

Abstract
Background
Peer group deviance (PGD) is strongly linked to liability to drug use including cannabis. Our aim was to model the genetic and environmental association, including direction of causation, between PGD and cannabis use (CU).
Method
Results were based on 1753 adult males from the Mid-Atlantic Twin Registry with complete CU and PGD data measured retrospectively at three time intervals between 15 and 25 years using a life-history calendar.
Results
At all ages, multivariate modeling showed that familial aggregation in PGD was explained by a combination of additive genetic and shared environmental effects, Moreover the significant PGD-CU association was best explained by a CU to PGD causal model in which large portions of the additive genetic (50% to 78%) and shared environmental variance (25% to 73%) in PGD were explained by CU.
Conclusions
Until recently PGD was assumed to be an environmental, upstream risk factor for CU. Our data are not consistent with this hypothesis. Rather, they suggest that the liability to affiliate with deviant peers is better explained by a combination of genetic and environmental factors that are indexed by CU which sits as a “risk indicator” in the causal pathway between genetic and environmental risks and the expression of PGD. This is consistent with a process of social selection by which the genetic and environmental risks in CU largely drive the propensity to affiliate with deviant peers.
Keywords: Peers, drugs, cannabis, risks, genes
A substantial body of literature has linked peer group deviance (PGD) and liability to drug use (1–12). In a meta-analysis of 2700 papers, Allen and colleagues (11) found that increasing PGD predicts drug use in general (r=.30) and cannabis use (CU) even more strongly (r=0.38).
Although most reports do not test alternate causal models, the general consensus is that peers influence the risk of drug use (13). However, among those that have explored competing models, Farrell and colleagues found that associations between peer deviance and drug use are better explained by a reciprocal interaction (14). An alternate explanation is that the association is the result of correlated liabilities which increases both the risk of deviant peer affiliation and drug use. These might be environmental risks such as low parental monitoring or biological such as a prefrontal cortex dysfunction leading to behavioural disinhibition (15–17).
Although PGD as a risk factor has typically been considered `environmental', a number of behavior genetic studies have revealed that variation in PGD is attributable to a combination of environmental and genetic factors (18–26). Unfortunately, the relative contribution of genes and environment varies across studies, perhaps due to variations in measurement, age of sample, study design, and low statistical power. Recently, Kendler and colleagues (27) showed, using a large population based sample of male twins, that genetic effects on PGD increase steadily from ~30% to ~50% between the ages of 8 and 25 years, while shared environmental influences decline. This suggests that as adolescents mature and create their own social worlds, genetic factors become increasingly important in peer affiliation while common or shared environmental become progressively less influential.
More is known about the etiology of drug use. For instance, a number of twin studies support the hypothesis that both genetic and environmental effects generate variation in drug use, abuse and dependence for a variety of licit and illicit substances including cannabis (28–44). Our knowledge of the etiological mechanisms influencing the transition from initiation, to regular use and abuse is also improving (29, 42, 44). Recently, Gillespie and colleagues (45) have estimated that nearly half of the total genetic variation in the symptoms of cannabis abuse can be explained by genetic effects underpinning variation in the liability to initiate cannabis.
Now, the challenge for twin studies is to move beyond estimating heritabilities and begin to identify the causal pathways to drug use and other complex behaviors. Rather than being entirely attributable to “within the skin” genetic effects (via for example, brain neurochemical systems), a proportion of the observed genetic risk to CU may be mediated by “outside the skin” genetic pathways via active genotype by environment correlations. Such correlations arise when individuals create or evoke environments as a result of their genetically influenced dispositions (46). In particular, individuals at risk for drug problems may seek out and help create deviant social environments which in turn exacerbate the risk of substance use.
To date no twin studies have examined the nature of the association between PGD and quantitative measures of CU. Our prediction is that if genetic risk for CU is mediated through self-selection into deviant peer groups then we would expect to see significant genetic contributions in the PGD-CU association. In order to test this hypothesis as well as determine the nature of the causal relationship, we model the PGD-CU association using data from three epochs between the ages of 15 to 25 years. The key issues are (i) to what extent is the covariance between PGD and CU explained by shared genetic and environmental liabilities, (ii) how does the relative contribution of these shared genetic and environmental liabilities change over time, and (iii) what is the direction of causation between PGD and CU?
Methods
Subjects
As part of on ongoing study of adult male twins from the Virginia Adult Twin Study of Psychiatric and Substance Use Disorders (VATSPSUD) this report is based on data collected from a 2nd and 3rd wave of interviews between 1994 and 2004. The VATSPSUD is described in detail elsewhere (47). Briefly, twins were eligible for participation in this study if one or both twins were successfully matched to birth records, were a member of a multiple birth with at least one male, were Caucasian, and were born between 1940 and 1974. Of 9,417 eligible individuals for the first wave (1993–1996), 6,814 (72.4%) completed the initial interviews. At least one year later, we contacted those who had completed the initial interview to schedule a second interview. The second interview (1994–1998) was completed by 5,629 individuals or 82.6% of those who had completed the 1st interview. The third interview wave (1998–2004), was completed solely by members of male-male twin pairs. Individuals were eligible for this study if they came from a male-male pair, and if both had been interviewed in wave 2.
The third interview included measures of retrospectively assessed peer group deviance (PGD) at five age period between 8 to 11, 12 to 14, 15 to 17, 18 to 21, and 22 to 25 years. The importance of these time periods is underscored by observations that (i) the mean onset or initiating age for most drugs use in the U.S. general population is between 12 and 20 years (45, 47, 48), (ii) cessation of drug use occurs normally by 29 years, while initiation rarely occurs after this age (49), and (iii) incidence of drug use, abuse and dependence peaks from age 15 to 25 (45, 50, 51). The PGD data were based on 10 items obtained from two validated instruments(52, 53) which assessed the proportion of the respondent's friends, at each particular epoch, who engaged in specific behaviors. Friends were defined as “…people who you would have seen regularly and spent time with in school and outside of school.” The 10 items were: (1) smoked cigarettes; (2) drunk alcohol; (3) got drunk; (4) had problems with alcohol; (5) been in trouble with the law; (6) stole or damaged property on purpose; (7) smoked marijuana; (8) used inhalants; (9) used other drugs like cocaine, downers or LSD; and (10) sold or gave drugs to other kids. The 5 response options were: (1) none; (2) a few; (3) some; (4) most; and (5) all. As an alternate to using raw sum scores and because measurement error and item specific variance are known to produce biased estimates in causal modeling (54) we estimated individual maximum likelihood factor scores for the PGD items at each time point based on the factor loadings and item thresholds calculated under a uni-dimensional factor structure in the Mx (55) software package.
