, frontal cortex; C, cerebellum; H, hippocampus; L, liver.
Identification of Forebrain Transgenic Mice by PCR
The presence of MAO A cDNA in forebrain MAO A transgenic mice was shown by PCR using a pair of primers that are complementary to parts of human MAO A exon 3 (E3F, 5′-GATTACGTAGATGTTGGTGGAGC-3′; 254–276) and exon 10 (E10R 5′-GATGGCAGGCAGTGACCCATCAG-3′; 1120 to 1098). A 0.87-kb PCR product is expected when human MAO A cDNA is expressed. The PCR conditions were as follows: 94 °C for 1 min, 60 °C for 1 min, and 72 °C for 2 min, 30 cycles (Fig. 1B).
Anchor PCR for Determination of Transgene Insertion Site
Genomic DNA isolated from the transgenic MAO A KO mouse tail was partially digested by Sau3AI enzyme to create sticky ends for ligation into the BamHI site of the pUC19 vector. The primer was designed according to the most 5′ end of mouse CaMKIIα promoter sequence in the antisense direction (5′-AGA AGG GTG CGG ACT ACA TCG-3′). The anchor primer was from pU19 flanking the BamHI site (5′-cgg ctc gta tgtt gtg tgg-3′). PCR was run under the following conditions: 94 °C 3 min; 94 °C 30 s, 58 °C 30 s, 72 °C 2 min, for 32 cycles; the last extension at 72 °C was for 10 min. The PCR product was checked by 1% agarose gel to ensure a product larger than 1 kb was present. The PCR product was cloned into TA cloning kit (Invitrogen), and single colonies were isolated. Clones containing inserts were sequenced. Sequences were subjected to National Center for Biotechnology Information VecScreen and BLAT analysis using the University of California, Santa Cruz, Genome Bioinformatics server (mouse genome assembly Feb. 2006).
Demonstration of Forebrain-specific Expression of Human MAO A Protein and RNA by Western Blot and RT-PCR
Western blot was done by using anti-human MAO A antibody. Tissue homogenates were isolated from various tissues of WT, MAO A KO, and forebrain transgenic mice, separated in a 7% SDS-polyacrylamide gel, and transferred to a polyvinylidene difluoride membrane. The membrane was immunoblotted by 1:1,000 diluted rabbit anti-human MAO A antibody for 1 h at room temperature, washed three times, and then incubated with 1:10,000 diluted goat anti-rabbit antibody for another 30 min, and washed three times. The membrane was incubated with ECL for 1 min and exposed to x-ray film. The total RNAs were isolated from WT, MAO A KO, and forebrain transgenic mice and reverse-transcripted by random primers. The specific primers were designed from the human MAO A gene (F1, 5′-GGT GGA TCT GGT CAAGTG AG-3′; R1, 5′-CTT TGT GGC AGT CTC TGT GC-3′), mouse MAO B gene (F as F1, R, 5′-GGGAGG GAA GTA GGT TGT GT-3′) and 18 S rRNA gene as the control (F, 5′-CGCCGC TAG AGG TGAAAT TC-3′;R, 5′-CGA ACC TCC GAC TTT CGT TC-3′). Amplification was carried out in a 25-μl volume by iCycler (Bio-Rad): 94 °C 3 min; 94 °C 30 s, 62 °C 30 s, and 72 °C for 1 min for 40 cycles.
Determination of MAO A Activity in Forebrain Transgenic Mice
Adult male wild type (C3H strain), MAO A KO, and forebrain transgenic mice aged 1–4 months were used in all studies. The mice were housed individually and were allowed free access to food and water. They were housed in an air-conditioned unit with controlled temperature (20–22 °C) and humidity (50–60%). Lighting was maintained on a 12-h lightdark cycle (lights on 0600–1800).
The mice were sacrificed, and their whole brains were rapidly removed and placed in a brain matrix (ASI Instruments) embedded in ice. 2-mm thick sections were cut, and samples from frontal cortex (3 per side from 3.2 to 1.2 mm), striatum (4 per side from 0.2 to −1.8 mm), hippocampus (3 per side from −1.8 to −3.8 mm), and cerebellum (4 per side from −5.8 to −7.8 mm) were punched out from the sections with a blunt 18-gauge needle. Punched samples were homogenized in 50 volumes of 50 mm sodium phosphate buffer, pH 7.4, used for MAO A activity determination as described previously (17). In some cases, mitochondria were isolated from different brain regions, and MAO A activity was determined in the mitochondria pellets.
Autoradiography
The mice were sacrificed, and their brains were removed and frozen in isopentane. The brains were stored at −70 °C for no longer than 1 month before sectioning. For autoradiographic mapping, 12-μm frozen coronal sections were cut in duplicate (500 μm apart) in a cryostat at −20 °C and were thaw-mounted onto precleaned, superfrost/plus ice-cold microscope slides and dried by using anhydrous CaSO4 for 2 h at 4 °C followed by 1 week at −20 °C. For the determination of total and nonspecific binding with [3H]Ro 41-1049, adjacent sections were cut from 3.20-mm bregma, which was identified according to the mouse brain atlas of Franklin and Paxinos (29). [3H]Ro 41-1049 was a kind gift from Dr. G. Richards at Hoffmann-La Roche.
For autoradiographic visualization of MAO A-binding sites, incubations were carried out in parallel for sections from wild type, MAO A KO, and forebrain transgenic mice with 15 nm [3H]Ro 41-1049. Prior to binding, slides were thawed for 30 min at room temperature. Slides for the determination of total binding were incubated in 50 mm Tris-HCl containing 120 mm NaCl, 1 mm MgCl2, 5mm KCl, and 0.5 mm EDTA, pH 7.4 (1 ml for each slide), for 60 min at 37 °C. Nonspecific binding was determined by simultaneously treating a parallel set of slides, under identical incubation conditions with the addition of 1 μm clorgyline. The detailed procedure was published previously (30).
Quantitative analysis of MAO A binding was carried out by video-based computerized densitometry using a Xenix image analyzer. Tissue equivalents (nCi per mg of tissue) for MAO A labeling were derived from 3H microscale standard-based calibrations laid down with each film after subtraction of nonspecific binding images. Quantified measures were taken from both the left and right sides of sections from the whole of the frontal cortex (3.2 to −1.8 mm), striatum (1.7 to −1.3 mm), hippocampus (−1.3 to −3.8 mm), and cerebellum (−5.8 to −7.3 mm) in successive sections 500 μm apart.
