

Abstract
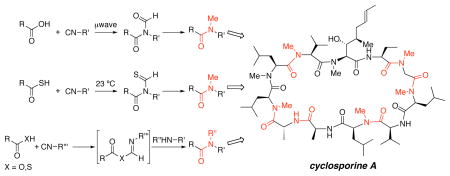
Recent developments in the use of isonitriles to furnish secondary and tertiary amide bond formations are applied to a novel total synthesis of the important cyclic polypeptide cyclosporine A. Specifically, the disclosed synthetic route demonstrates the utility of microwave-mediated carboxylic acid isonitrile couplings, thioacid isonitrile couplings at ambient temperature, and isonitrile-mediated couplings of carboxylic acids and thioacids with amines to form challenging amide bonds.
Recently, we described a series of findings using isonitriles in the formation of amide bonds.1,2,3,4 These early works suggested that isonitrile-mediated bond constructions might be applicable to the synthesis of tertiary amides. There is currently a great deal of interest in such systems, particularly with respect to improving the pharmaco-availability of biologically active polypeptides.5
We thought that our findings in this area were sufficiently promising that it would be appropriate to explore various ways for generating tertiary amides in the context of a total synthesis of a challenging target system. Given these considerations, it was only natural to turn to cyclosporine A as a worthy goal. Cyclosporine A (1), a reversible inhibitor of cytokines in T helper cells,6 was isolated from the fungus Tolypocladium inflatum gams.7 Its structure was confirmed by chemical degradation, NMR, and X-ray crystallographic studies,8 and its total synthesis was reported by Wenger in 1984.9 The immunosuppressive properties of cyclosporine A, which enable otherwise non-sustainable transplantations, are very well established.10 In addition, cyclosporine A has been applied to the treatment of Beçhet’s syndrome, endogenous uveitis, psoriasis, atopic dermatitis, rheumatoid arthritis, active Chron’s disease, and nephrotic syndrome.10 Indeed, its bioavailability with respect to proteolysis is highly dependent on the pattern of N-methylation present in 7 of the 11 amino acid residues of the cyclic polypeptide. As established by Wenger, Rich, and Galpin through analog studies,11 the unusual amino acid (2S,3R,4R,6E)-3-hydroxy-4-methyl-2-methylamino-6-octenoic acid (MeBmt) is of particular importance to the biological activity of 1, as well as the sequence MeBmt, Abu, Sar, MeLeu. Herein, we demonstrate the emerging versatility and power of isonitrile chemistry in the context of a total synthesis of cyclosporine A.
Retrosynthetic planning began by disconnecting the macrocycle at the Ala–(D-Ala) junction and on either side of the MeBmt residue, thus revealing MeBmt acetonide 2, tetrapeptide 3, and hexapeptide 4 (Scheme 1). The protected MeBmt residue 2 is a known compound, obtainable via the route charted by Evans and co-workers.12 Tetrapeptide 3 was to be assembled from its component dipeptides, each of which could be reached via isonitrile mediated coupling methods. Hexapeptide 4 was seen as arising from dipeptide and tetrapeptide fragments.
Scheme 1.

We began by targeting dipeptide 8, en route to tetrapeptide 3. Leucine-derived thioacid 513 was smoothly coupled with valine isonitrile 614 to afford the corresponding N-thioformyl amide (Scheme 2). Subsequent conversion to the N-methyl amide was challenging. Treatment with Raney-Ni as reported by Chupp and co-workers15 was unsuccessful, presumably owing to the base-sensitive nature of the thioformyl amide. Ultimately, it was found that the reduction proceeded well under free radical conditions. Thus, treatment of the crude N-thioformyl amide with Bu3SnH and AIBN at 100 °C in toluene afforded the desired N-methyl dipeptide 7 in 62% yield over two steps. Following cleavage of the carbamate, the elegant retro aza-Diels-Alder methylation approach described by Greico et. al16 provided access to the corresponding N-methylated derivative 8, without epimerization, in 52% yield over three steps.
Scheme 2.

