

Summary
Tetherin is an interferon-induced protein whose expression blocks the release of HIV-1 and other enveloped viral particles. The underlying mechanism by which tetherin functions, and whether it directly or indirectly causes virion retention are unknown. Here, we elucidate the mechanism by which tetherin exerts its antiviral activity. We demonstrate, through mutational analyses and domain replacement experiments, that tetherin configuration rather than primary sequence is critical for antiviral activity. These findings allowed the design of a completely artificial protein, lacking sequence homology with native tetherin, that nevertheless mimicked its antiviral activity. We further show that tetherin is incorporated into HIV-1 particles as a parallel homodimer using either of its two membrane anchors. These results indicate that tetherin functions autonomously and directly, and that infiltration of virion envelopes by one or both of tetherin's membrane anchors is necessary, and likely sufficient, to tether enveloped virus particles that bud through plasma membrane.
Introduction
Eukaryotic cells have evolved various mechanisms of defense against viral infections. Indeed, many components of the type I interferon (IFN)-induced innate immune system are thought to directly inhibit the replication of various viruses. However, while >900 genes are known to be induced by type-1 IFN (http://www.lerner.ccf.org/labs/williams/isgd.cgi), the mechanisms by which IFN-induced gene products inhibit virus replication are known for only a few (Samuel, 2001). Recently, we and others identified an IFN-induced protein, termed tetherin (also referred to as BST-2 or CD17), whose expression can cause inhibition of HIV-1 particle release from infected cells (Neil et al., 2008; Van Damme et al., 2008). Notably, HIV-1 encodes an accessory protein, Vpu, that counteracts tetherin activity. It is not known how tetherin expression causes the retention of viral particles, but the retained viral particles are mature and have lipid bilayers that are discontinuous with host cell membranes (Neil et al., 2006; Neil et al., 2007). While particles accumulate at both in endosomes and at the plasma membrane, endosomal accumulation is the result of internalization of virions that were initially trapped at the cell surface and is not required for their retention. Indeed, the virions can be released from cell surfaces by protease treatment, indicating that tetherin either forms or induces protein based tethers that cause virion entrapment (Neil et al., 2006; Neil et al., 2008). This mechanism of virion retention is distinct from that found in ‘late-domain’ mutants, where viral proteins fail to recruit the ESCRT machinery and the virion lipid envelope remains continuous with the host cell plasma membrane (Morita and Sundquist, 2004).
Tetherin is a type II single-pass transmembrane protein with a highly unusual arrangement, encoding both a transmembrane anchor towards its N-terminus, and a putative glycophosphatidylinositol (GPI) lipid anchor at its C-terminus (Kupzig et al., 2003). These membrane anchors are linked by an extracellular domain that is predicted to form a coiled-coil. What role each domain plays, and how this topology impacts the biological activity of tetherin is unknown. In principle, the tetherin protein itself could be necessary and sufficient to assemble into protease sensitive tethers that retain virions on the surface of infected cells. Alternatively, it is possible that tetherin is a component of the protease sensitive tethers, but requires specific virion or cellular interacting components to induce tethering. A third possibility is that tetherin is not directly involved in tethering at all, and instead is a signaling molecule, sensor or inducer, that triggers the expression of some other set of factors that are responsible for tethering. In this regard, a recent report identified a second protein, CAML, whose expression has the same consequence as tetherin expression, namely retention of HIV-1 particles (Varthakavi et al., 2008). It is difficult to reconcile these findings, unless one protein induces the expression of another that is actually responsible for tethering.
Tetherin expression induces retention of virus-like particles (VLPs) assembled using the structural proteins of a range of enveloped viruses (Jouvenet et al., 2009; Kaletsky et al., 2009; Sakuma et al., 2009). Moreover, tetherin colocalizes with retroviral Gag puncta at the plasma membrane (Jouvenet et al., 2009; Neil et al., 2008). This is consistent with the notion that it may be directly involved in tethering nascent virions. However, there is currently no direct evidence that tetherin is directly responsible for virion tethering.
Here, we show that tetherin is indeed directly responsible, and likely sufficient, for tethering of nascent enveloped virions to infected cell surfaces. First, we delineate features of tetherin that are required for antiviral activity and show that while the overall configuration of tetherin is important, there is little specific sequence requirement for antiviral activity. These findings allowed the design of a completely artificial tetherin protein, assembled from fragments of heterologous proteins. Despite lacking significant sequence homology to natural tetherins, this artificial tetherin causes tethering of two widely divergent enveloped viruses (HIV-1 and Ebola virus). We also show that either the transmembrane domain or the GPI modified C-terminus can drive insertion of tetherin into the lipid envelope of HIV-1 particles as a parallel homodimer. Together, these results strongly suggest the dual membrane anchors, configured as they are in tetherin proteins, are directly responsible, and likely sufficient, to cause retention of enveloped virions that bud through the plasma membrane.
Results
A putative topology of tetherin is depicted schematically in Figure 1A. To determine the accuracy of this configuration, and its role in antiviral activity, point mutations and deletions were introduced at various positions. These mutations targeted residues that are putatively involved in disulfide bond formation or glycosylation. Alternatively, major features of tetherin were deleted or replaced with heterologous protein domains.
Figure 1. Schematic representation of tetherin and analysis of post-translational modifications.
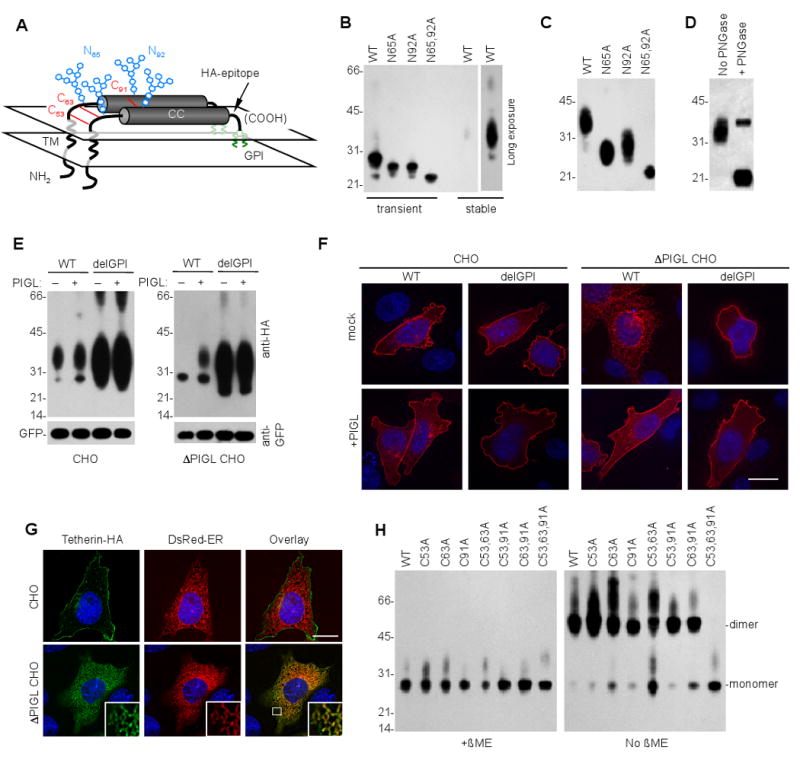
(A) Schematic representation of the tetherin dimer, indicating the transmembrane (TM) and coiled coil (CC) domains as well as glycophosphatidylinositol (GPI, green), glycosylation (blue) and disulfide bond (red) modifications. A tetherin derivative that incorporated an extracellular HA tag was used throughout, unless otherwise indicated.
(B) Western blot analysis (anti-HA) of 293T cells transiently transfected with 200ng of plasmids expressing WT tetherin and variants bearing mutations at putative glycosylation sites. Additionally, lysates of 293T cells stably expressing WT-tetherin were run on the same gel. A longer exposure of this lane is also shown.
