

Abstract
Purpose
IFN-α is a pleiotropic cytokine possessing immunomodulatory properties that may improve the efficacy of therapeutic cancer vaccines. The aim of this study was to evaluate the effectiveness and compatibility of combining recombinant IFN-α with poxvirus vaccines targeting the human carcinoembryonic antigen (CEA) in murine models of colorectal and pancreatic adenocarcinomas, where CEA is a self-antigen.
Experimental Design
The phenotypic and functional effects of IFN-α were evaluated in the draining inguinal lymph nodes of tumor-free mice. We studied the effect of the site of IFN-α administration (local versus distal) on antigen-specific immune responses to poxvirus vaccination. Mechanistic studies were conducted to assess the efficacy of IFN-α and CEA-directed poxvirus vaccines in tumor-bearing CEA transgenic mice.
Results
We identified a dose and schedule of IFN-α that induced a locoregional expansion of the draining inguinal lymph nodes and improved cellular cytotoxicity (natural killer and CD8+) and antigen presentation. Suppression of the vaccinia virus was avoided by administering IFN-α distal to the site of vaccination. The combination of IFN-α and vaccine inhibited tumor growth, improved survival, and elicited CEA-specific CTL responses in mice with CEA+ adenocarcinomas. In mice with pancreatic tumors, IFN-α slowed tumor growth, induced CTL activity, and increased CD8+ tumor-infiltrating lymphocytes.
Conclusions
These data suggest that IFN-α can be used as a biological response modifier with antigen-directed poxvirus vaccines to yield significant therapeutic antitumor immune responses. This study provides the rationale and mechanistic insights to support a clinical trial of this immunotherapeutic strategy in patients with CEA-expressing carcinomas.
Tumor cells, through either malignant transformation or immunoediting, develop adaptations to avoid immune surveillance within their immunocompetent hosts including down-regulation of MHC class I expression and other cellular components of the antigen presentation machinery (1). The loss of MHC class I expression, an observation reported in a wide range of carcinomas (2, 3), is inversely correlated with the degree of tumor differentiation (4) and positively correlated with metastatic potential (1, 5–7). To improve the efficacy of therapeutic cancer vaccines, combinatorial strategies are necessary not only to elicit robust antigen-specific immune responses but also to render tumors more susceptible to CTL-mediated tumor cell lysis.
The type I IFNs (IFN-α and IFN-β) show clinical efficacy and are Food and Drug Administration approved for the treatment of chronic hepatitis B and C, AIDS-related Kaposi's sarcoma, malignant melanoma, follicular lymphoma, chronic myeloid leukemia, and hairy cell leukemia. Although initially identified for their antiviral properties (8), the type I IFNs are pleiotropic cytokines that bridge innate and adaptive immunity. The type I IFNs possess direct antitumor and immunomodulatory properties that may improve the efficacy of antitumor vaccines by several reported mechanisms (9, 10): (a) inhibition of tumor cell growth, (b) suppression of tumor angiogenesis, (c) improved natural killer (NK) cell activity, (d) maturation of dendritic cells and cross-priming of CD8+ T-cell responses, (e) enhanced humoral immunity, (f) up-regulation of MHC expression, and (g) enhanced expression of tumor associated antigens such as carcinoembryonic antigen (CEA).
The use of genetically modified tumor cells expressing IFN-α has yielded significant antitumor activity (11, 12) even in IFN-resistant tumor cells (13). In related studies, IFN-α was shown to significantly improve the efficacy of granulocyte/macrophage-colony stimulating factor (GM-CSF)–secreting cellular vaccines (14, 15). Moreover, IFN-α has been used successfully as a vaccine adjuvant in studies using soluble protein (16), peptides (17), DNA plasmids (18), and whole tumor cell preparations (19, 20).
Translational Relevance
Tumor cells avoid immune surveillance by several mechanisms including down-regulation of the MHC, an observation that is inversely correlated with the degree of tumor differentiation and positively correlated with metastatic potential. This research uses IFN-α in a multimodal strategy to improve the efficacy of therapeutic cancer vaccines and render tumors more susceptible to immune cell recognition and lysis. IFN-α is a Food and Drug Administration – approved cytokine reported to up-regulate expression of the carcinoembryonic antigen (CEA) and MHC on tumor cells. The poxvirus vaccines, vaccinia and fowlpox, are novel vaccine platforms that have been used safely and effectively to elicit antigen-specific immune responses in cancer patients. This research addresses the need for combination strategies in clinical cancer immunotherapy by providing phenotypic, functional, and mechanistic data supporting the use of IFN-α as an immune adjuvant in conjunction with CEA-directed poxvirus vaccines in patients with CEA-expressing carcinomas.