A similar strategy was applied for the second key dipeptide fragment. Thus, D-alanine thioacid 913 underwent coupling with leucine isonitrile 1014 in CHCl3 at ambient temperature, followed by radical reduction, to afford the desired N-methylated dipeptide 11 in 59% yield over 2 steps (Scheme 3). At this point, we were poised to join the two dipeptide fragments. Hydrogenolysis of the benzyl ester afforded the corresponding acid, and subsequent coupling with fragment 8 was accomplished upon treatment with DEPBT17 and Hünig’s base in THF. Using these optimized conditions,18 we were able to synthesize the desired tetrapeptide 3 in 90% yield (over the two steps) with no observable epimerization.
Scheme 3.

With the tetrapeptide fragment (3) in hand, we turned our attention to the synthesis of hexapeptide 4. Microwave irradiation of azido acid 1219 and leucine isonitrile 1314 afforded N-formyl amide 14 in 85% yield (Scheme 4). We reasoned that the presence of the N-formyl group could be exploited to decrease the extent of oxazolone formation, which might otherwise lead to epimerization during the C-terminal extension with a suitably protected alanine.1c–d Maintenance of the N-formyl group during acidic removal of the t-butyl ester and during coupling with alanine benzyl ester was possible, thereby providing tripeptide 15 in 81% yield and >20:1 dr. The reduction of the N-formyl amide was very challenging because of its propensity to undergo either non-selective reduction (resulting in destruction of the peptide) or deformylation, resulting in formation of the native amide bond. Substantial optimization was required to identify conditions under which the required transformation could be performed chemoselectively. Ultimately, a combination of lithium borohydride and 0.5 equivalents of acetic anhydride in a mixture of CH2Cl2 and n-propanol at −50 °C was used to provide the relatively unstable N-hydroxymethyl intermediate. Immediate reduction of the crude product with triflic anhydride and triethylsilane, in CH2Cl2 afforded the desired N-methylated tripeptide 16 in 62% yield over two steps. Selective reduction of the azide functionality was readily accomplished by treatment of 16 with triphenylphosphine in the presence of water, forming 17. The final methyl-leucine residue was attached via a HATU coupling. Acidic removal of the Boc protecting group afforded TFA salt 18, bearing the secondary amide bond at the valine residue, in 85% yield over 2 steps.
Scheme 4.

We were now well-positioned to test additional isonitrile-mediated amide bond formations.4 Thus, thioacid 19 was treated with a “sacrificial isonitrile”4 to provide a putative thio-FCMA (Formimidate (thio)Carboxylate Mixed Anhydride), which was intercepted by amine 20, thereby producing dipeptide 21 in 80% yield (Scheme 5). Hydrogenolysis of the benzyl ester was achieved quantitatively, providing known dipeptide 22. We recently disclosed a method by which carboxylic acids could be treated with Lawesson’s reagent under microwave conditions or at room temperature to afford the corresponding thioacids.20 Employing this approach, 22 was converted (65% yield) to thioacid 23, which was then coupled with secondary amine 18. Following cleavage of the Boc group, the hexapeptide 24 was obtained in 63% yield.
Scheme 5.

We next began the final series of fragment couplings of the seco system. The protected MeBmt residue, 2, was coupled to hexapeptide 24 using DCC and HOBt. Subsequent treatment with aqueous HCl provided heptapeptide 25 in 75% yield over 2 steps.
At this point, we sought to couple the final two fragments: tetrapeptide 3 and heptapeptide 25. Hydrogenolysis of the C terminus of 3 followed by smooth coupling with fragment 25 afforded undecapeptide 26 in 52% yield (Scheme 6). Basic hydrolysis of 26 exposed the C terminus and subsequent acidic treatment cleaved the t-butoxy carbamate from the N terminus.
Scheme 6.