(C) Western blot analysis (anti-HA) of 293T cells stably transduced with LHCX-based retroviral vectors expressing WT tetherin and tetherin variants bearing mutations at putative glycosylation sites.
(D) Western blot analysis (anti-HA) of tetherin following deglycosylation. Lysates of 293T ells stably expressing HA tagged tetherin were incubated in the presence or absence of peptide-N-glycosidase-F (PNGase), as described in the supplemental information, prior to analysis.
(E) Western blot analysis (anti-HA) of parental (CHO) and PIGL-defective (ΔPIGL CHO) cells transfected with plasmids (200ng) expressing WT or delGPI tetherin in the absence (-) or presence (+) of 100ng of a plasmid expressing rat PIGL. A GFP expression plasmid (100ng) was cotransfected and blots were probed with anti-GFP (lower panel) verify equal transfection efficiency and gel loading.
(F) Immunofluorescence analysis (anti-HA, red) of parental (CHO) and PIGL-defective (ΔPIGL CHO) cells transfected with plasmids expressing WT or delGPI tetherin in the absence (mock) or presence (+PIGL) of a plasmid expressing rat PIGL. Cells were transfected as in (E) and nuclei were stained with DAPI (blue) Scale bar =10μm.
(G) Immunofluorescence analysis (anti-HA, green) of parental (CHO) and PIGL-defective (ΔPIGL CHO) cells transfected with plasmids expressing WT tetherin and an ER-localized DsRed2 fluorescent protein. Cells were transfected as in (E) and nuclei were stained with DAPI (blue). Scale bar =10μm. The inset shows an expanded portion of the image indicated by the white square in the lower right image. For (F) and (G), at least 50 cells were inspected and all exhibited similar patterns of tetherin localization.
(H) Western blot analysis (anti-HA) of 293T cells transiently transfected with 200ng of plasmids expressing WT tetherin or tetherin variants bearing mutations at the indicated cysteine residues. Samples were untreated or treated with β-mercaptoethanol (β-ME) prior to analysis. Numbers to the left of panels (B), (C), (D), (E) and (H) represent the positions and sizes (in kDa) of molecular weight markers.
Role of tetherin configuration and post translational modifications in antiviral activity
Tetherin migrates as a number of species upon SDS PAGE analysis, presumably due to heterogeneity in post-translational modifications (Goffinet et al., 2009; Kaletsky et al., 2009; McNatt et al., 2009; Miyagi et al., 2009; Neil et al., 2007). When transiently overexpressed in 293T cells, human tetherin that is untagged, or bears an HA epitope insertion at residue 154 in the extracellular domain (Figure 1A), migrates primarily as a ∼28kD species, sometimes accompanied by a higher molecular weight smear of ∼32-38 kDa (Figure 1B). Conversely, when stably expressed at lower levels, in the identical cell type, only the ∼32-38 kDa smear is observed (Figure 1B, C). This suggests that the addition of complex carbohydrates to tetherin occurs more slowly than translation under conditions of transient expression in 293T cells. Individual mutation of each of the two putative N-linked glycosylation sites (N65A and N92A) reduced the molecular weight of ∼28kD transiently expressed and the ∼32-38 kDa stably expressed tetherin by a few kDa, while mutation at both positions reduced the apparent molecular weight of both transiently and stably expressed tetherin to ∼21kDa (Figure 1B,C). Treatment of the stably expressed ∼32-38 kDa tetherin protein with peptide-N-glycosidase-F reduced its molecular weight to a similar degree as did mutation of both glycosylation sites (Figure. 1D).
A second post-translational modification of tetherin that is thought to occur is the addition of a GPI anchor at its C-terminus (Kupzig et al., 2003). To confirm that tetherin is indeed modified in this way, and to determine its effect on tetherin trafficking and maturation, we used a CHO-derived cell line lacking PIGL, an ER-resident enzyme required for the addition of GPI anchors. Proteins that harbor a GPI modification signal as their only membrane anchor are inserted into ER membrane but remain trapped there in the absence of PIGL (Nakamura et al., 1997). It was not known whether the same restriction would apply tetherin, which also encodes a transmembrane domain and cytoplasmic tail. Nonetheless, removal of the GPI modification signal (e.g. in tetherin delGPI) should allow escape from this block, if it occurs.
In CHO cells, both WT and delGPI tetherin forms were expressed at significantly lower levels than in transiently transfected 293T cells and, presumably as a consequence of this, appeared primarily in their fully glycosylated ∼32-38 kDa forms (Figure 1E). They localized primarily at the plasma membrane (Figure 1F). Conversely, in PIGL-defective CHO cells, WT tetherin was expressed in its incompletely glycosylated (28kD) form (Figure 1E), adopted a thread-like intracellular distribution, and was localized primarily in the ER (Figure 1F, G). Reconstitution of the PIGL defect, by transient expression of rat PIGL, allowed WT tetherin to achieve mature glycosylation and reach the plasma membrane (Figure 1E,F). Removal of the signal for GPI modification from tetherin (in delGPI) enhanced expression, but more importantly, abolished the requirement for PIGL in completing tetherin glycosylation and transport to the cell surface (Figure 1E,F).
Tetherin forms disulfide-linked dimers (Ohtomo et al., 1999), but which of the three available cysteines that are present in the tetherin extracellular domain participate in intra or intermolecular bond formation is not known. A triple cysteine mutant (C53,63,91A) abolished the ability of tetherin to form β-mercaptoethanol-sensitive dimers while mutations at individual cysteines or at any combination of two of the three cysteines did not (Figure 1H). Thus, all three cysteines form disulfide bonds with an orthologous cysteine in a tetherin dimer. Overall, the aforementioned experiments indicate that the tetherin molecule is configured, in schematic terms, approximately as depicted in Figure 1A.
To determine the importance of this configuration for the antiviral activity of tetherin, 293T cells were transfected with HIV-1(WT) or HIV-1(delVpu) proviral plasmids, along with varying amounts of plasmids expressing WT or one of a panel of mutant tetherin proteins (Figure 2A). Virion release was evaluated using infectious particle and western blot assays. As expected, the release of HIV-1(WT) was only slightly affected by human tetherin, while the release of HIV-1 (delVpu) was strongly inhibited (Figure 2B, C).
Figure 2. Tetherin features required for antiviral activity.