Cannabis use (CU) data were based on average monthly use. CU was measured in individual drug units and one joint was considered one dose. Therefore, if a subject smoked 3 joints per day every day, then monthly use was recorded as 90. So as to coincide with the fixed PGD measures, average monthly CU was calculated for the same five PGD age periods.
In order to improve the quality of the PGD and CU retrospective measures which have the potential for recall bias and telescoping effects (56), the interview utilized a Life History Calendar format developed by Thornton (57). This method has been empirically shown to improve the accuracy of retrospective reporting by providing multiple cues to improve the chances of accurate recall (57, 58). This makes the task more akin to the accurate and well-retained process of recognition than to the less reliable task of free recall.
Due to the sparseness in the cannabis use data at earlier ages, we limited our analyses to data between 15 and 25 years. In order to correct for skew, both the CU and the latent factor PGD scores at each time period were recoded onto three and five point ordinal scales respectively.
There were 1738, 1768, and 1761 male twins with complete latent factor PGD scores at times 1 through 3 respectively which represented 73% to 75% of the eligible sample from the previous interview (N=2368). Complete CU data were available from 1781 subjects at each time period. Age ranged from 24 to 62 years (μ = 40.3 years, σ2 = 9.1 years). Standardized Cronbach alpha coefficients for the PGD items at times 1 through 3 were 0.90, 0.87 and 0.87 respectively. As reported elsewhere (27), PGD test-retest correlations for the three time periods (based on 141 subjects interviewed on average of 29 days apart) were 0.81, 0.78 and 0.73 respectively. Age adjusted retest correlations for CU were 0.97, 0.94 and 0.94 at times 1 through 3 respectively.
Zygosity & Interview Protocol
Zygosity was diagnosed using a combination of self-report measures, photographs and DNA analysis(33). In both interviews, most subjects (~90%) were interviewed by telephone. A small number were interviewed in person because of subject preference, residence in an institutional setting (usually jail), or not having a telephone. The project was approved by the Virginia Commonwealth University institutional review board. Subjects were informed about the goals of the study and provided informed consent before interviews. Interviewers had a Master's degree in a mental health-related field or a Bachelor's degree in this area plus two years of clinical experience. The two members of a twin pair were each interviewed by different interviewers.
Statistical Analyses
We used the raw ordinal analysis method in Mx (55) to analyze the twin data. This approach is based on the Central Limit Theorem which assumes that ordered categories reflect an imprecise measure of an underlying, normal liability distribution, and that this distribution has one or more threshold values which discriminate between categories (59, 60). All analyses were corrected for the linear effects of age at interview to remove age and cohort effects which are confounded in these data.
In studies of monozygotic (MZ) and dizygotic (DZ) twins reared together, phenotypic variation can be explained by additive genetic (A), common environment (C) and random environment (E) variance components. With multivariate analysis, the additional information in the cross-twin cross-trait correlations can allow us to determine the extent to which genetic and environmental influences are shared in common or are variable specific (61).
Decomposing the Covariance
Multivariate analysis makes use of the information in the cross-twin cross trait correlations to permit us to determine the extent to which two or more measured phenotypes can be explained by common genetic and environmental influences (61). Since our first aim was to determine how much of the covariance between PGD and CU can explained by shared genetic and environmental liabilities, we fitted a Cholesky decomposition to the data (54). Illustrated in Figure 1, this is a method of triangular decomposition where the first variable is assumed to be caused by a latent factor that can also explain some or all of the variance in the remaining variable(s). This pattern continues until the final observed variable is explained by a latent variable, which is uncorrelated with all preceding factors and influences only one variable (i.e. a factor specific to one variable). The same factor structure is repeated for the sources of variance described above (A, C, and E). In order to estimate the variance in CU explained by PGD, we first entered PGD followed by CU.
Figure 1.
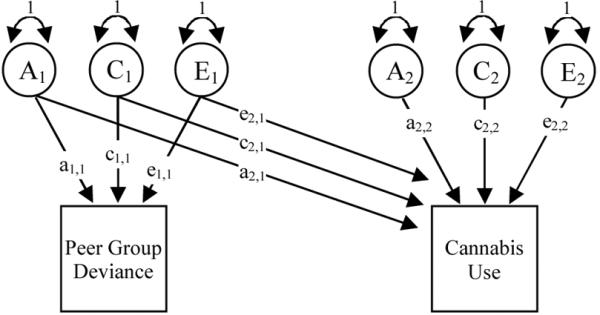
Cholesky decomposition to model the association between peer group deviance (PGD) and cannabis use (CU) by decomposing the source of covariance between PGD and CU into shared genetic (a2,1) and environmental (c2,1 & e2,1) effects. This approach also models the genetic (a2,2) and environmental (c2,2 & e2,2) effects which are unique to CU.
Note: A, C & E = latent additive genetic, shared and non-shared environmental effects for PGD and CU
Modeling Direction of Causation
Under the Cholesky decomposition model any covariance between PGD and CU is attributable to unmeasured correlated, latent liabilities. This model is agnostic in so far it does make predictions about the direction of causation. It is possible to use the same cross-twin cross-trait correlations to test hypotheses about the direction of causation at the phenotypic level between variables measured at the same time. This form of modeling assumes that (see 62) sibling cooperation or rivalry is absent, the relationship between PGD and CU is equivalent for twin 1 and twin 2, the twin pair correlations are different for PGD and CU (63), and there are no unmeasured variables which influence both measures thereby inflating the correlations arising through the causal influence of one variable on the other. Based on the methods described elsewhere (44, 62, 64) we fitted a series of uni-directional and reciprocal causation models to the twin data illustrated in Figure 2. Heath (62) has shown that the uni-directional and reciprocal causation models are nested within the Cholesky decomposition which permits model comparisons using goodness of fit statistics.
Figure 2.
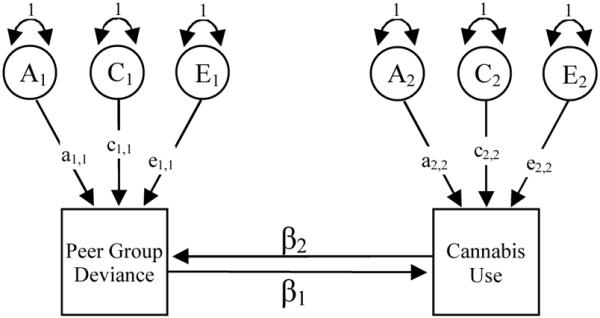
Modeling direction of causation between peer group deviance (PGD) and cannabis use (CU). This approach predicts the relationship between PGD and CU is explained by a reciprocal interaction at the phenotypic level. In the uni-directional PGD-to-CU and CU-to-PGD models, the β2 and β1 pathways are set to zero.