Immunocytochemistry
Animal procedures were conducted in strict compliance with approved institutional protocols and in accordance with the provisions for animal care and use described in the European Communities Council Directive of 24 November 1986 (86/609/EEC). Experiments were carried out on P7 and adult mice. The day of birth was counted as P0.
Anesthetized animals were transcardially perfused with saline followed by 4% paraformaldehyde in 0.12 m phosphate buffer, pH 7.4 (PB). Serial sections were cut on a freezing microtome (50 μm). Alternate series of sections were used for immunostaining and Nissl (0.05% thionine in acetate buffer, pH 5.5). For immunostaining, rabbit polyclonal antibodies to 5-HT (1:5000; Sigma), the 5-hydroxytryptamine transporter (1:5000; Calbiochem), the mGluR5 (1:2000; Chemicon, Temecula, CA), the MAO A (1:1000; this study), and MAO B (1:2000) were diluted in 0.02 m PB saline with 0.2% gelatin and 0.25% Triton X-100 (PBS+). Sections were washed in PB and incubated for 1 h in PBS+. Sections were incubated sequentially with the primary antibodies (24 h at 4 °C), PBS+ (30 min), secondary antibodies (biotinylated goat anti-rat; biotinylated swine antirabbit; 1:200; DAKO, Denmark) (2 h at room temperature), PBS+, streptavidin-biotin-peroxidase complex (1:200; Amersham Biosciences) (2 h at room temperature) and reacted with a solution containing 0.02% diaminobenzidine, 0.6% nickel ammonium sulfate (Carlo Erba), and 0.003% H2O2 in 0.05 m Tris buffer, pH 7.6. All sections were mounted on TESPA-coated slides (Sigma), air-dried overnight, dehydrated, and coverslipped in DePeX/CO. For visualization, sections were sequentially incubated in phosphate buffer (0.1 m, pH 7.4) with 10% sucrose, 0.2% cobalt chloride (10 min), phosphate-buffered sucrose, 0.007% cytochrome c, 0.002% catalase, 0.02% dimethyl sulfoxide, and 0.05% diaminobenzidine in phosphate-buffered sucrose (all products from Sigma), and phosphate buffer.
HPLC Determination of the Levels of NE, DA, Dihydroxyphenylacetic Acid, 5-HT, and 5-Hydroxyindolacetic Acid (5-HIAA) in Forebrain Regions of Forebrain Transgenic Mice
Mice were sacrificed, and brain samples were quickly removed and immediately frozen in isopentane. Brain samples were homogenized in 100 μl of a solution containing 0.1 m trichloroacetic acid, 10 mm sodium acetate, and 0.1 mm EDTA, pH 2.0. The homogenates were sonicated using a Fisher sonic dismembrator (model 550) with the probe sonicator at setting 2 in 10 volumes of buffer at 4 °C and centrifuged for 10 min at 12,000 × g. The protein concentrations of the pellets were determined using the BCA kit (Pierce). The supernatant was centrifuged and stored at −70 °C until HPLC analysis. HPLC analysis was described previously (18).
Resident-Intruder Confrontations of Forebrain Transgenic Mice
All mice were housed individually for at least 1 week in transparent Makrolon cages measuring 29 × 13 × 13 cm. Confrontations were between mice of the same strain and of similar ages and weights. They were organized in a Latin Square arrangement, and all sessions were at least 2 days apart to prevent animal fatigue. An intruder mouse was placed in the cage of the resident mouse. The mice were allowed to interact for 10 min after the first attack. The interactions of the mice were videotaped for later analysis. The analysis procedure was described previously (31). The following five behaviors were assessed: (i) nonsocial (absence of exploration of other mouse); (ii) investigative (subject actively investigates cage, mainly by sniffing); (iii) defensive (subject actively defends or shields itself from opponent, e.g. standing on hind legs with forearms in the air); (iv) aggressive (subject engages in physical fight with opponent, e.g. biting or kicking attack); and (v) locomotive (subject is rapidly roaming cage).
RESULTS
Generation of Forebrain-specific MAO A Transgenic Mice from MAO A KO Mice
To produce forebrain-specific MAO A transgenic mice, a full-length human MAO A cDNA clone was ligated to the CaMKIIα promoter to confer expression in specific brain regions where the CaMKIIα promoter is normally active. The orientation of the insert was determined by digestion with HindIII. Fig. 1A illustrates the construction schema and final construct used to create the transgenic mice by micro-injection of the linearized plasmid DNA into fertilized eggs of MAO A KO mice. After birth, the introduction of human MAO A was assayed by PCR and confirmed. A specific 0.87-kb PCR product was detected in genomic DNA prepared from the tails of forebrain transgenic mice but not in the wild type or in the MAO A KO mice (Fig. 1B). This confirms that we were able to successfully integrate the human MAO A cDNA into the mice genomes.
Sequences derived from anchor PCR using a CaMKIIα promoter indicated insertion took place in chromosome seven (7qE3 region) and in chromosome four (4qC7 region). In each case chromosomal sequences contiguous with CaMKIIα promoter-based PCR primer corresponding to genomic sequences flanking the 5′ end of the transgenic insertion were identified by BLAT analysis for both forward and reverse CaMKIIα primers with these primer sequences aligning with chromosome 18 in the upstream promoter region of the CaMKIIα gene. The chromosome seven anchor PCR-derived sequences spanned 763 bases with 99.5% identity from 105031154 to 105031916 (same results with forward and reverse primers) and included partial overlap with an intronless olfactory receptor gene, Olfr684. Chromosome four aligned with derived anchor PCR sequences over a span of 290 bases from 106111874 to 106112163 with 99% identity. This integration site fell in to the fourth intron of the uncharacterized BC055111 gene.