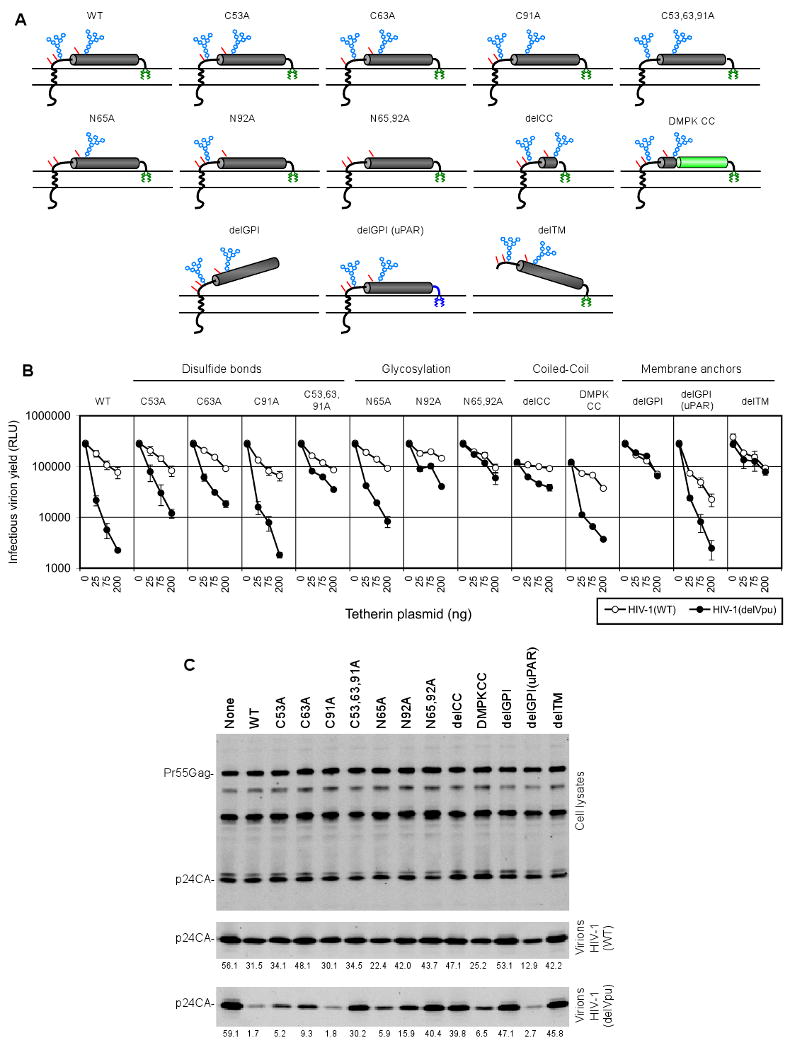
(A) Schematic representation of the tetherin mutants panel, indicating the positions of glycophosphatidylinositol (GPI, green), glycosylation (blue) and disulfide bond (red) modifications. The tetherin proteins bore point mutations at cysteine residues involved in disulfide bonding (C53, C63 and/or C91) or N-linked glycosylation sites (N65 and/or N92). Alternatively, the tetherin coiled-coil was deleted (delCC) or replaced with a coiled-coil from dystrophia myotonica protein kinase (DMPK CC). The membrane anchors were removed (delGPI and delTM) or replaced by the the C-terminal GPI modification signal from urokinase Plasminogen Activator Receptor (delGPI(uPAR)).
(B), (C) Effect of WT and mutant tetherin proteins on HIV-1 release. 293T cells were transfected with HIV-1(WT) or HIV-1(delVpu) proviral plasmids and increasing amount of plasmids expressing WT or mutant tetherin proteins (0, 25, 75 and 200ng for each). Infectious virion yield, measured using TZM-Bl indicator cells is given in relative light units (RLU). Error bars indicate the range of duplicate determinations and are representative of 2 to 5 experiments for each tetherin mutant. (C) Quantitative western blot analysis (LICOR, anti-p24) of 293T cells and corresponding virions after transfection with HIV-1(WT) or HIV-1(delVpu) proviral plasmids and 50ng of the WT and mutant tetherin plasmids. Numbers below each lane indicate the amounts of p24 in virion pellets, in arbitrary units and are representative of three experiments.
In general, tetherin mutations affecting disulfide bonding or glycosylation had variable effects on antiviral activity. Disruption of individual intermolecular disulfide bonds by mutation of cysteine residues (C53A, C63A, or C91A) was quite well tolerated. The C53A and C63A mutations reduced but did not eliminate antiviral activity while the C91A mutation had no effect (Figure 2B, C). Mutation of all three cysteines (C53,63,91A), dramatically reduced tetherin activity, without inhibiting cell surface expression (Figure S1), suggesting that disulfide bond mediated maintenance or stabilization of tetherin conformation was important for activity. Nonetheless, no individual disulfide bond was essential and even mutation of all three cysteine residues resulted in a tetherin protein that retained weak activity (Figure 2B).
Analysis of tetherin glycosylation site mutants revealed that the N65A mutation had rather small effects on tetherin activity while the N92A mutation markedly impaired activity (Figure 2B,C). The N65,92A double mutant was almost completely inactive. However, this mutant was less well expressed at the cell surface than WT and other mutant tetherins (Figure S1), and a secreted form of tetherin (delTM/delGPI), lacking both membrane anchors was dependent on glycosylation for efficient secretion (Figure S2). These results suggested that glycosylation was required for the correct transport, and perhaps folding of the tetherin molecule, rather than playing a specific role in tethering activity, although the latter possibility could not be excluded.
A prominent feature of tetherin protein is a coiled-coil that incorporates the majority of the extracellular protein sequence. Deletion of the bulk of the coiled-coil domain (in delCC) strongly inhibited antiviral activity, although a low residual level of Vpu-reversible particle release inhibition was observed (Figure 2B,C). The delCC protein was also well expressed at the cell surface (Figure S1) and formed disulfide linked homodimers (data not shown), demonstrating that the coiled-coil is important for reasons other than dimer formation.
A mutant tetherin protein, named delTM, that lacked the N-terminal transmembrane domain and was retained in the cell membrane only by its GPI anchor (Figure 2A) did not block particle release (Figure 2B,C). Similarly, removal of the GPI anchor signal from tetherin (delGPI) also abolished antiviral activity. Despite their inactivity, these truncated proteins were well expressed at the cell surface (Figure S1). Thus, the major structural features of tetherin, namely N- and C- terminal membrane anchors positioned at either end of a coiled-coil domain were required for antiviral activity.
Notably, the reintroduction of a heterologous coiled-coil from a constitutively dimeric cytoplasmic protein, dystrophia myotonica protein kinase (DMPK), into the poorly active delCC tetherin protein (Figure 2A) substantially restored activity (Figure 2B,C). Similarly, appending the delGPI tetherin protein with a C-terminal sequence from a heterologous GPI-linked protein, urokinase plasminogen activator receptor (uPAR), fully rescued activity (Figure 2B,C). These reintroduced protein domains exhibited little, if any, sequence homology with tetherin. Thus, the presence of these structural features appeared more important for activity than did their specific primary sequence.
An artificial tetherin-like protein assembled from heterologous protein domains has virion tethering activity
Because critically important features of the tetherin molecule could be functionally replaced by heterologous protein domains, we attempted to construct a completely artificial tetherin-like protein (art-tetherin). This designed protein incorporated domains from proteins that were ‘tetherin-like’ based on size, topology and post-translational modifications but were otherwise unrelated and lacked sequence homology with tetherin (Figure 3A). The N-terminus of art-tetherin consisted of a fragment of the N-terminus of transferrin receptor (TfR) comprising the membrane proximal ∼20 aa residues of its cytoplasmic tail (residues 43-62), the transmembrane domain (residues 63-88) and part of the extracellular stalk (residues 89-121). Like tetherin, TfR is a dimeric type-II transmembrane protein, and the extracellular portion incorporated into art-tetherin, contains two cysteines that form intermolecular disulfide bonds. This TfR-derived N-terminus was linked to the ∼75 residue coiled-coil from the cytoplasmic dimeric protein DMPK. An HA epitope tag was inserted immediately C-terminal to the DMPK coiled-coil for detection purposes and the C-terminus of art-tetherin was from uPAR, including a signal for GPI modification (Figure 3A).
Figure 3. An artificial tetherin-like protein has potent antiviral activity.