Note: A, C & E = latent additive genetic, shared and non-shared environmental effects for PGD and CU
We chose a priori to retain all parameters in our best fitting multivariate model. Sullivan and Eaves (65) have reported that in analyses based on discreet traits, estimates from the full ACE model will be more accurate and that attempts at parsimony result in oversimplification of the models rather than a simpler and more accurate representation of the data. This will likely occur in cases such as ours which involve more complex multivariate modeling and where the sample is not large enough to make definitive conclusions. Removing all parameters with lower bounds spanning zero, including parameters with small point estimates i.e. < 0.10, assumes that the component of variance is known to be zero without any error variance, and if this argument is incorrect, then future research might ignore an important source of variance (65).
Results
Phenotypic & Cross-Twin Cross-Trait Correlations
The cross-trait correlations between the latent factor PGD scores and CU at 15 to 17 years, 18 to 21 years and 22 to 25 years were 0.65, 0.64 and 0.61 respectively. The monozygotic (MZ) and dizygotic (DZ) cross-twin cross-trait correlations at each epoch are shown in Table 1. Based on the 95% confidence intervals, all correlations were significantly different from zero, suggesting that familial aggregation accounts for some of the covariance between PGD and CU. All of the DZ twin pair cross-trait correlations were greater than half of the MZ twin pair cross-trait correlations which suggests that a combination of genetic and shared environmental likely explains the familial covariance.
Table 1.
Comparison of monozygotic (MZ) and dizygotic (DZ) cross-twin cross-trait peer group deviance and cannabis use polychoric correlations with 95% confidence intervals for each time period at 15 to 17 years, 18 to 21 years and 22 to 25 years.
| 15–17 years | 18–21 years | 22–25 years | |
|---|---|---|---|
| MZ | 0.52 (0.45 0.59) | 0.48 (0.41 0.55) | 0.44 (0.36 0.51) |
| DZ | 0.38 (0.27 0.47) | 0.36 (0.25 0.45) | 0.29 (0.18 0.39) |
Multivariate Analyses
We next compared the fit the Cholesky decomposition to the two uni-directional (PGD→CU & CU→PGD) and reciprocal causation (PGD↔CU) models. As shown in Table 2, the PGD→CU model could be rejected at all ages. The causal CU→PGD and reciprocal PGD↔CU models both provided a good fit to the data as judged by the non-significant changes in log likelihood and lowest sample size adjusted Bayesian Information Criterion (BIC) [which has been shown to outperform the more traditionally used Akaike Information Criterion (66)]. In the reciprocal interaction model, the standardized causal pathways (β1) from PGD→CU at each time period were negative and therefore more likely to be artifactual than substantive. They were also small (−0.14, −0.27 and −0.17) which meant that PGD explained very little variance in CU, so the results more closely resemble the CU→PGD model. Therefore, the more parsimonious CU→PGD causal model was chosen as the best fitting.
Table 2.
Comparison of the (i) full Cholesky decomposition, (ii) uni-directional Peer Group Deviance causes Cannabis Use (PGD→CU), (iii) uni-directional Cannabis Use causes Peer Group Deviance (CU→PD), and (iv) reciprocal causation (PGD↔CU) models at 15 to 17 years, 18 to 21 years and 22 to 25 years.
| 15–17 years | −2LL | df | Δ−2LL | Δdf | p | BIC |
|---|---|---|---|---|---|---|
| Cholesky | 6921.31 | 3546 | −3214.81 | |||
| PGD→CU | 6935.70 | 3548 | 14.39 | 2 | <0.001 | −3211.38 |
| CU→PGD | 6923.23 | 3548 | 1.92 | 2 | 0.38 | −3217.62 |
| PGD↔CU | 6921.34 | 3547 | 0.03 | 1 | 0.86 | −3216.68 |
| 18–21 years | LL | df | Δ2LL | Δdf | p | BIC |
|---|---|---|---|---|---|---|
| Cholesky | 7722.99 | 3541 | −2804.56 | |||
| PGD→CU | 7752.65 | 3543 | 29.66 | 2 | <0.001 | −2793.50 |
| CU→PGD | 7726.97 | 3543 | 3.98 | 2 | 0.14 | −2806.33 |
| PGD↔CU | 7723.28 | 3542 | 0.29 | 1 | 0.59 | −2806.30 |
| 22–25 years | −2LL | df | Δ2LL | Δdf | p | BIC |
|---|---|---|---|---|---|---|
| Cholesky | 7725.38 | 3542 | −2805.25 | |||
| PGD→CU | 7744.32 | 3544 | 18.94 | 2.00 | <0.001 | −2799.54 |
| CU→PGD | 7729.08 | 3544 | 3.70 | 2.00 | 0.16 | −2807.16 |
| PGD↔CU | 7728.00 | 3543 | 2.62 | 1.00 | 0.11 | −2805.82 |
−2LL = log-likelihood
Δ−2LL=change in log-likelihood which is asymptotically distributed as a chi-square
BIC = sample size adjusted Bayesian Information Criterion
All PGD and CU thresholds are adjusted for the linear effects of age at interview
Standardized path coefficients and their 95% confidence intervals (CI) for all parameters in the CU→PGD causal model are shown in Table 3. Although we have retained all parameters in the model, the 95% CIs for the additive genetic and shared environmental pathways from PGD span zero at all ages. The same table also includes the CU→PGD causal parameters which are large, significant and range from 0.60 to 0.67.
Table 3.
Standardized path coefficients and 95% confidence intervals from the latent genetic, shared and non-shared environmental factors based on the best fitting Cannabis Use to Peer Group Deviance causal model. Also included are the standardized causal pathways (β2, in Figure 1) at each time period.
| Peer Group Deviance | Cannabis Use | ||||||
|---|---|---|---|---|---|---|---|
| a1 | c1 | E1 | ← β 2— | a2 | c2 | e2 | |
| 15–17 years | 0.33 (−0.50 0.50) | 0.27 (−0.46 0.46) | 0.61 (0.56 0.67) | 0.67 (0.62 0.71) | 0.57 (0.27 0.78) | 0.67 (0.44 0.82) | 0.47 (0.40 0.55) |
| 18–21 years | 0.37 (−0.54 0.54) | 0.30 (−0.49 0.49) | 0.58 (0.53 0.64) | 0.66 (0.61 0.69) | 0.57 (0.25 0.78) | 0.63 (0.37 0.79) | 0.53 (0.47 0.60) |
| 22–25 years | 0.23 (−0.53 0.53) | 0.42 (−0.53 0.53) | 0.63 (0.57 0.68) | 0.61 (0.56 0.65) | 0.73 (0.47 0.87) | 0.40 (−0.65 0.65) | 0.55 (0.48 0.63) |
a1, c1 & c1 = path coefficients from latent genetic environmental latent effects which explain variance in PGD
a2, c2 & c2 = path coefficients from latent genetic environmental latent effects which explain variance in CU
Based on the path coefficients in Table 3, standardized variance components were estimated and appear in Table 4. The proportion of total variance in CU attributable to additive genetic effects was steady at 32% to 33% between 15 to 21 years but then increased to 54% between 22 to 25 years. By contrast, the proportion of shared environmental variance over the same period declined from 45% to 16%. Although the standardized additive genetic and shared environmental variance in PGD appeared stable over time, the proportion of additive genetic variance explained by CU increased; it ranged from 50% to 58% between 15 to 21 years and 78% between 22 to 25 years. This coincided with a decline in the proportion of shared environmental variance explained by CU which equaled 73% between 15 to 17 years and 25% by 22 to 25 years. Most of the non-shared environmental variance (74% to 79%) in PGD was unique and not attributable to CU.