Forebrain-specific expression of the introduced human MAO A transgene was evaluated by reverse transcription PCR and Western blotting using anti-human MAO A antibody. Both evaluations indicated a brain region-specific expression was achieved in the transgenic mice. Fig. 1C illustrates Western blot results and RT-PCR results for the human MAO A transcript and protein, respectively. Transcript and protein are observed in specific brain regions or tissue. Frontal cortex and hippocampus both expressed MAO A, but cerebellum and liver did not. Negative controls were wild type mouse, which do not express human MAO A and the MAO A KO strain used to create this transgenic mouse strain. As expected there was no detectable human MAO A mRNA or protein present in MAO A KO mice (Fig. 1C).
This line of transgenic mice was viable and fertile. They were intercrossed to obtain the needed number of animals for behavioral and biochemical studies. Healthy forebrain transgenic mice were obtained at the expected frequency and weight. No changes in overall brain structure or appearance compared with MAO A KO and wild type mice could be detected.
MAO A Catalytic Activity and Autoradiography Show the Expression of MAO A Specifically in the Forebrain of Transgenic Mice
Catalytic activity of MAO A using 5-HT as substrate was measured in the whole homogenates or mitochondria of the frontal cortex, striatum, hippocampus, and cerebellum of wild type, MAO A KO, and the forebrain transgenic mice (Table 1). No activity was detectable in MAO A KO mice as expected. Significant MAO A activity was detected in the frontal cortex, striatum, and hippocampus but not in the cerebellum of the transgenic mice. This suggests that MAO A is indeed expressed in the specific forebrain regions under the control of the CaMKIIα promoter. Furthermore, MAO A activity was found to be associated with the mitochondria, and this result indicates that the expressed MAO A are located on mitochondria, as is wild type for MAO. This result indicates that the human C-terminal sequence retains this targeting ability (32-34) when expressed in mice. Thus, MAO A is expressed in a forebrain-specific manner, with mitochondrial localization; however, the MAO A activity is only 2–5% of the wild type. The MAO A KO mice into which the human MAO A was introduced have essentially zero MAO A activity. The low MAO A activity in these transgenic mice points toward low CaMKIIα promoter activity compared with the endogenous MAO A promoter.
TABLE 1. MAO A catalytic activity with 5-HT as the substrate in forebrain transgenic, MAO A KO, and wild type mice.
Values are expressed in nmol/20 min/mg of protein and represent the mean ± S.E. n represents the number of mice. A 5-HT oxidation was 30 cpm above a blank that had no homogenate, thus no detectable MAO A. Multiway comparisons (one-way analysis of variance and Duncan’s test) for wild type versus MAO A KO mice, wild type versus forebrain transgenic mice, and MAO A KO versus forebrain transgenic mice.
| Genotype | MAO A activity (nmol/20 min/mg of protein) |
|||
|---|---|---|---|---|
| Frontal cortex | Striatum | Hippocampus | Cerebellum | |
| Wild type (n = 4) | 26.5 ± 0.1 | 37.2 ± 1.2 | 47.6 ± 4.8 | 39.8 ± 2.6 |
| MAO A KO (n = 5) | 0.3 ± 0.1** | 0.4 ± 0.01** | 0.4 ± 0.1** | 0.3 ± 0.1** |
| Forebrain transgenic (n = 5) | 1.3 ± 0.1##++ | 1.0 ± 0.2##++ | 1.3 ± 0.2##++ | 0.3 ± 0.1** |
p < 0.01
p < 0.01
p < 0.01
Ro 41-1049 is a specific inhibitor of MAO A (Kd value in low nanomolar range), which can be used as a radioligand for MAO A. Autoradiographic visualization of MAO A-binding sites in the brain sections using [3H]Ro 41-1049 was done to assess regional specific MAO A expression in the brain of the forebrain transgenic mice compared with MAO A KO and wild type mice. Results are shown in Fig. 2. High levels of [3H]Ro 41-1049 labeling in the frontal cortex, striatum, hippocampus, and cerebellum in wild type mice was observed (and was abolished by 1 μm clorgyline; data not shown) demonstrating that [3H]Ro 41-1049 selectively labels MAO A. [3H]Ro 41-1049 labeling in MAO A KO mice was at background levels confirming the absence of MAO A as expected in MAO KO mice. On the other hand, significant levels of [3H]Ro 41-1049 labeling in the frontal cortex, striatum, and hippocampus but not in the cerebellum were observed in the forebrain transgenic mice. Taken together, these data (Table 1 and Fig. 2) suggest that MAO A was specifically expressed in the frontal cortex, striatum, and hippocampus but not in cerebellum of forebrain transgenic mice, which is consistent with the expression of CaMKIIα (17-20) and is the expected outcome of expressing MAO A under the control of this promoter (24-27).
FIGURE 2. Autoradiography of MAO A in forebrain transgenic, MAO KO, and wild type mice.
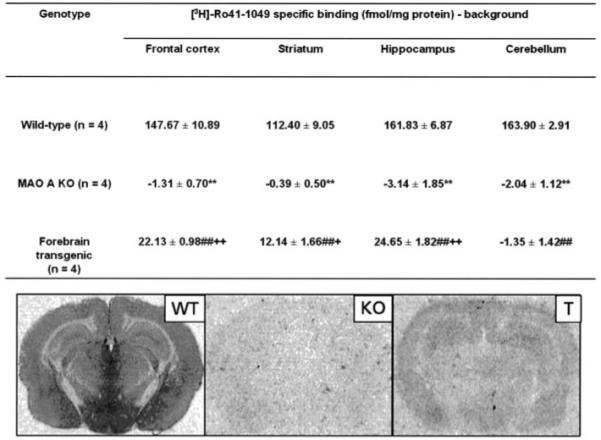
MAO A was labeled with [3H]Ro 41-1049 in 12-μm coronal sections. See “Experimental Procedures” for details. Upper panel, values are expressed in fmol/mg protein and represent the mean ± S.E. n represents the number of mice. Regional determinations were averaged from both left and right sides up to 11 sections, 500 μm apart. Multiway comparisons (one-way analysis of variance and Duncan’s test) for wild type versus MAO A KO mice (**, p < 0.01), wild type versus forebrain transgenic mice (##, p < 0.01), and MAO A KO versus forebrain transgenic mice (+, p < 0.05; ++, p < 0.01). Lower panel, visualization of [3H]Ro 41-1049 binding. Abbreviations used are as follows: WT, wild type mice; T, forebrain transgenic mice; KO, MAO A knock-out mice.