The stage was now set for applying the logic of our recently developed4 coupling method to a macrolactamization. This was no minor extension of our earlier work because, in the case at hand, we would be attempting to apply the logic to a C-terminal acid rather than to a thioacid.4 In the event, exposure of the unprotected undecapeptide 27 to 10 equivalents of cyclohexyl isonitrile at 70 °C under microwave radiation, resulted in isolation of cyclosporine (1) in 30% yield over three steps.
We note that, ordinarily, with carboxylic acids (as opposed to thioacids), amide bond formation by 1,3-O→N acyl transfer does not occur below ca. 130 °C under microwave mediation. We had conjectured that the FCMA actually forms at lower temperature, but that the usual 1,3-O→N acyl transfer requires higher temperature for efficient rearrangement. In the case at hand, the substrate is likely pre-organized due to intra-strand hydrogen bonding.21 Thus, the FCMA of the seco-acid, once formed, has an increased proclivity for cyclization, and macrolactamization is achieved at 70 °C, with only a trace amount of 1,3-O→N acyl transfer visible by LCMS of the crude reaction mixture.
While this finding was encouraging, the yield was, nonetheless, disappointing. Fortunately, the yield could be significantly increased through the addition of HOBt (1.5 equiv) to the cyclization medium. Actually, the reaction seemed very clean and appeared to proceed with high conversion. However, the isolated yield of cyclosporine A is, at this time, 54%. It is tempting to suppose, but certainly not proven, that an initial FCMA, formed from 27 and cyclohexylisonitrile, is intercepted with HOBt, thereby generating an active HOBt ester. The latter could well be the actual intermediate for macrolactamization, thereby attenuating the tendency for either 1,3-O→N acyl transfer or FCMA hydrolysis. Importantly, no dimerization is observed with either the HOBt or HOBt-free cyclization reactions.
In summary, we have shown how the chemistry of isonitriles can be applied to the construction of a variety of tertiary amides. These findings allowed for a total synthesis of cyclosporine A in a fashion that allows for a more detailed mapping of its SAR.1a Further applications of isonitrile logic in peptide, cyclic peptide, and glycopeptide settings will be disclosed in due course.
Supplementary Material
Acknowledgments
Support was provided by the NIH (CA28824 to S.J.D.). The authors thank Dr. George Sukenick, Hui Fang, and Sylvi Rusli of SKI’s NMR core facility for mass spectral and NMR assistance, and Rebecca Wilson and Dana Ryan for assistance with the preparation of the manuscript. We also thank Dr. Pavel Nagorny, Dr. Cindy Kan, and Dr. Karthik Iyer for helpful discussions. We thank Professor Gregory Verdine for suggesting cyclosporine A as a methodology-challenging molecule.
Footnotes
Supporting Information Available Experimental procedures, copies of spectral data, and characterization data are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Li X, Danishefsky SJ. J Am Chem Soc. 2008;130:5446–5448. doi: 10.1021/ja800612r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li X, Danishefsky S. J Nat Prot. 2008;3:1666–1670. doi: 10.1038/nprot.2008.153. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li X, Yuan Y, Berkowitz WF, Todaro LJ, Danishefsky S. J J Am Chem Soc. 2008;130:13222–13224. doi: 10.1021/ja8047078. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Li X, Yuan Y, Kan C, Danishefsky SJ. J Am Chem Soc. 2008;130:13225–13227. doi: 10.1021/ja804709s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu X, Li X, Danishefsky SJ. Tetrahedron Lett. 2009;50:1523–1525. doi: 10.1016/j.tetlet.2009.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yuan Y, Zhu J, Li X, Wu X, Danishefsky SJ. Tetrahedron Lett. 2009;50:2329–2333. doi: 10.1016/j.tetlet.2009.02.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rao Y, Li X, Danishefsky SJ. J Am Chem Soc. 2009;131:12924–12926. doi: 10.1021/ja906005j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatterjee J, Gilon C, Hoffman A, Kessler H. Acc Chemm Res. 2008;41:1331–1342. doi: 10.1021/ar8000603. [DOI] [PubMed] [Google Scholar]