Table 4.
Standardized proportions of additive genetic (A), shared (C) and non-shared (E) environmental variance in Cannabis Use (CU) and Peer Group Deviance (PGD) including the proportions of variance in PGD which can be explained by CU as well as variance which is unique to PGD.
| 15–17 years | 18–21 years | 22–25 years | ||||
|---|---|---|---|---|---|---|
| PGD | CU | PGD | CU | PGD | CU | |
| Total A variance (%)→ | 25 | 33 | 28 | 32 | 25 | 54 |
| A Explained by CU | 58% | - | 50% | - | 78% | - |
| A Unique to PGD | 42% | - | 50% | - | 22% | - |
| Total C variance (%) → | 27 | 45 | 26 | 39 | 24 | 16 |
| C Explained by CU | 73% | - | 66% | - | 25% | - |
| C Unique to PGD | 27% | - | 34% | - | 75% | - |
| Total E variance (%) → | 47 | 22 | 46 | 28 | 51 | 30 |
| E Explained by CU | 21% | - | 26% | - | 22% | - |
| E Unique to PGD | 79% | - | 74% | - | 78% | - |
Because secular trends in the use of cannabis are unlikely to be linear, we re-ran our models with linear and quadratic age adjustments on the PGD and CU thresholds. We found an identical pattern of results; the uni-directional PGD→CU model was rejected while the CU→PGD causal model provided the best fit to the data.
Discussion
This is the first study to examine the nature of the genetic and environmental association between the liability to affiliate with deviant peers and cannabis use. Our prediction was that part of the genetic risk for CU would be mediated through selection into deviant peer groups. Although there was a significant genetic contribution in the PGD-CU association we found no evidence that genetic or environmental risks in PGD increase or mediate the risk of cannabis use. Instead, our results support the hypothesis that the association arises because of a causal pathway from CU to PGD. Between the ages 15 and 25 years, CU explained between one half and three quarters of the genetic variance in PGD. Although declining over time, large proportions of the shared environmental variance in PGD were likewise attributable to CU. CU can therefore be understood as the “risk indicator” for the liability to affiliate with deviant peers because it appears to sit in the causal pathway between genetic and environmental risks on the one hand and the expression of PGD on the other.
The significant association between PGD and CU is consistent with the seminal research by Dishion (67) who found that although early problem behaviors, poor peer relations, and family management practices were correlated with drug use, these effects were non-significant when deviant peer affiliation was included. The question is whether socialization or social / self selection provides a better explanation for the observed PGD-CU association.
The dominant socialization model (68–70) is well supported in the literature and has been considered by some to be primarily responsible for the relationship between PGD and liability to drug use (7, 71–75). However, this hypothesis, captured by our PGD→CU causal and reciprocal interaction models is not well supported by our findings. Our results are also inconsistent with Wills' transactional model (70) which predicts that childhood temperament, family environment and peer effects all influence, and precede, the development of self-control, which in turn mediates the liability to drug initiation, regular use and abuse.
Our findings are instead more consistent with social or self selection processes which drive and underpin deviant peer affiliation (76). Snyder (6) has argued that individuals when operating in open environments offering elective relationships select and affiliate with others who are behaviourally similar. A number of mechanisms including temperament and maladaptive externalizing behaviours have been proposed to make individuals more likely to affiliate with deviant peers (5, 77, 78) and biometrical modeling has also shown that variation in the sorts of friends we choose and affiliate with can be partly explained by genes (79, 80) which is consistent with our data.
Social and self selection processes are closely related to genotype-environmental correlations in biology which describe non random distributions of environments among different genotypes. In other words, `at risk' genotypes get or create more than their fair share of `at risk' environments. If expanded to include at risk `phenotypes', then this concept is in line with the causal CU→PDG model in which genetic and environmental risks in CU also increase the risk of individuals seeking out and affiliating with similarly inclined peers.
We still do not know if the `A', `C' and `E' in CU are the causal components in the relationship with CU or whether they index more distal phenotypic or broader genetic risks which predispose individuals to drug use which in turn mediates the risk of affiliating with deviant peers. However, we do know that the `A' and `C' risks are shared in common with the genetic and shared environmental risk for using and abusing cocaine, hallucinogens, sedatives, stimulants and opiates (81, 82), and that a number of other risks which CU might index have also been shown to elevate the risk of affiliating with deviant and drug using peers (5, 76, 78, 83). Environmental variables such as family structure (84–86) and adverse family environments (78) are predictive of PGD but the extent to which these are correlated with `A', `C' and `E' in CU remains unclear. Combined, our findings support the interpretation that the environmental and genetic risks explaining average cannabis use also mediate the risk of affiliating with deviant peers. This conclusion has implications for intervention and harm reduction strategies. If PGD is a `downstream' consequence of CU, then earlier targeted interventions based on data which have clearly identified the `A', `C' and `E' risks in CU are required in order to reduce mean levels and variation in maladaptive forms of both CU and PGD.
Limitations
Our findings must be interpreted in the context of three potential limitations. First, our data were drawn from white Virginian males. Males have a higher prevalence of drug use (87–90) and although previous analyses using the same data suggest it is broadly representative of US males and does not differ from the general population in rates of psychopathologic conditions, including illicit substance use, abuse and dependence (33), our results cannot be extrapolated to females. Second, w did not model cannabis initiation. However because there is converging evidence showing how initiation, regular use and progression to abuse and dependence can be explained by common genetic and environmental processes (29, 45) we would expect to see a similar pattern of results using binary measures of initiation. Finally, the latent PGD factor scores were assumed to take a uni-dimensional factor structure. The first three eigen values at 15 to 17 years (6.95, 1.12, 0.86), 18 to 21 years (5.56, 1.35, 0.80), and 22 to 25 years (5.85, 1.31, 0.73) followed by a comparison of the 1 and 2 factor solutions, suggested that a 2 factor solution provides a marginally better fit to the data. These 2 factors were interpreted as `General Peer Group Deviance' and `Peer Alcohol and Cigarette Use' dimensions. Between 18 to 25 years, the General Peer Group Deviance factor also included illicit drug items. The Peer Alcohol and Cigarette Use factor included items for alcohol related problems at all ages, as well as peers' use of cannabis at 21 to 25 years. Modeling the two latent factor scores separately revealed an almost identical pattern of results. Regardless of whether the PGD construct was divided into peers' licit versus illicit drug use, or modeled as a predominately delinquency factor, the CU→PGD causal model still provided the best fit to the data.