The direct visualization of MAO A protein in the forebrain transgenic mice was achieved by using polyclonal antibodies raised against purified human MAO A. Immunocytochemistry was carried out using the anti-human MAO A antibodies. The specificity and efficacy of staining was first assessed using wild type and MAO A KO mice. In normal mice MAO A is expressed abundantly in the intralaminar nuclei of the thalamus, noradrenergic neurons of the locus coeruleus, and adrenergic neurons (A1–A3) of the brainstem. Fig. 3, A and B, illustrates specific staining using these antibodies to the locus coeruleus and brainstem, respectively, whereas Fig. 3C shows the absence of MAO A reactivity in the A3 region of the brainstem in MAO A KO mice. These experiments on control mice verified the specificity of the antibody, and staining of the forebrain transgenic mice was then assessed.
FIGURE 3. MAO A immunoreactivity (ir) in forebrain transgenic, MAO A KO, and wild type mice.
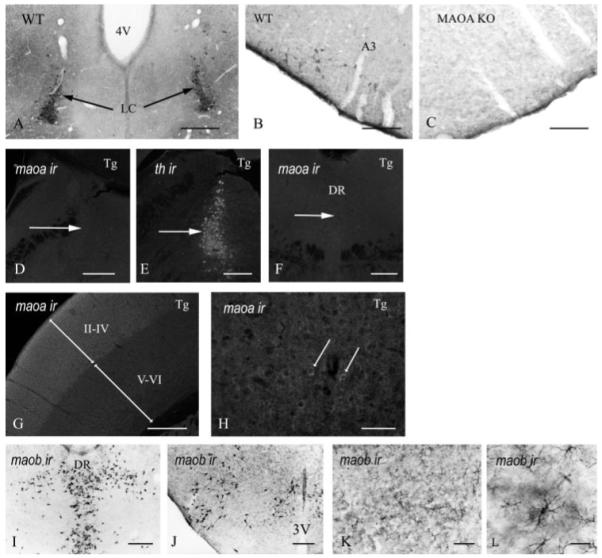
MAO A IR was detected in noradrenergic neurons of the locus coeruleus (A) and adrenergic neurons of the brainstem (B). In mice lacking MAO A, MAO A IR was absent of adrenergic neurons of the brainstem (C). In forebrain transgenic (Tg) mice, MAO A IR was absent in noradrenergic neurons (D) that express tyrosine hydroxylase (E) and serotoninergic neurons (F). In the somatosensory cortex of forebrain transgenic, MAO A IR was detected in the supragranular layers (II–IV) and in the infragranular layers (V–VI) (G). In the supragranular layers, MAO A IR was arranged in punctate clusters around the nucleus (arrows in H). In forebrain transgenic mice, MAO B IR was normally expressed in the serotoninergic neurons (I), histaminergic neurons (J), and cortical astrocytes (K and L). A3, adrenergic neurons of the brainstem; 3V, third ventricle; DR, dorsal raphe; LC, locus coeruleus. Scale bars: A and G = 175 μm; B and C = 350 μm; F and I–K = 400 μm; H and L = 85 μm.
Fig. 3, D–H, illustrates the regional specificities of expression obtained by using the CaMKIIα promoter to drive human MAO A expression in this transgenic system. Similar to MAO A KO mice, MAO A expression was absent in noradrenergic, tyrosine hydroxylase neurons of the locus coeruleus (Fig. 3, D and E), and serotoninergic neurons of the raphe nucleus (Fig. 3F) of forebrain transgenic mice. Similar to the WT mice, the somatosensory cortex showed strong MAO A expression (Fig. 3G) in the supragranular layers II–IV and expression in the infragranular layers V–VI, albeit of lower intensity than in layers II–IV. These results suggest that MAO A was specifically expressed in the forebrain. Individual neuron staining in the supragranular layers can be visible at higher magnification (arrows in Fig. 3H). The staining pattern observed in Fig. 3H is consistent with MAO A mitochondrial expression.
Fig. 3, I–L, presents immunocytochemistry using anti-MAO B antibodies to label MAO B in the transgenic mice. MAO B immunoreactivity is present and consistent with that of normal wild type mice in serotoninergic neurons (Fig. 3I), histaminergic neurons (J), and cortical astrocytes (K and L).
Taken together, these data (Table 1, Fig. 2, and Fig. 3) suggest that MAO A was specifically expressed in the somatosensory cortex, not in norepinephrine neurons in locus coeruleus or cerebellum of forebrain transgenic mice, and this is consistent with the expression of CaMKIIα (24 -27).
MAO A Substrates 5-HT, NE, and DA Decreased and MAO A Metabolite 5-HIAA in Forebrain of Transgenic Mice Increased Compared with MAO A KO Mice
To understand the consequences of the presence of MAO A on the neurotransmission in forebrain regions, steady-state levels of monoamines were determined in the frontal cortex, hippocampus, striatum, and cerebellum of transgenic, MAO A KO, and wild type mice (Fig. 4). MAO A KO mice have about a 2-fold increase in levels of MAO A substrates NE and 5-HT in all brain regions studied (frontal cortex, hippocampus, striatum, and cerebellum) than wild types in accordance with Kim et al. (18). The levels of 5-HIAA were concomitantly and significantly lower in all regions, which reflects the absence of oxidation of 5-HT in the brain of MAO A KO mice. The levels of DA in MAO A KO mice were significantly higher than wild types in the striatum and were slightly higher in the frontal cortex, the hippocampus, and the cerebellum. The levels of dihydroxyphenylacetic acid were concomitantly lower in all regions.
FIGURE 4. Levels of neurotransmitters in brain regions of forebrain transgenic, MAO KO, and wild type mice.