- 6.Faulds D, Goa KL, Benfield P. Drugs. 1993;45:953–1040. doi: 10.2165/00003495-199345060-00007. [DOI] [PubMed] [Google Scholar]
- 7.Rüegger A, Kuhn H, Lichti HR, Loosi R, Huguenin R, Quiquerez A, von Wartburg A. Helv Chim Acta. 1976;59:1075–1092. doi: 10.1002/hlca.19760590412. [DOI] [PubMed] [Google Scholar]
- 8.(a) Kessler H, Loosli HR, Oschkinat H. Helv Chim Acta. 1985;68:661–681. [Google Scholar]; (b) Loosli HR, Kessler H, Oschkinat H, Weber HP, Petcher TJ, Widmer A. Helv Chim Acta. 1985;68:682–704. [Google Scholar]
- 9.(a) Wenger RM. Helv Chim Acta. 1984;67:502–525. [Google Scholar]; (b) Wenger RM. Helv Chim Acta. 1983;66:2308–2321. [Google Scholar]; (c) Wenger RM. Helv Chim Acta. 1983;66:2672–2702. [Google Scholar]
- 10.(a) Laupacis A, Keown PA, Ulan RA, McKenzie N, Stiller CR. Can Med Assoc J. 1982;126:1041–1046. [PMC free article] [PubMed] [Google Scholar]; (b) Dreyfuss M, Harri E, Hofmann H, Kobel H, Pache W, Tscherter H. Eur J Appl Microbiol. 1976;3:125–133. [Google Scholar]; (c) Bueding E, Hawkins J, Cha Y-N. Agents and Actions. 1981:380–383. doi: 10.1007/BF01982474. [DOI] [PubMed] [Google Scholar]
- 11.(a) Wenger RM. Angew Chem, Int Ed Engl. 1985;24:77–138. [Google Scholar]; (b) Colucci WJ, Tung RD, Petri JA, Rich DH. J Org Chem. 1990;55:2895–2903. [Google Scholar]; (c) Rich DH, Dhaon MK, Dunlap B, Miller SPF. J Med Chem. 1986;29:978–984. doi: 10.1021/jm00156a014. [DOI] [PubMed] [Google Scholar]; (d) Rich DH, Sun C-Q, Guillaume D, Evans DA, Weber AE. J Med Chem. 1989;32:1982–1987. doi: 10.1021/jm00128a048. [DOI] [PubMed] [Google Scholar]; (e) Galpin IJ, Mohammed AKA, Patel A. Tetrahedron. 1988;44:1783–1794. [Google Scholar]
- 12.(a) Aebi JD, Dhaon MK, Rich DH. J Org Chem. 1987;52:2881–2886. [Google Scholar]; (b) Tung RD, Rich DH. Tetrahedron Lett. 1987;28:1139–1142. [Google Scholar]; (c) Evans DA, Weber AE. J Am Chem Soc. 1986;108:6757–6761. [Google Scholar]
- 13.Fu X, Jiang S, Li C, Xin J, Yang Y, Ji R. Bioorg Med Chem Lett. 2007;17:465–470. doi: 10.1016/j.bmcl.2006.10.021. [DOI] [PubMed] [Google Scholar]
- 14.Zhu J, Wu X, Danishefsky SJ. Tetrahedron Lett. 2009;50:577–579. doi: 10.1016/j.tetlet.2008.11.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chupp JP, Lechinsky KL. J Org Chem. 1975;40:66–71. [Google Scholar]
- 16.Greico PA, Bahas A. J Org Chem. 1987;52:5746–5749. [Google Scholar]
- 17.Nagalingam AC, Radford SE, Warriner SL. Synlett. 2007;16:2517–2520.DEPBT = 3-(diethoxy-phosphoryloxy)-3H-benzo[d][1,2,3]triazin-4-one.
- 18.Standard coupling conditions including HATU/DIPEA/DMF, PyBOP/NMM/CH2Cl2, and DCC/HOBt/NMM/THF led to low yields (40–45%) and high levels of epimerization (1:1 to 2:1 ratios of diastereomers).
- 19.Prepared according to: Goddard-Borger ED, Stick RV. Org Lett. 2007;9:3797–3800. doi: 10.1021/ol701581g.
- 20.Rao Y, Li X, Nagorny P, Hayashida J, Danishefsky SJ. Tetrahedron Lett. 2009;50:6684–6686. doi: 10.1016/j.tetlet.2009.09.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.This pre-organization was originally proposed by Wenger in Ref 9a. Our 1H NMR data support this hypothesis. The resolution of the 7 N-Me groups demonstrates that 26 exists largely as one rotamer. The acetonide-protected precursor to heptapeptide 25, which would possess only 2 potential intra-strand hydrogen bonds, exists as a mixture of rotamers. See the Supporting Information for images of these spectra.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.