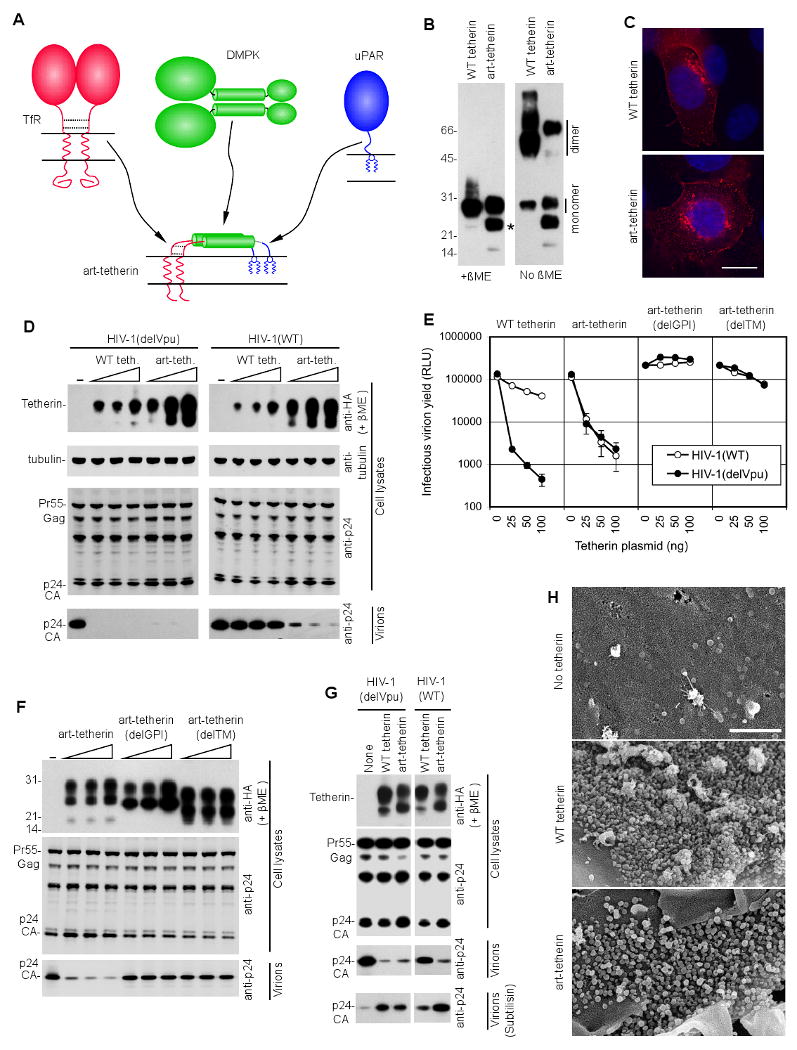
(A) Design of an artificial tetherin (art-tetherin) protein using domains of similar configuration from the transferrin receptor (TfR), dystrophia myotonica protein kinase (DMPK) and urokinase Plasminogen Activator Receptor (uPAR). An HA epitope tag was inserted between the DMPK and uPAR domains.
(B) Western blot analysis (anti-HA) of 293T cells transfected with plasmids expressing WT tetherin or art-tetherin and subjected to SDS PAGE in the absence or presence of β-mercaptoethanol. A presumed degradation product from art-tetherin is indicated by an asterisk.
(C) Immunofluorescence analysis (anti-HA, red) of HT1080 cells transfected with plasmids expressing WT tetherin or art-tetherin. Nuclei were stained with DAPI (blue) and the images are representative of at least 50 cells that were inspected for each expressed protein. Scale bar = 10μm
(D) Western blot analysis (anti-HA and anti-tubulin, upper panels and anti-p24, lower panels) of cell lysates and corresponding released virions following transfection of 293T cells with HIV-1(WT) or HIV-1(delVpu) proviral plasmids and increasing amounts (0ng 25ng, 50ng, 100ng) of plasmids expressing WT tetherin or art-tetherin.
(E) Infectious virion release, measured using TZM-Bl indicator cells, following transfection of 293T cells with HIV-1(WT) or HIV-1(delVpu) proviral plasmids and increasing amounts (0ng 25ng, 50ng, 100ng) of plasmids expressing WT tetherin, art-tetherin or art-tetherin mutants. Error bars indicate the range of duplicate determinations and are representative of 2 to 5 experiments for each protein.
(F) Western blot analysis (anti-HA upper panels and anti-p24, center and lower panels) of cell lysates and corresponding released virions following transfection of 293T cells with an HIV-1(delVpu) proviral plasmid and increasing amounts (0ng 25ng, 50ng, 100ng) of plasmids expressing or art-tetherin and mutant derivatives.
Numbers to the left of panels (B) and (F) represent the positions and sizes (in kDa) of molecular weight markers.
(G) Western blot analysis of 293T cells (anti-p24 and anti-HA) and virions (anti-p24) following transfection with HIV-1(WT) or HIV-1(delVpu) and plasmids expressing WT tetherin or art-tetherin. Virions that were constitutively released, or released following incubation of the cells with subtilisin (bottom panels), were pelleted through sucrose prior to analysis.
(H) Scanning electron micrograph of HT1080 cells transfected with plasmids expressing HIV-1 Gag in the presence or absence of either WT or art-tetherin. Scale bar indicates 1μm. At least ten particle expressing cells were evaluated in each of two independent experiments and representative images are shown.
The art-tetherin protein was well expressed in 293T cells and formed β-mercaptoethanol-sensitive dimers (Figure 3B). Immunofluorescence (Figure 3C) and FACS (Figure S3) analyses revealed that art-tetherin was expressed at the plasma membrane, as well as at intracellular locations, adopting a distribution that resembled that of native tetherin (Figure 3C).
Strikingly, art-tetherin potently inhibited the release of extracellular HIV-1 particles (Figure 3D,E). Inhibition occurred without effects on Gag protein expression (Figure 3D). As expected, art-tetherin was not antagonized by HIV-1 Vpu, unlike native tetherin. The mechanism by which art-tetherin inhibited particle release mimicked that of native tetherin. Specifically, mutant forms of art-tetherin that lacked either the N-terminal transmembrane domain, or the C-terminal GPI anchor were devoid of antiviral activity (Figure 3E, F), indicating that both membrane anchors were essential for art-tetherin function. Moreover, when particle release was inhibited by either art-tetherin or native tetherin, particles could be recovered from cell surfaces by subtilisin treatment (Figure 3G). Scanning EM analysis revealed that art-tetherin, like WT tetherin, induced the accumulation of HIV-1 particles on cell surfaces (Figure 3H). Art-tetherin also inhibited the release of VLPs assembled using the Ebola VP40 protein, although it appeared somewhat less potent than native tetherin (Figure S4). Overall, the fact that it proved rather straightforward to design an active artificial tetherin, that has no significant sequence homology to native tetherin, makes it unlikely that tetherin functions through indirect mechanisms, or requires specifically associated cofactors.
Tetherin incorporation into HIV-1 particles
If tetherin acts directly, and without cofactors, to block the release of virion particles then a possible mechanism of action could involve incorporation of one, or both, of the membrane anchors into virion membranes. Thereafter, the other membrane anchor, or one of a pair of molecules in a tetherin dimer, could remain embedded in the host cell membrane. This would inevitably result in the formation of a tether, composed of the tetherin protein itself.
To determine whether tetherin is incorporated into virion envelopes, we employed both biochemical and electron microscopic assays. Transmission EM analysis of HT1080 cells expressing tetherin-HA and infected with either HIV-1(WT) or HIV-1(delVpu) revealed that only HIV-1(delVpu) infection resulted in significant numbers of virions tethered at the cell surface. Immunogold labeling of cell surfaces with anti-HA antibodies revealed that tetherin-HA was frequently associated with virions (Figure 4A).
Figure 4. Incorporation of tetherin variants into HIV-1 particles.