Conclusion
Although our modeling was not exhaustive, we have demonstrated how genetic and environmental risks in CU and PGD are related. Until recently PGD was assumed to be an environmental (27) upstream risk factor for CU. The current data are not consistent with this hypothesis. Rather, the liability to affiliate with deviant peers was better explained by a combination of genetic and environmental factors for which CU could be understood as a causal “risk indicator”. This is consistent with a social or self selection process by which the genetic and environmental risks in CU largely underpin and drive the likelihood of affiliating with deviant peers.
Acknowledgments
The project was supported by grants from the US National Institutes of Health (DA-11287, MH/AA/DA-49492, DA-18673, MH-01458, and AA-00236). Funding was also received from US National Institute on Drug Abuse (1K99DA023549-01A2). We thank Indrani Ray for database assistance. We also thank Dr. Linda Corey for assistance with the ascertainment of twins from the Virginia Twin Registry, now part of the Mid-Atlantic Twin Registry (MATR), directed by Dr. Judy Silberg. The registry has received support from NIH, the Carman Trust, and the W.M. Keck, John Templeton, and Robert Wood Johnson Foundations.
References
- 1.Patterson GR, DeBaryshe BD, Ramsey E. A developmental perspective on antisocial behavior. American Psychologist. 1989;44:329–335. doi: 10.1037//0003-066x.44.2.329. [DOI] [PubMed] [Google Scholar]
- 2.Petraitis J, Flay BR, Miller TQ, Torpy EJ, Greiner B. Illicit substance use among adolescents: a matrix of prospective predictors. Substance Use and Misuse. 1998;33:2561–2604. doi: 10.3109/10826089809059341. [DOI] [PubMed] [Google Scholar]
- 3.Dishion TJ, Bullock BM, Granic I. Pragmatism in modeling peer influence: dynamics, outcomes, and change processes. Development and Psychopathology. 2002;14:969–981. doi: 10.1017/s0954579402004169. [DOI] [PubMed] [Google Scholar]
- 4.Patterson GR, Dishion TJ, Yoerger K. Adolescent growth in new forms of problem behavior: macro- and micro-peer dynamics. Prevention Science. 2000;1:3–13. doi: 10.1023/a:1010019915400. [DOI] [PubMed] [Google Scholar]
- 5.Granic I, Patterson GR. Toward a comprehensive model of antisocial development: a dynamic systems approach. Psychological Review. 2006;113:101–131. doi: 10.1037/0033-295X.113.1.101. [DOI] [PubMed] [Google Scholar]
- 6.Snyder J, Reid JB, Patterson GR, Snyder J. Antisocial Behavior in Children and Adolescents: A Developmental Analysis and Model for Intervention. American Psychological Association; Washington DC: 2002. Reinforcement and Coercion Mechanisms in the Development of Antisocial Behavior: Peer Relationships; p. 101. [Google Scholar]
- 7.Kandel DB, Kessler RC, Margulies RS. Antecedents of adolescent intitiation into stages of drug use: a developmental analysis. Journal of Youth and Adolescence. 1978;7:13. doi: 10.1007/BF01538684. [DOI] [PubMed] [Google Scholar]
- 8.van den Bree MB, Pickworth WB. Risk factors predicting changes in marijuana involvement in teenagers. Archives of General Psychiatry. 2005;62:311–319. doi: 10.1001/archpsyc.62.3.311. [DOI] [PubMed] [Google Scholar]
- 9.Ary DV, Duncan TE, Duncan SC, Hops H. Adolescent problem behavior: the influence of parents and peers. Behaviour Research and Therapy. 1999;37:217–230. doi: 10.1016/s0005-7967(98)00133-8. [DOI] [PubMed] [Google Scholar]
- 10.Biglan A, Duncan TE, Ary DV, Smolkowski K. Peer and parental influences on adolescent tobacco use. Journal of Behavioral Medicine. 1995;18:315–330. doi: 10.1007/BF01857657. [DOI] [PubMed] [Google Scholar]
- 11.Allen M, Donohue WA, Griffin A, Ryan D, Turner MM. Comparing the Influence of Parents and Peers on the Choice to Use Drugs. Criminal Justice and Behavior. 2003;30:163–186. [Google Scholar]
- 12.Feske U, Tarter RE, Kirisci L, Gao Z, Reynolds M, Vanyukov M. Peer environment mediates parental history and individual risk in the etiology of cannabis use disorder in boys: a 10-year prospective study. American Journal of Drug and Alcohol Abuse. 2008;34:307–320. doi: 10.1080/00952990802013631. [DOI] [PubMed] [Google Scholar]
- 13.Guxens M, Nebot M, Ariza C, Ochoa D. Factors associated with the onset of cannabis use: a systematic review of cohort studies. Gaceta Sanitaria. 2007;21:252–260. doi: 10.1157/13106811. [DOI] [PubMed] [Google Scholar]
- 14.Farrell AD, Danish SJ. Peer drug associations and emotional restraint: causes or consequences of adolescents' drug use? Journal of Consulting and Clinical Psychology. 1993;61:327–334. doi: 10.1037//0022-006x.61.2.327. [DOI] [PubMed] [Google Scholar]
- 15.Mezzich AC, Tarter RE, Feske U, Kirisci L, McNamee RL, Day BS. Assessment of risk for substance use disorder consequent to consumption of illegal drugs: psychometric validation of the neurobehavior disinhibition trait. Psychology of Addictive Behaviors. 2007;21:508–515. doi: 10.1037/0893-164X.21.4.508. [DOI] [PubMed] [Google Scholar]
- 16.Tarter RE, Kirisci L, Kirillova GP, Gavaler J, Giancola P, Vanyukov MM. Social dominance mediates the association of testosterone and neurobehavioral disinhibition with risk for substance use disorder. Psychology of Addictive Behaviors. 2007;21:462–8. doi: 10.1037/0893-164X.21.4.462. [DOI] [PubMed] [Google Scholar]
- 17.Tarter RE, Kirisci L, Habeych M, Reynolds M, Vanyukov M. Neurobehavior disinhibition in childhood predisposes boys to substance use disorder by young adulthood: direct and mediated etiologic pathways. Drug and Alcohol Dependence. 2004;73:121–132. doi: 10.1016/j.drugalcdep.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 18.Bullock BM, Deater-Deckard K, Leve LD. Deviant peer affiliation and problem behavior: a test of genetic and environmental influences. Journal of Abnormal Child Psychology. 2006;34:29–41. doi: 10.1007/s10802-005-9004-9. [DOI] [PubMed] [Google Scholar]