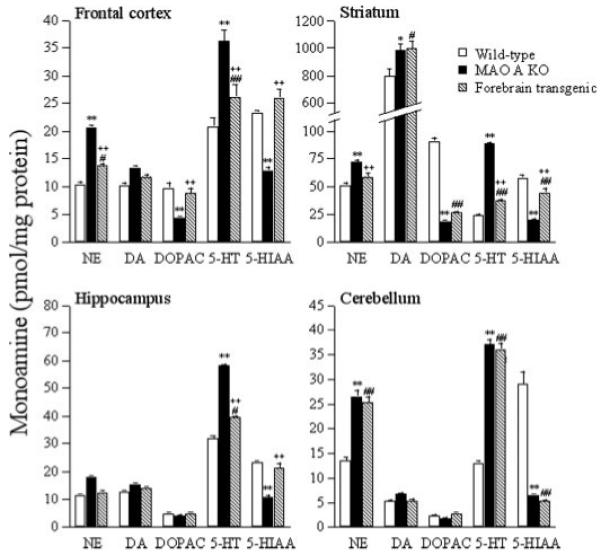
Values are expressed in pmol/mg protein and represent the mean ± S.E. Wild type (WT), MAO A KO (KO), and forebrain transgenic (T) mice n = 4. n represents the number of mice. Multiway comparisons (one-way analysis of variance and Duncan’s test) for wild type versus MAO A KO mice (**, p < 0.01; *, p < 0.05), wild type versus forebrain transgenic mice (##, p < 0.01; #, p < 0.05), and MAO A KO versus forebrain transgenic mice (++, p < 0.01; +, p < 0.05). DOPAC, dihydroxyphenylacetic acid.
The levels of NE and 5-HT in the frontal cortex (~32%), striatum (52%), and hippocampus (35%) of forebrain transgenic mice were lower than MAO A KO and were higher than wild types (frontal cortex, ~25%; striatum 56%; hippocampus 22%). These changes in monoamines suggest that MAO A was expressed in these regions of forebrain transgenic mice. In contrast, as a control, the levels of NE, DA, 5-HT, and 5-HIAA in the cerebellum of forebrain transgenic mice were similar to MAO A KO suggesting that MAO A was not expressed in the cerebellum of forebrain transgenic mice. DA levels in the frontal cortex and hippocampus of forebrain transgenic mice were lower than MAO A KOs, suggesting DA is oxidized by MAO A. In the striatum, the levels of DA in forebrain transgenic mice were similar to MAO A KO and were significantly higher than wild types. This suggests that expressed MAO A in the striatum (about 3% by activity compared with wild type) is inadequate to oxidize the exceptionally high amount of DA in this brain region.
Somatosensory Cortex Organization in Forebrain Transgenic Mice
Fig. 5 illustrates 5-HT immunolabeling in wild type, MAO A KO, and forebrain transgenic mice in substantia nigra (Fig. 5, A–C, respectively) and in the thalamus (Fig. 5, D–F, respectively). Darker regions of 5-HT immunostaining because of higher 5-HT levels are clearly visible in the MAO A KO compared with wild type and the forebrain transgenic mice where MAO A activity is present. Comparisons of the labeled substantia nigra and median geniculate nucleus in the substantia nigra region (Fig. 5, A–C) and somatosensory and visual thalamus regions (Fig. 5, D–F) illustrate well the decreases in 5-HT compared with MAO A KO. These axons target the forebrain; thus, this is another indicator that the forebrain transgenic mice express active MAO A.
FIGURE 5. 5-HT accumulation in midbrain dopaminergic neurons in postnatal day 7.
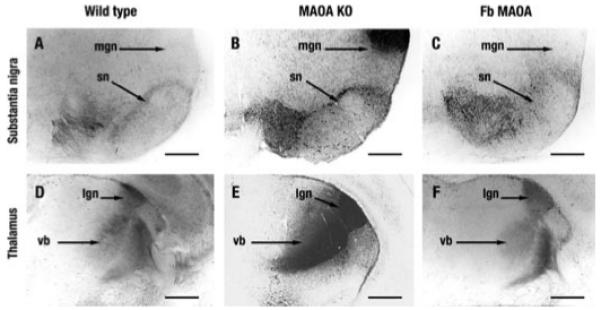
A, in wild type mice 5-HT IR labels varicose fibers at the level of the median forebrain bundle and soma in the substantia nigra (sn). B, MAO-A KO. C, in forebrain (Fb) transgenic mice 5-HT accumulates in dopaminergic neurons of substantia nigra. Note also the disappearance of 5-HT accumulation in the median geniculate nucleus (mgn), the auditory sensory thalamus. D–F, 5-HT accumulation in the somatosensory (vb) and visual (lgn) thalamus. D, in wild type mice 5-HT accumulates in cell bodies and thalamocortical axons passing through the reticular thalamic nucleus and the internal capsule. E, in MAO-A KO mice the 5-HT staining is considerably increased. F, in contrast forebrain transgenic mice display a rather normal 5-HT IR in somatosensory (vb) and visual (lgn) thalamus. Scale bars = 400 μm.
MAO A KO mice display permanent alterations in layer IV of the primary somatosensory cortex (S1); the thalamocortical axons and the granular neurons form a homogeneous band instead of being clustered into barrels (23). Restoration of MAO A function in the forebrain transgenic mice should therefore result in a restoration or similarity of cortical structure between wild and forebrain transgenic mice, compared with MAO A KO. Three experiments addressed this issue as follows: (i) analysis of thalamocortical axon segregation using 5-HT immunoreactivity (Fig. 6, A–C); (ii) dendritic differentiation using immunoreactivity of the metabotropic glutamate receptor 5 (mGluR5); and (iii) differentiation of cytoarchitecture via Nissl substance staining.
FIGURE 6. Cortical alterations in S1 of MAO-A KO and forebrain transgenic mice at postnatal day 7.