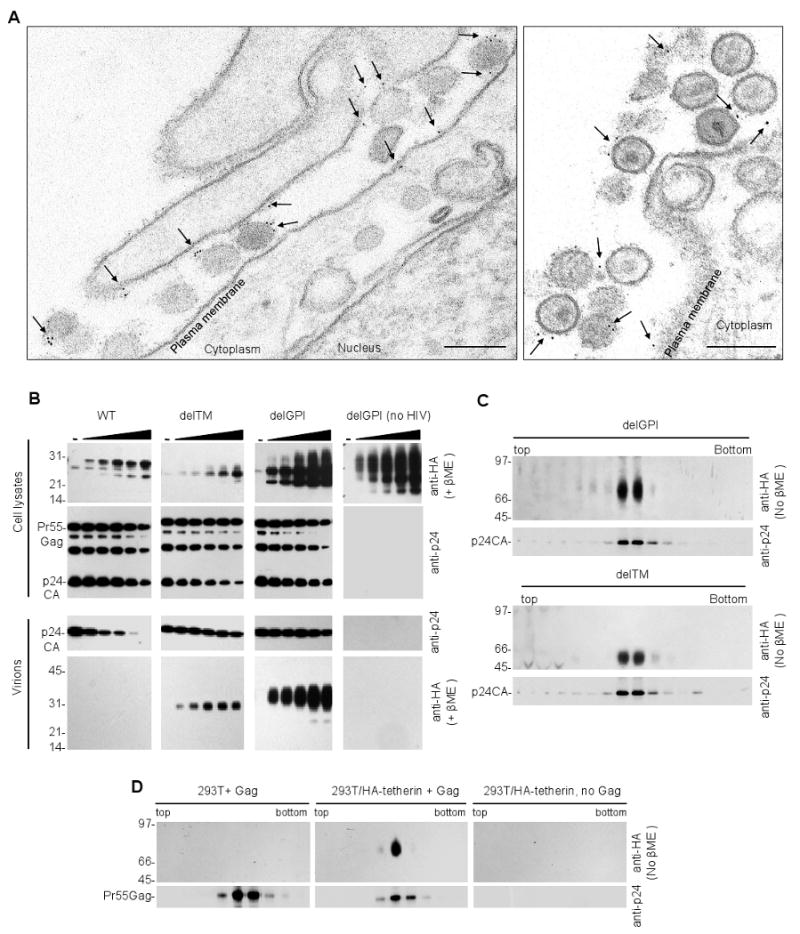
(A) Transmission electron microscopic analysis of HT1080 cells stably expressing tetherin-HA and infected with HIV-1(delVpu). Infected cells were fixed and stained with anti-HA and anti mouse-IgG-gold particles prior to sectioning. Scale bars = 200nm.
(B) Analysis of mutant tetherin incorporation into HIV-1 particles. 293T cells and corresponding released virions were harvested following transfection with HIV-1(delVpu) and increasing amounts of plasmids (0, 3.125, 6.25, 12.5, 25 and 50ng) expressing HA-tagged WT and mutant tetherin proteins. Virions were pelleted through sucrose prior to analysis. Cells and virions were subjected to Western blot analysis (anti-p24 and anti-HA, as indicated).
(C) Western blot analysis (anti-p24 and anti-HA) of virions derived from HIV-1(delVpu) infected 293T cells stably expressing delTM or delGPI tetherin proteins. Virions were purified on Optiprep gradients and sixteen fractions were collected for western blot analysis of p24 and tetherin content.
(D) Analysis of WT tetherin incorporation into virions. Unmodified 293T cells (left panels), or 293T cells stably expressing full-length tetherin-HA (center panels), were transfected with a plasmid expressing codon-optimized HIV-1 Gag. VLPs were pelleted through sucrose and purified on linear (10-30%) optiprep gradient. Ten fractions were collected, precipitated and subjected to a western blot analysis using anti-p24 and anti-HA antibodies. As a control, mock transfected 293T cells stably expressing full-length tetherin-HA, were subjected to the same procedure (right panels).
Wild-type tetherin blocked HIV-1 particle release and, therefore, it was difficult to determine whether it was incorporated into virions using western blot assays (Figure 4B). Indeed, if a given virion incorporated molecules that cause tethering it would be unlikely to be released. Therefore, we generated HIV-1(delVpu) particles in the presence of inactive tetherin mutants. Deletion of either one of the two of the tetherin membrane anchors (in the delTM and delGPI mutants) resulted in proteins that did not impair virion release but were efficiently incorporated into virions (Figure 4B). Importantly, the apparent incorporation of tetherin into pelleted virions was specific, because no pelletable tetherin was observed in the absence of virion production (Figure 4B). Other poorly, but partly, active tetherin mutants, for example (C53,63,91A), that retained both membrane anchors were also incorporated into particles, albeit at significantly reduced levels compared to delGPI (Figure S5).
To further demonstrate that delGPI and delTM tetherin proteins were incorporated into HIV-1 particles, 293T stably expressing either delTM or delGPI tetherin proteins were infected with a HIV-1(delVpu) and progeny virions purified on Optiprep gradients. Analysis of gradient fractions showed a precise concordance between those that contained virions and those that contained delGPI and delTM tetherin proteins (Figure 4C).
In contrast to the delGPI, delTM and C53,63,91A mutant tetherin proteins, full-length tetherin was not found in virion particles when transiently expressed with HIV-1(delVpu) proviruses, or in single cycle infection assays (Figure 4B, S5 and data not shown), likely because virions that encounter tetherin are not efficiently released. However, we reasoned that high levels of Gag expression and modest levels of tetherin expression might ‘dilute’ the available cell surface tetherin among a large number of nascent particles, thereby reducing its potency and perhaps enabling its detection in released virions. Indeed, when 293T cells stably expressing HA-tagged tetherin were transiently transfected with a codon-optimized Gag expression plasmid that generates large numbers of particles, inhibition of particle release by tetherin was incomplete (Figure S6). Under these conditions, disulfide linked tetherin dimers were detected in VLPs purified on Optiprep gradients (Figure 4D). Thus tetherin was incorporated into virion particles, and either of its two membrane anchors was capable of driving incorporation.
Configuration of tetherin in virions and exclusion by Vpu
To determine the topology of tetherin with respect to the virion envelope HIV-1(delVpu) virions were harvested from 293T stably expressing delGPI (HA-tagged at the N-terminus) or delTM (HA-tagged at the C-terminus) and purified virions were either untreated or treated with subtilisin A. Both proteins were incorporated into HIV-1(delVpu) virions as dimers (Figure 5A) and an N-terminal fragment of the delGPI tetherin protein that migrated at ∼25 kDa (under non-reducing conditions) was protected from subtilisin digestion (Figure 5A). Under reducing conditions, this fragment migrated at approximately half this molecular weight (∼13 kDa) (Figure 5A), strongly suggesting that delGPI is incorporated in particles as a parallel disufide linked dimer with both N-termini inside the virion particle. Conversely, the delTM tetherin protein was also incorporated in particles as a dimer but was not protected from subtilisin digestion, indicating that the entire delTM tetherin protein was on the outside of virion envelopes, anchored therein by GPI at its C-terminus (Figure 5A).
Figure 5. Configuration of tetherin in virions and exclusion by Vpu.
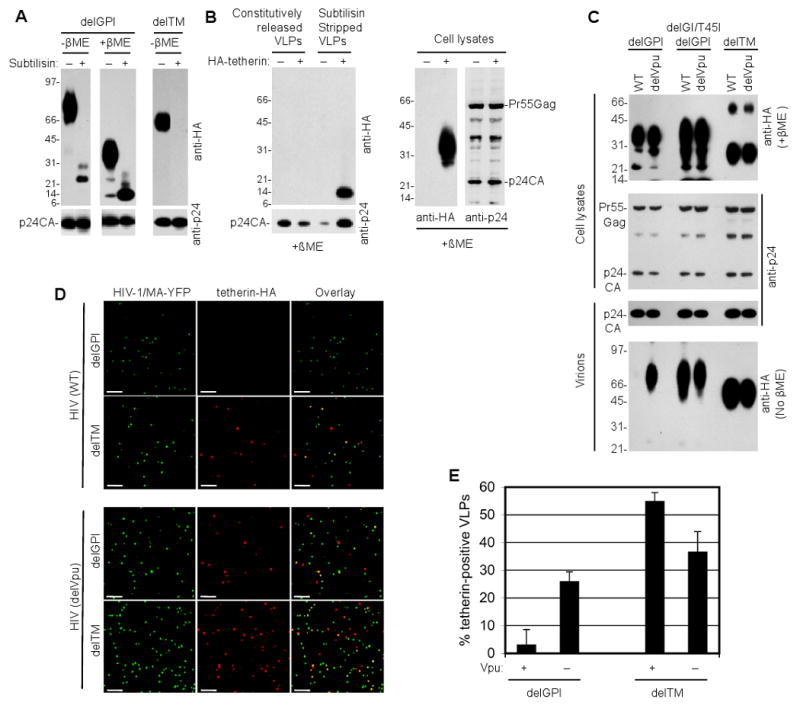
(A) Analysis of virions derived from HIV-1(delVpu) infected 293T cells stably expressing N-terminally HA-tagged delGPI, and C-terminally HA-tagged delTM tetherin proteins. Virions were pelleted through sucrose, treated with (+) or without (-) subtilisin, pelleted again though sucrose and subjected to SDS PAGE in the presence or absence of β-mercaptoethanol prior to Western blot analysis with anti-p24 and anti-HA antibodies.