- 19.Daniels D, Dunn J, Furstenberg FF, Jr., Plomin R. Environmental differences within the family and adjustment differences within pairs of adolescent siblings. Child Development. 1985;56:764–774. [PubMed] [Google Scholar]
- 20.Baker LA, Daniels D. Nonshared environmental influences and personality differences in adult twins. Journal of Personality and Social Psychology. 1990;58:103–110. doi: 10.1037//0022-3514.58.1.103. [DOI] [PubMed] [Google Scholar]
- 21.Pike A, Manke B, Reiss D, Plomin R. A genetic analysis of differential experiences of adolescent siblings across three years. Social Development. 2000;9:96–114. [Google Scholar]
- 22.Manke B, McGuire S, Reiss D, Hetherington EM, Plomin R. Genetic contributions to Adolescents extrafamilial social interactions - Teachers, best friends, and peers. Social Development. 1995;4:238–256. [Google Scholar]
- 23.Iervolino AC, Pike A, Manke B, Reiss D, Hetherington EM, Plomin R. Genetic and environmental influences in adolescent peer socialization: evidence from two genetically sensitive designs. Child Development. 2002;73:162–174. doi: 10.1111/1467-8624.00398. [DOI] [PubMed] [Google Scholar]
- 24.Walden B, McGue M, Iacono WG, Burt SA, Elkins I. Identifying shared environment contributions to early susbstance use: the importance of peers versus parents. Journal of Abnormal Psychology. 2004;113:440–450. doi: 10.1037/0021-843X.113.3.440. [DOI] [PubMed] [Google Scholar]
- 25.Cleveland HH, Wiebe RP, Rowe DC. Sources of exposure to smoking and drinking friends among adolescents. Journal of Genetic Psychology. 2005;166:153–169. [PubMed] [Google Scholar]
- 26.Rose RJ, Pulkkinen L, Avshalom C. Paths to successful development: Personality in the life course. Cambridge University Press; Cambridge, UK: 2002. How do adolescents select their friends? A behavior-genetic perspective. [Google Scholar]
- 27.Kendler KS, Gardner CO, Jr, Gillespie NA, Jacobson K, Aggen SH, Prescott CA. Creating a social world: A developmental twin study of peer group deviance. Annals of General Psychiatry. 2007;64:958–965. doi: 10.1001/archpsyc.64.8.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agrawal A, Neale MC, Prescott CA, Kendler KS. A twin study of early cannabis use and subsequent use and abuse/dependence of other illicit drugs. Psychological Medicine. 2004;34:1227–1237. doi: 10.1017/s0033291704002545. [DOI] [PubMed] [Google Scholar]
- 29.Agrawal A, Neale MC, Jacobson KC, Prescott CA, Kendler KS. Illicit drug use and abuse/dependence: modeling of two-stage variables using the CCC approach. Addictive Behaviors. 2005;30:1043–1048. doi: 10.1016/j.addbeh.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 30.Heath AC, Martin NG. Teenage alcohol use in the Australian twin register: genetic and social determinants of starting to drink. Alcoholism, Clinical and Experimental Research. 1988;12:735–741. doi: 10.1111/j.1530-0277.1988.tb01337.x. [DOI] [PubMed] [Google Scholar]
- 31.Kendler KS, Karkowski LM, Corey LA, Prescott CA, Neale MC. Genetic and environmental risk factors in the aetiology of illicit drug initiation and subsequent misuse in women. British Journal of Psychiatry. 1999;175:351–356. doi: 10.1192/bjp.175.4.351. [DOI] [PubMed] [Google Scholar]
- 32.Kendler KS, Neale MC, Sullivan P, Corey LA, Gardner CO, Prescott CA. A population-based twin study in women of smoking initiation and nicotine dependence. Psychological Medicine. 1999;29:299–308. doi: 10.1017/s0033291798008022. [DOI] [PubMed] [Google Scholar]
- 33.Kendler KS, Karkowski LM, Neale MC, Prescott CA. Illicit psychoactive substance use, heavy use, abuse, and dependence in a US population-based sample of male twins. Archives of General Psychiatry. 2000;57:261–269. doi: 10.1001/archpsyc.57.3.261. [DOI] [PubMed] [Google Scholar]
- 34.Kendler KS, Bulik CM, Silberg J, Hettema JM, Myers J, Prescott CA. Childhood sexual abuse and adult psychiatric and substance use disorders in women: an epidemiological and cotwin control analysis. Archives of General Psychiatry. 2000;57:953–959. doi: 10.1001/archpsyc.57.10.953. [DOI] [PubMed] [Google Scholar]
- 35.Lynskey MT, Heath AC, Bucholz KK, Slutske WS, Madden PA, Nelson EC, et al. Escalation of drug use in early-onset cannabis users vs co-twin controls. Journal of the American Medical Association. 2003;289:427–433. doi: 10.1001/jama.289.4.427. [DOI] [PubMed] [Google Scholar]
- 36.McGue M, Elkins I, Iacono WG. Genetic and environmental influences on adolescent substance use and abuse. American Journal of Medical Genetics. 2000;96:671–677. doi: 10.1002/1096-8628(20001009)96:5<671::aid-ajmg14>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 37.Pickens RW, Svikis DS, McGue M, LaBuda MC. Common genetic mechanisms in alcohol, drug, and mental disorder comorbidity. Drug and Alcohol Dependence. 1995;39:129–138. doi: 10.1016/0376-8716(95)01151-n. [DOI] [PubMed] [Google Scholar]
- 38.Prescott CA, Hewitt JK, Heath AC, Truett KR, Neale MC, Eaves LJ. Environmental and genetic influences on alcohol use in a volunteer sample of older twins. Journal of Studies on Alcohol. 1994;55:18–33. doi: 10.15288/jsa.1994.55.18. [DOI] [PubMed] [Google Scholar]
- 39.Rhee SH, Hewitt JK, Young SE, Corley RP, Crowley TJ, Stallings MC. Genetic and environmental influences on substance initiation, use, and problem use in adolescents. Archives of General Psychiatry. 2003;60:1256–1264. doi: 10.1001/archpsyc.60.12.1256. [DOI] [PubMed] [Google Scholar]
- 40.Sullivan PF, Kendler KS. The genetic epidemiology of smoking. Nicotine and Tobacco Research. 1999;1(Suppl 2):S51–7. doi: 10.1080/14622299050011811. Discussion S69-70. [DOI] [PubMed] [Google Scholar]