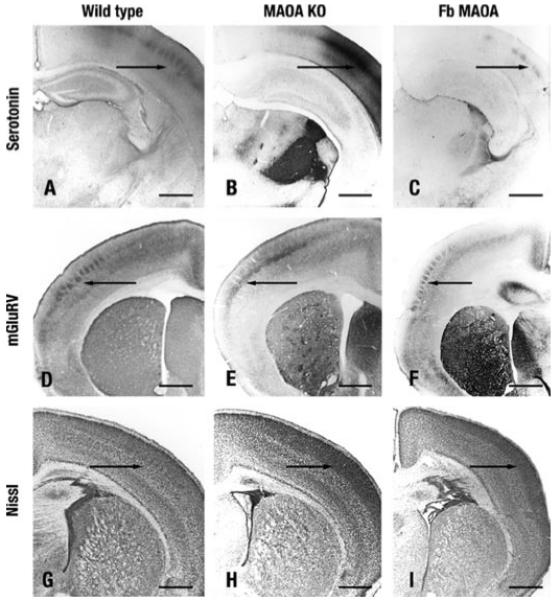
A–C, thalamocortical axon segregation visualized with 5-HT IR. A, wild type. C, forebrain (Fb) transgenic mice 5-HT IR forms whisker-related patches in layer IV of S1 (arrow). B, in MAO-A KO mice thalamocortical axons do not form patches. D–F, dendritic differentiation visualized with mGluR5 IR. D, wild type. F, in forebrain transgenic mice, mGluR5 IR labels barrels in layer IV (arrow), whereas they do not form in MAO-A KO mice (E). G–I, cytoarchitectonic differentiation. G, in wild type mice granular neurons form barrels in layer IV (arrow). H, MAO-A KO. I, forebrain transgenic mice granular neurons do not form barrels (arrow). Scale bars = 560 μm.
Fig. 6, A–C, shows thalamocortical axon segregation visualized with serotonin immunoreactivity. Whisker-related patches of S1 layer IV are found in wild type mice, abrogated in MAO A KO mice, but are observed here in the forebrain transgenic mice. Fig. 6, D–F, clearly indicates the barrel formation resulting from mGluR labeling is abrogated in MAO A KO mice but is observed in the transgenic mice with very similar patterning as wild type. Finally, Nissl body staining is illustrated in Fig. 6, G–I, showing the granular neuron barrels of layer IV of wild type mice are also present in the forebrain transgenic mice but are absent in the MAO A KO.
Aggression Reduced in Transgenic Mice
Behavioral characterization (nonsocial, investigative, defensive, aggressive, and locomotive behavior) of forebrain transgenic, MAO A KO, and wild type mice is presented in Table 2. MAO A KO mice showed higher levels of aggressive behavior than wild types in accordance with Cases et al. (17) (Table 2). The duration of aggressive behavior of forebrain transgenic mice was reduced to wild type levels, and they spent more time in investigative behavior (Table 2). They have similar locomotive behavior to MAO A KOs. No significant difference in nonsocial behavior was found in all mice.
TABLE 2. Aggressive behavior of forebrain transgenic, MAO A KO, and wild type mice.
The duration of aggressive behavior of the more aggressive mouse from each pair is reported, and values are expressed in seconds and represent the mean ± S.E. n represents the number of mice. Multiway comparisons (one-way ANOVA and Tukey-Kramer test) are shown for wild type versus MAO A KO mice, wild type versus forebrain transgenic mice, and MAO A KO versus forebrain transgenic mice. The animal care was in accordance with institutional guidelines.
| Genotype | Duration of behavior (s/10 min) |
||||
|---|---|---|---|---|---|
| Nonsocial | Investigative | Defensive | Aggressive | Locomotive | |
| Wild type (n = 10) | 234 ± 22 | 159 ± 20 | 20 ± 8 | 52 ± 7 | 136 ± 31 |
| MAO A KO (n = 12) | 250 ± 33 | 108 ± 31 | 26 ± 17 | 125 ± 19** | 99 ± 29 |
| Forebrain transgenic (n = 20) | 243 ± 24 | 252 ± 24++ | 2 ± 1 | 30 ± 7++ | 78 ± 12## |
p < 0.01
p < 0.01
p < 0.001
DISCUSSION
One line of forebrain-specific MAO A transgenic mice has been generated from MAO A KO mice by using the promoter of CaMKIIα (Fig. 1A). The presence of the human MAO A transgene (Fig. 1B) was shown by PCR of the genomic DNA, and its expression was verified by RT-PCR and Western blots using various regions of the tissues (Fig. 1C).
Radioautography using the MAO A-specific radioligand [3H]Ro 41-1049 (Fig. 2), MAO A antibody (Fig. 3), or 5-HT immunostain (Fig. 5) provides evidence for the forebrain-specific expression of the human MAO A. Compared with MAO A KO mice, lower levels of 5-HT, NE, and DA and higher levels of the MAO A metabolite 5-HIAA were found in the forebrain regions but not in the cerebellum of the forebrain transgenic mice (Fig. 4). Taken together, these results suggest that the human MAO was indeed specifically expressed in the forebrain of forebrain transgenic mice.
It is intriguing that in vitro assay showed only about 5% of the wild type MAO A is expressed in forebrain transgenic mice. However, it is capable of producing significant changes in the steady-state levels of monoamines (22–56%). These results suggest that there may be an abundant excess of MAO A in vivo. This result is consistent with the previous finding that MAO inhibitors are only effective as antidepressants when inhibiting MAO activity by 80–90% (34). It is also possible that the newly expressed human MAO A may have a different microenvironment on the mitochondria, thus the catalytic activity may have been underestimated. Nevertheless, these results confirm a direct role of MAO A in aggression rather than a secondary effect related to the MAO A KO.
These results suggest that the increased levels of NE, 5-HT, and possibly DA in the forebrain of MAO A KO mice may underlie their aggressive behavior. Our analysis of 5-HT immunolabeling correlates with the HPLC study (Fig. 4). MAO rescue in Fb transgenic mice of 5-HT and NE metabolism in postsynaptic regions such as cortex and striatum implies 5-HT is transported onto these cells. It has been shown that there are atypical locations of 5-HT containing neurons in cortical, hippocampal, or amygdaloid areas of MAO A KO mice and that the thalamic neurons transiently express the serotonin transporter on axonal terminals in early postnatal development (35). Similarly, we have found 5-HT immunolabeling in the substantia nigra region (Fig. 5, A–C), also in somatosensory and visual thalamus regions (Fig. 5, D–F). The dopamine transporter may play a role in this. Previous work has shown that in substantia nigra neurons the dopamine transporter acts to cause uptake of 5-HT in mice that are doubly deficient in MAO and the 5-HT transporter (36). Importantly, assessment of the rescue of 5-HT metabolism in Fb transgenic mice was consistent as assessed by autoradiography and by biochemical means; both the intensity of 5-HT immunolabeling (Fig. 5) and the levels of 5-HT determined by HPLC (Fig. 4) were highest in MAO A KO mice, whereas forebrain transgenic mice were reduced to WT type levels.