(B) Analysis of virions recovered from the surface of 293T cells stably expressing N-terminally HA-tagged WT tetherin. Cells were infected with HIV-1(delVpu) and virions that were constitutively released, or released following incubation of the cells with subtilisin, were pelleted through sucrose prior to analysis. Virions and corresponding cell lysates were subjected to western blot analysis with anti-p24 and anti-HA antibodies.
(C) Analysis of the effect of Vpu on virions derived from HIV-1 infected 293T cells stably expressing HA-tagged delGPI, delGI/T45I/delGPI, or delTM tetherin proteins. Cells were infected with HIV-1 (WT) or HIV-1(delVpu) and virions were pelleted through sucrose prior to analysis. Virions and corresponding cell lysates were subjected to western blot analysis with anti-p24 and anti-HA antibodies.
Numbers to the left of panels (A), (B), and (C) represent the positions and sizes (in kDa) of molecular weight markers.
(D) Immunofluorescence analysis (anti-HA) of virions harvested from HIV-1/MA-YFP(WT) or HIV-1/MA-YFP(delVpu) infected 293T cells stably expressing delTM and delGPI tetherin proteins. Virions were pelleted, applied to poly-D-lysine coated coverslips prior to analysis. Scale bars = 2μm. A proportion of representative fields observed in each of two experiments is shown.
(E) Quantitative analysis of tetherin incorporation into virions. The proportion of HIV-1/MA-YFP(WT) or HIV-1/MA-YFP(delVpu) virions that were positive for delTM and delGPI tetherin proteins was quantified. The mean and standard deviation of the percentage of YFP-positive virions that were positive for tetherin is shown. Three fields containing a total of at least 600 YFP-positive virions for each condition were evaluated, in each of two independent experiments.
We also analyzed virions trapped at the cell surface by N-terminally tagged WT tetherin (Figure 5B). In this case, virions were recovered from the cell surface by subtilisin stripping. These subtilisin stripped virions contained an N-terminal tetherin fragment that migrated in a manner that was indistinguishable from that of fragment recovered from subtilisin treated, constitutively released virons containing the N-terminally tagged delGPI tetherin protein. Thus, that at least some of the virions trapped at the cell surface by WT tetherin incorporated tetherin into the particles with the tetherin N-terminus in the virion interior.
The HIV-1 Vpu protein antagonizes the antiviral activity of the human tetherin protein via poorly understood mechanisms. However, the differential sensitivity of human, monkey and chimeric tetherin proteins to antagonism, suggests that Vpu targets the tetherin transmembrane domain to neutralize its activity (McNatt et al., 2009). We reasoned that if tetherin incorporation into virions was relevant to its antiviral function, then HIV-1 Vpu should be able to impair the incorporation of delGPI tetherin into viral particles.
Analysis of virions harvested from HIV-1(WT) or HIV-1(delVpu) infected 293T cells stably expressing delTM tetherin (anchored in membrane by the GPI modification) revealed that delTM tetherin incorporation was unaffected by Vpu (Figure 5C). In contrast, delGPI tetherin dimers (anchored in membranes by the TM domain) was efficiently incorporated into HIV-1(delVpu) but excluded from HIV-1(WT) virions (Figure 5C). Mutations in the tetherin transmembrane domain (delGI/T45I) can confer complete resistance to antagonism by Vpu (McNatt et al., 2009). Concordantly, this mutant delGPI tetherin protein was equally well incorporated into virions in the presence or absence of Vpu (Figure 5C).
To further analyze tetherin incorporation into virions and its antagonism by Vpu, 293T cells stably expressing delGPI or delTM tetherin proteins were infected with HIV-1(WT) or HIV-1(delVpu) derivatives that harbor YFP inserted into the stalk region of matrix, such that infected cells generate fluorescent virion particles (Jouvenet et al., 2006). Immunofluorescence analysis of these virions, fixed to coverslips, revealed that the HIV-1(WT) particles incorporated delTM but excluded the delGPI tetherin whereas HIV-1(delVpu) particles incorporated both delGPI and delTM tetherin proteins (Figure 5D,E). Altogether, these results indicate that Vpu excludes tetherin from virions by targeting its transmembrane domain.
Tetherin association with budding virions
To visualize the infiltration of nascent virions by tetherin in greater detail, we conducted scanning EM experiments with gold immunolabeling and backscatter electron detection. Virions that were in the act of, but had not completed, budding were generated in 293T cells transiently expressing either WT tetherin-HA or the delTM or delGPI mutants by simultaneously expressing HIV-1 Gag carrying a mutation in the PTAP L-domain. This manipulation arrests budding before completion, and enables reliable detection of nascent virion particles on cell surfaces. However, virions are retained at the cell surface whether or not they encounter tetherin during budding, and regardless of whether the tetherin protein blocks release. Thus, this approach carries the disadvantage that does not allow the observation of tetherin-dependent tethering events, but the advantage that the interaction between inactive tetherin deletion mutants (delTM and delGPI) and budding particles could be observed.
The WT and delTM tetherin proteins were found to be associated with many budding virions, but there was not strong enrichment at budding sites (Figure 6A). In contrast, the delGPI tetherin protein preferentially localized at sites of particle budding. In many cases, the delGPI protein appeared to adopt a ring-like arrangement at viral budding sites (Figure 6). Overall, the full length tetherin protein, as well as derivatives containing only one of the two membrane anchors, were able to infiltrate the envelope of budding virions, and the N-terminal domain of tetherin appears to have a particular propensity to drive localization to virion buds.
Figure 6. Scanning electron microscopic analysis of tetherin interaction with budding virions and models for tetherin incorporation.
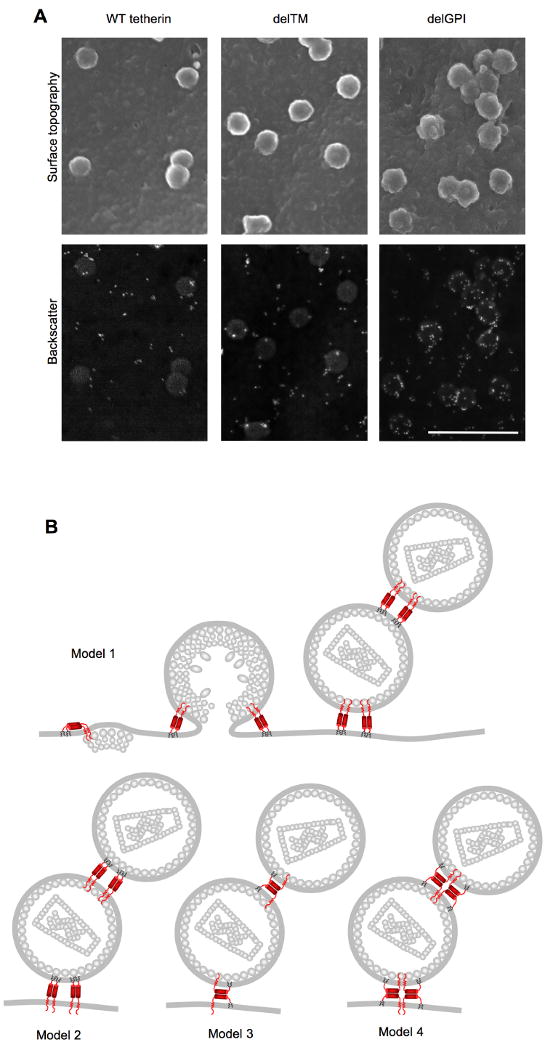
(A) Scanning electron microscopic analysis of 293T cells transiently expressing HA-tagged WT, delTM or delGPI tetherin proteins and HIV-1 Gag bearing mutations in the PTAP L-domain sequence. Cells were fixed and stained with anti-HA primary antibody and anti-mouse IgG-gold conjugate. Surface topography (upper panels) reveals virion particles, while backscatter electron detection (lower panels) reveals gold particles (white) marking the position of tetherin molecules. Scale bar = 500nm
(B) Models for incorporation and virion tethering by the tetherin protein. Several stages of HIV-1 assembly are depicted for model 1, and only tethered virions are shown for models 2-4. In model 1, the TM domains of a tetherin dimer are incorporated into the virion envelope, and the GPI anchors remain embedded in the host-cell membrane. In model 2, the reverse situation occurs. In model 3, only one of a pair of disulfide linked tetherin molecules has both membrane anchors incorporated into the virion envelope. In model 4, one disulfide linked tetherin dimer incorporated into the virion envelope interacts with another dimer in the host-cell membrane via coiled-coil based interactions.