- 41.Tsuang MT, Lyons MJ, Eisen SA, Goldberg J, True W, Lin N, et al. Genetic influences on DSM-III-R drug abuse and dependence: a study of 3,372 twin pairs. American Journal of Medical Genetics. 1996;67:473–477. doi: 10.1002/(SICI)1096-8628(19960920)67:5<473::AID-AJMG6>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 42.Tsuang MT, Lyons MJ, Harley RM, Xian H, Eisen S, Goldberg J, et al. Genetic and environmental influences on transitions in drug use. Behavior Genetics. 1999;29:473–479. doi: 10.1023/a:1021635223370. [DOI] [PubMed] [Google Scholar]
- 43.van den Bree MB, Johnson EO, Neale MC, Pickens RW. Genetic and environmental influences on drug use and abuse/dependence in male and female twins. Drug and Alcohol Dependence. 1998;52:231–241. doi: 10.1016/s0376-8716(98)00101-x. [DOI] [PubMed] [Google Scholar]
- 44.Neale MC, Harvey E, Maes HH, Sullivan PF, Kendler KS. Extensions to the modeling of initiation and progression: applications to substance use and abuse. Behavior Genetics. 2006;36:507–524. doi: 10.1007/s10519-006-9063-x. [DOI] [PubMed] [Google Scholar]
- 45.Gillespie NA, Neale MC, Kendler KS. (submitted) Pathways to cannabis abuse: A multi-stage model from cannabis availability, cannabis initiation, and progression to abuse. Addiction. doi: 10.1111/j.1360-0443.2008.02456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eaves L, Last KA, Martin NG, Jinks JL. A progressive approach to non-additivity and genotype-environmental covariance in the analysis of human differences. British Journal of Mathematics and Statistical Psychology. 1977;30:1–42. [Google Scholar]
- 47.Fergusson DM. Annotation: structural equation models in developmental research. Journal of Child Psychology and Psychiatry. 1997;38:877. doi: 10.1111/j.1469-7610.1997.tb01607.x. [DOI] [PubMed] [Google Scholar]
- 48.Feinberg M, Carroll BJ. Separation of subtypes of depression using discriminant analysis. I. Separation of unipolar endogenous depression from non-endogenous depression. British Journal of Psychiatry. 1982;140:384. doi: 10.1192/bjp.140.4.384. [DOI] [PubMed] [Google Scholar]
- 49.Farrell AD. Structural equation modeling with longitudinal data: strategies for examining group differences and reciprocal relationships. Journal of Consulting and Clinical Psychology. 1994;62:477. doi: 10.1037//0022-006x.62.3.477. [DOI] [PubMed] [Google Scholar]
- 50.Klerman GL, Leon AC, Wickramaratne P, Warshaw MG, Mueller TI, Weissman MM, et al. The role of drug and alcohol abuse in recent increases in depression in the US. Psychological Medicine. 1996;26:343. doi: 10.1017/s0033291700034735. [DOI] [PubMed] [Google Scholar]
- 51.Farmer A, Mahmood A, Redman K, Harris T, Sadler S, McGuffin P. A sib-pair study of the Temperament and Character Inventory scales in major depression. Archives of General Psychiatry. 2003;60:490. doi: 10.1001/archpsyc.60.5.490. [DOI] [PubMed] [Google Scholar]
- 52.Johnston LD, Bachman JG, O'Malley PM. Monitoring the future: Questionnaire responses from the nation's high school seniors, 1981. Institute for Social Research; Ann Arbor, MI: 1982. p. 286. [Google Scholar]
- 53.Tarter RE, Hegedus AM. The drug use screening inventory: It's application in the evaluation and treatment of alcohol and drug abuse. Alcohol Health and Research World. 1991;15:65–75. [Google Scholar]
- 54.Neale MC, Cardon LR. Methodology for Genetic Studies of Twins and Families. Kluwer Academic Publishers; Dordrecht: 1992. [Google Scholar]
- 55.Neale MC. Mx: Statistical Modelling. 5ed. Department of Psychiatry; Richmond, VA 23298: 1999. Box 126 MCV. [Google Scholar]
- 56.Pickles A, Neale M, Simonoff E, Rutter M, Hewitt J, Meyer J, et al. A simple method for censored age-of-onset data subject to recall bias: mothers' reports of age of puberty in male twins. Behavior Genetics. 1994;24:457–468. doi: 10.1007/BF01076181. [DOI] [PubMed] [Google Scholar]
- 57.Freedman D, Thornton A, Camburn D, Alwin D, Young-demarco L. The life history calendar: a technique for collecting retrospective data. Sociological Methodology. 1988;18:37–68. [PubMed] [Google Scholar]
- 58.Belli RF. The structure of autobiographical memory and the event history calendar: potential improvements in the quality of retrospective reports in surveys. Memory. 1998;6:383–406. doi: 10.1080/741942610. [DOI] [PubMed] [Google Scholar]
- 59.Tallis GM. The maximum likelihood estimation of correlation from contingency tables. Biometrics. 1962;18:342–353. [Google Scholar]
- 60.Jöreskog K, Sörbom D. New features in PRELIS 2. Scientific Software International; Chicago: 1993. [Google Scholar]
- 61.Heath AC, Cloninger CR, Martin NG. Testing a model for the genetic structure of personality: a comparison of the personality systems of Cloninger and Eysenck. Journal of Personality and Social Psychology. 1994;66:762–775. doi: 10.1037//0022-3514.66.4.762. [DOI] [PubMed] [Google Scholar]
- 62.Heath AC, Kessler RC, Neale MC, Hewitt JK, Eaves LJ, Kendler KS. Testing hypotheses about direction of causation using cross-sectional family data. Behavior Genetics. 1993;23:29–50. doi: 10.1007/BF01067552. [DOI] [PubMed] [Google Scholar]
- 63.Neale MC, Duffy DL, Martin NG. Direction of causation: reply to commentaries. Genetic Epidemiology. 1994;11:463–472. [Google Scholar]
- 64.Gillespie NG, Zhu G, Neale MC, Heath AC, Martin NG. Direction of causation modeling between measures of distress and parental bonding. Behavior Genetics. 2003;33:383–396. doi: 10.1023/a:1025365325016. [DOI] [PubMed] [Google Scholar]
- 65.Sullivan PF, Eaves LJ. Evaluation of analyses of univariate discrete twin data. Behavior Genetics. 2002;32:221–227. doi: 10.1023/a:1016025229858. [DOI] [PubMed] [Google Scholar]