MAO A KO mice display permanent alterations of the somatosensory cortex, as shown by 5-HT immunolabeling (Fig. 6), Interestingly, axonal, cellular, and dendritic patterning are restored in forebrain transgenic mice (Fig. 6). We also analyzed two general differentiation markers of the cortex, calretenin and calbindin, and found no alterations in their distribution (data not shown).
In summary, this study demonstrates that the somatosensory cortex organization, including thalamocortical axon segregation, barrel field structure, and cytoarchitecture, reverted to a phenotype with similarity to wild type mice and easily distinguishable for the MAO A KO mice that were transformed by introduction of the CaMKIIα promoter-driven expression of human MAO and consequent regional specific expression of human MAO A. Aggressive behavior of MAO A KO mice decreased with this forebrain-specific MAO A expression in the MAO A KO mice. Our results suggest that the increased levels of NE, 5-HT, and possibly DA in the forebrain of MAO A KO mice may trigger a complex chain of events in the brain, which lead to aggressive behavior. It is also possible that an increased 5-HT level in forebrain during the development of MAO A KO pups permanently disrupts the distribution of layer IV neurons in the somatosensory cortex (37), and thus resulted in behavioral changes in MAO A-deficient mice. Although the aggressive behavior seen in MAO A-deficient mice is consistent with the impulsive aggression in man with MAO A deficiency (10, 11), considerable caution has to be taken in extrapolating results obtained in mice to explain complex aggressive behavior in humans. Nevertheless, a mouse “knock-out” and “knock-in” model for the MAO A gene, as we have demonstrated in this study, provides a valuable basis for understanding the mode of its function in the brain. The region-specific alterations of brain neurotransmitters because of MAO A expression, as shown in forebrain transgenic mice, may contribute to the understanding and perhaps to the treatment of aggression.
Our findings that aggressive behavior was reduced in MAO A forebrain-specific expressed mice are consistent with literature reports in that the aggressive behavior in adults is because of early neuronal developmental effects on the brain structures (17). A functional polymorphism in humans with maltreatment in childhood increased the risk of antisocial and criminal behavior index. Brain structures are affected by the polymorphism in MAO A promoter (15). These studies and our current study demonstrated that MAO A may be an important enzyme that regulates the brain structure at a critical neuronal developmental stage, which is important for the development of brain structures and behaviors in adults. In addition, we have found a new function of MAO A and its novel repressor R1 (RAM2/CDCA7L/JPO2) in apoptosis and c-Myc-induced cell cycle signaling pathway (38). The various MAO KO animal models generated, including MAO A KO, MAO B KO, MAO AB double KO, and forebrain-specific human MAO A transgenic MAO A KO generated here, would be valuable for further studying the role of MAO A and neurotransmitters, 5-HT, NE, and DA, in neuronal development, brain structure, and behavior.
Acknowledgments
We are grateful to Dr. M. Mayford for the supply of CaMKIIα promoter constructs, Dr. G. Richards at Hoffmann-La Roche for 3[H]Ro 41-1049, and Rani Geha for determining MAO A activity.
Footnotes
This work was supported in part by National Institute for Mental Health Grants R37 MH39085 (MERIT Award) and RO1 MH067968 and the Boyd and Elsie Welin professorship award (to J. C. S.).
- MAO A
- monoamine oxidase A
- KO
- knock-out mice
- NE
- norepinephrine
- 5-HT
- serotonin
- 5-HIAA
- 5-hydroxyindoleacetic acid
- DA
- dopamine
- CaMKIIα
- calmodulin-dependent kinase IIα
- RT
- reverse transcription
- PBS
- phosphate-buffered saline
- HPLC
- high pressure liquid chromatography
- PEA
- phenylethylamine
- IR
- immunoreactivity
REFERENCES
- 1.Shih JC. Neuropsychopharmacology. 1991;4:1–7. [PubMed] [Google Scholar]
- 2.Thorpe LW, Westlund KN, Kochersperger LM, Abell CW, Denney RM. J. Histochem. Cytochem. 1987;35:23–32. doi: 10.1177/35.1.3025289. [DOI] [PubMed] [Google Scholar]
- 3.Johnston JP. Biochem. Pharmacol. 1968;17:1285–1297. doi: 10.1016/0006-2952(68)90066-x. [DOI] [PubMed] [Google Scholar]
- 4.Knoll J, Magyar K. Adv. Biochem. Psychopharmacol. 1972;5:393–408. [PubMed] [Google Scholar]
- 5.Shih JC, Chen K, Ridd MJ. Annu. Rev. Neurosci. 1999;22:197–217. doi: 10.1146/annurev.neuro.22.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bach AW, Lan NC, Johnson DL, Abell CW, Bembenek ME, Kwan SW, Seeburg PH, Shih JC. Proc. Natl. Acad. Sci. U. S. A. 1988;85:4934–4938. doi: 10.1073/pnas.85.13.4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grimsby J, Chen K, Wang LJ, Lan NC, Shih JC. Proc. Natl. Acad. Sci. U. S. A. 1991;88:3637–3641. doi: 10.1073/pnas.88.9.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lan NC, Heinzmann C, Gal A, Klisak I, Orth U, Lai E, Grimsby J, Sparkes RS, Mohandas T, Shih JC. Genomics. 1989;4:552–559. doi: 10.1016/0888-7543(89)90279-6. [DOI] [PubMed] [Google Scholar]