Discussion
In general, the mechanisms by which interferon induced gene products inhibit viral replication is poorly understood (Samuel, 2001). Here, we show that one such gene product, tetherin, inhibits enveloped virus release via a simple and direct mechanism. Identification of the key features of the tetherin molecule that were required for antiviral activity allowed the design of a completely artificial tetherin-like protein using domains from other proteins. Despite lacking significant sequence homology to native tetherin, the synthetic tetherin-like protein mimicked the biological activity of native tetherin. These data strongly suggest that tetherin activity does not involve interactions with specific cofactors, sensor function, signaling, or other indirect mechanisms. Rather, they suggest that tetherin itself directly tethers viral particles to cellular membranes.
Consistent with this notion, both of the tetherin membrane anchors were essential for tethering function (in both native and art-tetherin) and either of native tetherin's membrane anchors could drive incorporation into the envelope of HIV-1 particles. Because (i) all three cysteines that are situated towards the tetherin N-terminus form intermolecular disulfide bonds with the corresponding residue in the tetherin homodimer and (ii) the delGPI tetherin mutant was incorporated into released virions with N-termini protected from externally applied protease, these data strongly suggest that tetherin bridges viral and infected cell membranes by inserting either or both its N- or C-termini as a parallel homodimer into the viral membrane. Based on these findings, we favor the model for tetherin incorporation shown in Figure 6B (models 1, 2) whereby one pair of tetherin membrane anchors are incorporated into an assembling virion envelope, while the other pair remains embedded in the cell membrane. The finding that the delGPI tetherin molecule appears to have a particular propensity for localizing at sides of viral budding would tend to favor model 1. In these models, we speculate that the role of the coiled-coil and homotypic disulfide bonds might be to constrain the dimer in an extended conformation, thereby maintaining distance between the two pairs of membrane anchors. This spatial separation should increase the likelihood that one pair of membrane anchors is incorporated into the virion envelope while the other remains in the cell membrane. However, there are a few possible configurations that tetherin could adopt during virion tethering that are consistent with most of the data presented in this study (models 3,4, Figure 6B) and it is even possible that several different configurations are used. Additionally, tetherin could form higher order complexes through interactions between dimers. Nonetheless, the key features of each of these models are: (i) that tetherin itself appears necessary and sufficient to form tethers and (ii) that membrane anchors in a single tetherin dimer, or higher order complex, partition into both virion and host cell membranes. Earlier findings demonstrate that tetherin causes virions to be tethered to each other, as well as to the cell surface. Obviously this can only occur if both types of tetherin membrane anchor can be incorporated into virion membranes, and could result from the assembly of a virion at a site on the plasma membrane that is already occupied by a tethered virion.
Vpu causes exclusion of delGPI tetherin but not delTM tetherin from HIV-1 particles. Put another way, Vpu blocked the insertion of the tetherin transmembrane domain into virion envelopes. This effect was highly specific, as mutations in the human tetherin TM domain (delGI/T45I) that confer resistance to antagonism by Vpu (McNatt et al., 2009) also induced resistance to virion exclusion by Vpu. This suggests that exclusion of tetherin from assembling virions is central to the mechanism by which Vpu counteracts tetherin's antiviral activity, consistent with previous findings that Vpu prevented colocalization of tetherin with Gag puncta at the plasma membrane of VLP producing cells (Jouvenet et al., 2009).
Our conclusions contrast with those of with Miyagi and colleagues who failed to detect tetherin in virions that were released from HeLa cells following agitation, and suggested that tetherin does not directly tether virions to cells (Miyagi et al., 2009). However, it is not clear how effectively agitation releases bona fide tethered viruses from cells, as HIV-1(WT) and HIV-1(delVpu) were released from cells by agitation with only moderately differing efficiencies (Miyagi et al., 2009). We were clearly able to detect full-length tetherin in constitutively released virion particles under conditions where sufficient Gag was expressed to saturate the available tetherin and allow reasonable levels of particle release. Additionally, electron microscopic analyses showed clear association of tetherin with tethered and budding HIV-1 particles. Moreover, tetherin mutants bearing only one of the two membrane anchors were efficiently inserted into the envelope of released HIV-1 virions.
The studies described herein focused primarily on HIV-1 particle release. However, while HIV-1 normally replicates in T-cells and macrophages, type-1 interferon can also induce tetherin expression in a wide variety of other cell types that are not normally target cells of HIV-1, including those used in this study. Indeed, tetherin can induce essentially the same virion retention effect in a variety of cell contexts, and is also capable of tethering diverse enveloped viruses (Neil et al., 2007; Jouvenet et al., 2009; Kaletsky et al., 2009; Sakuma et al., 2009). The mechanism of action elucidated herein explains how tetherin is able to inhibit the release of a variety of enveloped viruses from a variety of cells, and thereby constitute a generalized IFN-induced antiviral defense. Because this mechanism constitutes simple bridging of host and viral lipid bilayers, then one can view the lipid envelope as the viral target of tetherin. Because the viral target is actually derived from the host cell, then it is difficult for a virus to evade tetherin via simple mutations, as would be the case if such an inhibitor targeted a viral protein. This is likely the reason why several viruses have independently evolved trans-acting tetherin antagonists such as HIV-1 Vpu (Neil et al., 2008; Van Damme et al., 2008), SIV Nef (Jia et al., 2009; Zhang et al., 2009), Ebola GP (Kaletsky et al., 2009), KSHV K5 (Bartee et al., 2006) and, possibly, HIV-2 Env. The mechanism by which these antagonists counteract tetherin activity is not yet defined, although tetherin downregulation from the cell surface, proteasome-dependent degradation and possibly retention at the trans-Golgi network have been observed (Bartee et al., 2006; Dube et al., 2009; Goffinet et al., 2009; Van Damme et al., 2008). The mechanism of tetherin action described herein suggests that simply relocating tetherin such that it is excluded from sites of enveloped viral budding should be sufficient to counteract its inhibitory effect on virion release.
Experimental procedures
Materials
Details of the cells and plasmid constructs used in this study are given in the supplemental information.
Tetherin activity assays
To measure tetherin activity, 293T cells in 24-well plates were transfected using polyethylenimine (PEI, PolySciences) with 500ng of HIV-1(WT), HIV-1(delVpu) proviral plasmids (Neil et al., 2006) or 400ng of a plasmid expressing myc-tagged Ebola virus VP40 (Martin-Serrano et al., 2001). Tetherin expression plasmids were included at 3 to 200ng per transfection. Cells were harvested at 40 hrs after transfection. Virion-containing supernatants were clarified by centrifugation and infectious HIV-1 release was determined using HeLa-TZM indicator cells as previously described (Martin-Serrano et al., 2001). The remainder of the supernatant (800 μl) was layered onto 600 μl of 20% sucrose in PBS and centrifuged at 20,000g for 90 minutes. Particle yield determined using western blot assays (see supplemental information).