- 66.Akaike H. Factor analysis and AIC. Psychometrika. 1987;52:317–332. [Google Scholar]
- 67.Dishion TJ, Loeber R. Adolescent marijuana and alcohol use: the role of parents and peers revisited. American Journal of Drug and Alcohol Abuse. 1985;11:11–25. doi: 10.3109/00952998509016846. [DOI] [PubMed] [Google Scholar]
- 68.Patterson GR. Coercive Family Process. Castalia; Eugene, OR: 1982. [Google Scholar]
- 69.Patterson GR. Family process: loops, levels, and linkages. In: Bolger N, Caspi A, editors. Persons in Context: Developmental Processes. Cambridge University Press; New York: 1988. [Google Scholar]
- 70.Wills TA, Dishion TJ. Temperament and adolescent substance use: a transactional analysis of emerging self-control. Journal of Clinical Child and Adolescent Psychiatry. 2004;33:69–81. doi: 10.1207/S15374424JCCP3301_7. [DOI] [PubMed] [Google Scholar]
- 71.Hawkins RO., Jr. Adolescent alcohol abuse: a review. Journal of Developmental and Behavioral Pediatrics. 1982;3:83–87. doi: 10.1097/00004703-198206000-00007. [DOI] [PubMed] [Google Scholar]
- 72.Thornberry TP, Krohn MD, Lizotte AJ, Chard-Wierschem D. The Role of Juvenile Gangs in Facilitating Delinquent Behavior. Journal of Research in Crime and Delinquency. 1993;30:55–87. [Google Scholar]
- 73.Wills TA, Cleary SD. Peer and adolescent substance use among 6th-9th graders: latent growth analyses of influence versus selection mechanisms. Health Psychology. 1999;18:453. doi: 10.1037//0278-6133.18.5.453. [DOI] [PubMed] [Google Scholar]
- 74.Gatti U, Tremblay RE, Vitaro F, McDuff P. Youth gangs, delinquency and drug use: a test of the selection, facilitation, and enhancement hypotheses. Journal of Child Psychology and Psychiatry. 2005;46:1178. doi: 10.1111/j.1469-7610.2005.00423.x. [DOI] [PubMed] [Google Scholar]
- 75.Steinberg L, Fletcher A, Darling N. Parental monitoring and peer influences on adolescent substance use. Pediatrics. 1994;93:1060–1064. [PubMed] [Google Scholar]
- 76.Kandel DB. The parental and peer contexts of adolescent deviance: an algebra of interpersonal influences. Journal of Drug Issues. 1996;26:289–315. [Google Scholar]
- 77.Gordon RA, Lahey BB, Kawai E, Loeber R, Stouthamer-Loeber M, Farrington DP. Antisocial behavior and youth gang membership: Selection and socialization. Criminology. 2004;42:55. [Google Scholar]
- 78.Lacourse E, Nagin DS, Vitaro F, Cote S, Arseneault L, Tremblay RE. Prediction of early-onset deviant peer group affiliation: a 12-year longitudinal study. Archives of General Psychiatry. 2006;63:562–568. doi: 10.1001/archpsyc.63.5.562. [DOI] [PubMed] [Google Scholar]
- 79.Rose R, Pulkkinen L, Caspi A. Paths to Successful Development: Personality in the Life Course. Cambridge University Press; 2001. How do adolescents select their friends? A behavior-genetic perspective; p. 106. [Google Scholar]
- 80.Rushton JP, Bons TA. Mate choice and friendship in twins: evidence for genetic similarity. Psychol Science. 2005;16:555. doi: 10.1111/j.0956-7976.2005.01574.x. [DOI] [PubMed] [Google Scholar]
- 81.Kendler KS, Myers J, Prescott CA. The specificity of genetic and environmental risk factors for symptoms of cannabis, cocaine, alcohol, caffeine and nicotine dependence. Annals of General Psychiatry. 2007;64:1313–1320. doi: 10.1001/archpsyc.64.11.1313. [DOI] [PubMed] [Google Scholar]
- 82.Kendler KS, Jacobson KC, Prescott CA, Neale MC. Specificity of genetic and environmental risk factors for use and abuse/dependence of cannabis, cocaine, hallucinogens, sedatives, stimulants, and opiates in male twins. American Journal of Psychiatry. 2003;160:687–495. doi: 10.1176/appi.ajp.160.4.687. [DOI] [PubMed] [Google Scholar]
- 83.Kandel DB. On processes of peer influences in adolescent drug use: a developmental perspective. Advances in Alcohol and Substance Abuse. 1985;4:139–163. doi: 10.1300/J251v04n03_07. [DOI] [PubMed] [Google Scholar]
- 84.Needle RH, Su SS, Doherty W. Divorce, remarriage, and adolescent substance use: a prospective longitudinal study. Journal of Marriage and the Family. 1990;52:157–169. [Google Scholar]
- 85.Turner RA, Irwin CE, Millstein SG. Family structure, family processes, and experimenting with substances during adolescence. Journal of Research on Adolescence. 1991;1:93–106. [Google Scholar]
- 86.Short JL. Predictors of substance use and mental health of children of divorce: A prospective analysis. Journal of Divorce and Remarriage. 1998;29:147–166. [Google Scholar]
- 87.Merikangas KR, Stolar M, Stevens DE, Goulet J, Preisig MA, Fenton B, et al. Familial transmission of substance use disorders. Arch Gen Psychiatry. 1998;55:973–9. doi: 10.1001/archpsyc.55.11.973. [DOI] [PubMed] [Google Scholar]
- 88.Bierut LJ, Dinwiddie SH, Begleiter H, Crowe RR, Hesselbrock V, Nurnberger JI, Jr., et al. Familial transmission of substance dependence: alcohol, marijuana, cocaine, and habitual smoking: a report from the Collaborative Study on the Genetics of Alcoholism. Archives of General Psychiatry. 1998;55:982–8. doi: 10.1001/archpsyc.55.11.982. [DOI] [PubMed] [Google Scholar]
- 89.Kendler KS, Neale MC, Thornton LM, Aggen SH, Gilman SE, Kessler RC. Cannabis use in the last year in a US national sample of twin and sibling pairs. Psychological Medicine. 2002;32:551–554. doi: 10.1017/s0033291701004950. [DOI] [PubMed] [Google Scholar]
- 90.Kendler KS, Prescott CA. Cannabis use, abuse, and dependence in a population-based sample of female twins. American Journal of Psychiatry. 1998;155:1016–1022. doi: 10.1176/ajp.155.8.1016. [DOI] [PubMed] [Google Scholar]
- 91.Wright S. The method of path coefficients. Annals of Mathematical Statistics. 1934;5 [Google Scholar]