- 9.Grimsby J, Lan NC, Neve R, Chen K, Shih JC. J. Neurochem. 1990;55:1166–1169. doi: 10.1111/j.1471-4159.1990.tb03121.x. [DOI] [PubMed] [Google Scholar]
- 10.Brunner HG, Nelen M, Breakefiel XO, Ropers HH, Van Oost BA. Science. 1993;262:578–580. doi: 10.1126/science.8211186. [DOI] [PubMed] [Google Scholar]
- 11.Brunner HG, Nelen MR, Van Zandvoort P, Abeling NGGM, Van Gennip AH, Wolters EC, Kuiper MA, Ropers HH, Van Oost BA. Am. J. Hum. Genet. 1993;52:1032–1039. [PMC free article] [PubMed] [Google Scholar]
- 12.Sabol SZ, Hu S, Hamer D. Hum. Genet. 1998;103:273–279. doi: 10.1007/s004390050816. [DOI] [PubMed] [Google Scholar]
- 13.Caspi A, McClay J, Moffitt TE, Mill J, Martin J, Craig IW, Taylor A, Poulton R. Science. 2002;297:851–854. doi: 10.1126/science.1072290. [DOI] [PubMed] [Google Scholar]
- 14.Kim-Cohen J, Caspi A, Taylor A, Williams B, Newcombe R, Craig IW, Moffitt TE. Mol. Psychiatry. 2006;11:903–913. doi: 10.1038/sj.mp.4001851. [DOI] [PubMed] [Google Scholar]
- 15.Meyer-Lindenberg A, Buckholtz JW, Kolachana BR, Hariri A, Pezawas L, Blasi G, Wabnitz A, Honea R, Verchinski B, Callicott JH, Egan M, Mattay V, Weinberger DR. Proc. Natl. Acad. Sci. U. S. A. 2006;103:6269–6274. doi: 10.1073/pnas.0511311103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Passamonti L, Fera F, Magariello A, Cerasa A, Gioia MC, Muglia M, Nicoletti G, Gallo O, Provinciali L, Quattrone A. Biol. Psychiatry. 2006;59:334–340. doi: 10.1016/j.biopsych.2005.07.027. [DOI] [PubMed] [Google Scholar]
- 17.Cases O, Seif I, Grimsby J, Gaspar P, Chen K, Pournin S, Müller U, Aguet M, Babinet C, Shih JC, De Maeyer E. Science. 1995;268:1763–1766. doi: 10.1126/science.7792602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim JJ, Shih JC, Chen K, Chen L, Bao S, Maren S, Anagnostaras SG, Faneslow MS, De Maeyer E, Seif I, Thompson RF. Proc. Natl. Acad. Sci. U. S. A. 1997;94:5929–5933. doi: 10.1073/pnas.94.11.5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grimsby J, Toth M, Chen K, Kumazawa T, Klaidman L, Adams J, Karoum F, Gal J, Shih JC. Nat. Genet. 1997;17:1–5. [Google Scholar]
- 20.Lenders JW, Eisenhofer G, Abeling NG, Berger W, Murphy DL, Konings CH, Bleeker CH, Wagemakers LM, Kopin IJ, Karoum F, Van Gennip AH, Brunner HG. J. Clin. Investig. 1996;97:1010–1019. doi: 10.1172/JCI118492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen K, Holschneider DP, Wu W, Rebrin I, Shih JC. J. Biol. Chem. 2004;279:39645–39652. doi: 10.1074/jbc.M405550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vitalis T, Cases O, Callebert J, Launay JM, Price DJ, Seif I, Gaspar P. J. Comp. Neurol. 1998;393:169–184. doi: 10.1002/(sici)1096-9861(19980406)393:2<169::aid-cne3>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 23.Rebsam A, Seif I, Gaspar P. Neuroscience. 2005;25:706–710. doi: 10.1523/JNEUROSCI.4191-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayford M, Bach ME, Huang Y-Y, Wang L, Hawkins RD, Kandel ER. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- 25.Miller SG, Kennedy MB. Cell. 1986;44:861–870. doi: 10.1016/0092-8674(86)90008-5. [DOI] [PubMed] [Google Scholar]
- 26.Wang H, Ferguson GD, Pineda VV, Cundiff PE, Storm DR. Nat. Neurosci. 2004;7:635–642. doi: 10.1038/nn1248. [DOI] [PubMed] [Google Scholar]
- 27.Packer MA, Hemish J, Mignone JL, John S, Pugach I, Enikolopov G. Cell. Mol. Biol. (Noisy-Le-Grand) 2005;51:269–277. [PubMed] [Google Scholar]
- 28.Choi T, Huang M, Gorman C, Jaenisch R. Mol. Cell. Biol. 1991;11:3070–3074. doi: 10.1128/mcb.11.6.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. Academic Press; New York: 1997. [Google Scholar]
- 30.Cesura AM, Bos M, Galva MD, Imof R, Da Prada M. Mol. Pharmacol. 1990;37:358–366. [PubMed] [Google Scholar]
- 31.Shih JC, Ridd MJ, Chen K, Meehan WP, Kung M-P, Seif I, De Mayer E. Brain Res. 1999;835:104–112. doi: 10.1016/s0006-8993(99)01478-x. [DOI] [PubMed] [Google Scholar]
- 32.Mitoma J, Ito A. J. Biochem. (Tokyo) 1992;111:20–24. doi: 10.1093/oxfordjournals.jbchem.a123712. [DOI] [PubMed] [Google Scholar]
- 33.Chen K, Wu HF, Shih JC. J. Neurochem. 1996;66:797–803. doi: 10.1046/j.1471-4159.1996.66020797.x. [DOI] [PubMed] [Google Scholar]
- 34.Rebrin I, Geha RM, Chen K, Shih JC. J. Biol. Chem. 2001;276:29499–29506. doi: 10.1074/jbc.M100431200. [DOI] [PubMed] [Google Scholar]
- 35.Cases O, Lebrand C, Giros B, Vitalis T, De Maeyer E, Caron MG, Price DJ, Gaspar P, Seif I. J. Neurosci. 1998;18:6914–6969. doi: 10.1523/JNEUROSCI.18-17-06914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mossner R, Simantov R, Marx A, Lesch KP, Seif I. Neurosci. Lett. 2006;401:49–54. doi: 10.1016/j.neulet.2006.02.081. [DOI] [PubMed] [Google Scholar]
- 37.Cases O, Vitalis T, Seif I, De Maeyer E, Sotelo C, Gaspar P. Neuron. 1996;16:297–307. doi: 10.1016/s0896-6273(00)80048-3. [DOI] [PubMed] [Google Scholar]
- 38.Ou X-M, Chen K, Shih JC. Proc. Natl. Acad. Sci. U. S. A. 2006;103:10923–10928. doi: 10.1073/pnas.0601515103. [DOI] [PMC free article] [PubMed] [Google Scholar]