Microscopy
Fluorescence and electron microscopy assay were carried out according to standard procedures as outlined in the supplemental information.
Tetherin incorporation assays
To assess incorporation of transiently expressed tetherin, virions generated as described above were subjected to western blot analysis using anti-HA antibody. Alternatively, 293T cells stably expressing delGPI or delTM tetherin mutants (5×105 in 6-well plates) were infected with VSV-G pseudotyped HIV-1(WT) or HIV-1(delVpu) at MOI of 1. Two days later, 2ml of supernatant was harvested and layered on top of an 8 ml linear Optiprep gradient (10%-30%) and centrifuged at 110,000 g for 1 hour. Sixteen gradient fractions were collected. Alternatively 293T stably expressing tetherin-HA were plated in 10 cm dishes and transfected with 10μg of a plasmid expressing codon-optimized Gag or a corresponding empty vector. Two days later, extracellular VLPs were pelleted through a 20% sucrose cushion at 90,000g for 90 min, resuspended in PBS, layered on top of a 4ml linear Optiprep gradient (10-30%) and centrifuged at 84,000g for 1h. Ten gradient fractions were collected. Proteins in each fraction were precipitated by adding 50μg of BSA as a protein carrier and TCA to 10%, and subjected to western blot analyses.
Subtilisin stripping of cells and virions
To recover virions tethered to cell surfaces, 293T cells in 35 mm dishes were cotransfected with 1μg of HIV-1(WT) or HIV-1(delVpu) proviral plasmids and 200 ng of WT-tetherin or art-tetherin expression plasmids. Two days later, constitutively released virions were collected from supernatants. The cells were washed twice with PBS, once with subtilisin A buffer (10mM Tris pH 8.0, 1 mM CaCl2, 150 mM NaCl) and treated with 500μl of 1 mg/ml of subtilisin A (Sigma) for 2-3 minutes at room temperature. Subtilisin treatment was stopped using DMEM containing 10% FCS, 5mM PMSF and 20mM EGTA. The ‘subtilisin-strip’ supernatant was layered on a 20% sucrose cushion and centrifuged at 20000 g for 90 minutes.
For subtilisin digestion of tetherin on the surface of virion particles, 293T cells stably expressing delGPI HA-tetherin or delTM tetherin-HA were infected with HIV-1(delVpu) at an MOI of 1. Two days later, virions were harvested by centrifugation through 20% sucrose. Pelleted virions were resuspended in subtilisin buffer and treated with subtilisin for 45 min at room temperature. Thereafter, subtilisin digestion was stopped as above, and virions were pelleted through sucrose for a second time prior to western blot analysis.
Supplementary Material
Acknowledgments
We thank members of the Bieniasz laboratory for helpful discussions and T. Kinoshita for PIGL-defective cells. This work was supported by NIH grant RO1AI50111. PDB is an Investigator of the Howard Hughes Medical Institute
References
- Bartee E, McCormack A, Fruh K. Quantitative membrane proteomics reveals new cellular targets of viral immune modulators. PLoS Pathog. 2006;2:e107. doi: 10.1371/journal.ppat.0020107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube M, Roy BB, Guiot-Guillain P, Mercier J, Binette J, Leung G, Cohen EA. Suppression of Tetherin-Restricting Activity on HIV-1 Particle Release Correlates with Localization of Vpu in the trans-Golgi Network. J Virol. 2009 doi: 10.1128/JVI.01800-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffinet C, Allespach I, Homann S, Tervo HM, Habermann A, Rupp D, Oberbremer L, Kern C, Tibroni N, Welsch S, et al. HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe. 2009;5:285–297. doi: 10.1016/j.chom.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Jia B, Serra-Moreno R, Neidermyer W, Rahmberg A, Mackey J, Fofana IB, Johnson WE, Westmoreland S, Evans DT. Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 2009;5:e1000429. doi: 10.1371/journal.ppat.1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenet N, Neil SJ, Bess C, Johnson MC, Virgen CA, Simon SM, Bieniasz PD. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 2006;4:e435. doi: 10.1371/journal.pbio.0040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenet N, Neil SJ, Zhadina M, Zang T, Kratovac Z, Lee Y, McNatt M, Hatziioannou T, Bieniasz PD. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J Virol. 2009;83:1837–1844. doi: 10.1128/JVI.02211-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaletsky RL, Francica JR, Agrawal-Gamse C, Bates P. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc Natl Acad Sci U S A. 2009;106:2886–2891. doi: 10.1073/pnas.0811014106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupzig S, Korolchuk V, Rollason R, Sugden A, Wilde A, Banting G. Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic. 2003;4:694–709. doi: 10.1034/j.1600-0854.2003.00129.x. [DOI] [PubMed] [Google Scholar]
- Martin-Serrano J, Zang T, Bieniasz PD. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat Med. 2001;7:1313–1319. doi: 10.1038/nm1201-1313. [DOI] [PubMed] [Google Scholar]
- McNatt MW, Zang T, Hatziioannou T, Bartlett M, Fofana IB, Johnson WE, Neil SJ, Bieniasz PD. Species-specific activity of HIV-1 Vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog. 2009;5:e1000300. doi: 10.1371/journal.ppat.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagi E, Andrew AJ, Kao S, Strebel K. Vpu enhances HIV-1 virus release in the absence of Bst-2 cell surface down-modulation and intracellular depletion. Proc Natl Acad Sci U S A. 2009;106:2868–2873. doi: 10.1073/pnas.0813223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita E, Sundquist WI. Retrovirus budding. Annu Rev Cell Dev Biol. 2004;20:395–425. doi: 10.1146/annurev.cellbio.20.010403.102350. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Inoue N, Watanabe R, Takahashi M, Takeda J, Stevens VL, Kinoshita T. Expression cloning of PIG-L, a candidate N-acetylglucosaminyl-phosphatidylinositol deacetylase. J Biol Chem. 1997;272:15834–15840. doi: 10.1074/jbc.272.25.15834. [DOI] [PubMed] [Google Scholar]
- Neil SJ, Eastman SW, Jouvenet N, Bieniasz PD. HIV-1 Vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Pathog. 2006;2:e39. doi: 10.1371/journal.ppat.0020039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil SJ, Sandrin V, Sundquist WI, Bieniasz PD. An interferon-alpha-induced tethering mechanism inhibits HIV-1 and Ebola virus particle release but is counteracted by the HIV-1 Vpu protein. Cell Host Microbe. 2007;2:193–203. doi: 10.1016/j.chom.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008;451:425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- Ohtomo T, Sugamata Y, Ozaki Y, Ono K, Yoshimura Y, Kawai S, Koishihara Y, Ozaki S, Kosaka M, Hirano T, et al. Molecular cloning and characterization of a surface antigen preferentially overexpressed on multiple myeloma cells. Biochem Biophys Res Commun. 1999;258:583–591. doi: 10.1006/bbrc.1999.0683. [DOI] [PubMed] [Google Scholar]
- Sakuma T, Noda T, Urata S, Kawaoka Y, Yasuda J. Inhibition of Lassa and Marburg virus production by tetherin. J Virol. 2009;83:2382–2385. doi: 10.1128/JVI.01607-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev. 2001;14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe. 2008;3:245–252. doi: 10.1016/j.chom.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varthakavi V, Heimann-Nichols E, Smith RM, Sun Y, Bram RJ, Ali S, Rose J, Ding L, Spearman P. Identification of calcium-modulating cyclophilin ligand as a human host restriction to HIV-1 release overcome by Vpu. Nat Med. 2008;14:641–647. doi: 10.1038/nm1778. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang F, Wilson SJ, Landford WC, Virgen B, Gregory D, Johnson MC, Munch J, Kirchhoff F, Bieniasz PD, Hatziioannou T. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe. 2009;6:54–67. doi: 10.1016/j.chom.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.