Our laboratory has previously reported that the use of a diversified prime [recombinant vaccinia (rV)] and boost [recombinant fowlpox (rF)] vaccination strategy is capable of inducing CEA-specific T-cell–mediated antitumor responses (21). The insertion of three T-cell costimulatory molecules (B7-1, intercellular adhesion molecule-1, and LFA-3, termed TRICOM) into these viral vectors significantly improved B-cell–and T-cell (CD4+ and CD8+)–mediated responses against CEA without inducing autoimmunity (22, 23). The recombinant fowlpox virus has also been used to express GM-CSF at the site of vaccination, thereby enriching the regional lymph nodes with professional antigen-presenting cells and further enhancing antigen-specific immune responses in mice (24, 25). This vaccine strategy has been successfully translated into clinical trials of patients with CEA-positive carcinomas (26, 27) resulting in the isolation of CD8+ MHC-restricted CTLs that are capable of lysing autologous tumors expressing CEA (28, 29).
IFN-α is effective at suppressing viral replication in vaccinia-infected mice (30) and therefore may not be a compatible adjuvant with poxvirus vaccines. Thus, the aim of this study was to evaluate the use of IFN-α in combination with CEA-directed poxvirus vaccines in murine models of colorectal and pancreatic adenocarcinomas. These studies show for the first time that (a) suppression of the vaccinia virus can be avoided by administering IFN-α distal to the site of vaccination, and (b) the combination of IFN-α and poxvirus vaccines can yield significant CEA-specific immune and antitumor responses in mice with CEA+ adenocarcinomas.
Materials and Methods
Tumor cell lines and cell culture conditions
The murine pancreatic adenocarcinoma cell line Panc02 was generously provided by Dr. Michael A. Hollingsworth (University of Nebraska Medical Center, Omaha, NE). The Panc02 cell line was established through the induction of pancreatic tumors with 3-methyl-cholanthrene and serial subcutaneous transplantation in C57BL/6 mice (31). The MC38 murine colon carcinoma cell line was supplied by Dr. Steven Rosenberg (National Cancer Institute, Bethesda, MD). The CEA-expressing Panc02 cells (Panc02.CEA) and MC38 cells (MC38.CEA) were produced by transducing the human CEA gene using the retroviral expression vector pBNC as previously described (32).
Functional assays for lymphocytes were carried out in RPMI 1640 containing 15 mmol/L HEPES (pH 7.4), 10% heat-inactivated fetal bovine serum, 2 mmol/L l-glutamine, 0.1 mmol/L nonessential amino acids, 1 mmol/L sodium pyruvate, 50 units/mL G418 sulfate, and 50 μmol/L β-mercaptoethanol. All cells were maintained in a humidified incubator at 37°C in a 5% CO2-95% air atmosphere. Unless otherwise indicated, all media and their components were purchased from Mediatech, Inc.
Animal care and use
All mice were housed in microisolator cages under pathogen-free conditions in accordance with the procedures outlined in The Guide for Care and Use of Laboratory Animals (National Research Council). Female C57BL/6 mice (H-2b) were purchased from Taconic Farms. Mice expressing the transgene for human CEA (CEA.Tg) were generously provided by Dr. John Shively (City of Hope, Duarte, CA). The mice were originally generated by microinjecting a 32.6-kb AatII restriction fragment containing the entire human CEA genomic region into a pronucleus of C57BL/6 zygotes (33). Homozygosity for CEA expression was tested and verified using PCR analysis of DNA isolated from the tails of progeny mice (34).
Vaccines, adjuvants, and injection schema
Recombinant murine IFN-α was purchased from PBL Biomedical Laboratories and contained <0.1 unit/μg of endotoxin. Consistent with its clinical use, IFN-α was administered in our studies s.c. thrice a week (MWF). Recombinant vaccinia (rV) and recombinant fowlpox (rF) vectors that express the transgenes for CEA and a triad of T-cell costimulatory molecules (TRICOM) were administered on the right inner thigh (s.c., ventral surface) at 108 plaque-forming units (pfu) admixed with 107 pfu of the recombinant fowlpox virus expressing murine GM-CSF as previously described (21, 24, 25, 35). In all immune and antitumor studies in which IFN-α was combined with the poxvirus vaccines, a 1-wk course of IFN-α (50,000 IU/injections, 3× a week, MWF) was initiated on the same day as vaccination with either vaccinia or fowlpox (Monday). As depicted in Supplementary Fig. S1, locally administered IFN-α was given at the site of vaccination (s.c., right, ventral surface) whereas distally administered IFN-α was given on the left side of the lower back (s.c., left, dorsal surface).
In the antitumor studies, 6- to 8-wk-old CEA.Tg mice were injected on the lower back (s.c., right, dorsal surface) with either 3 × 105 MC38.CEA or 1 × 106 Panc02.CEA tumor cells in 100 μL of HBSS (Supplementary Fig. S1). When tumors reached 0.3 to 0.6 cm3 (1 and 3 wk after tumor challenge for MC38.CEA and Panc02.CEA tumors, respectively), mice were randomized to one of six treatment groups: (a) untreated (controls), (b) non-CEA vaccine (108 pfu rV/F-TRICOM + 107 pfu rF-GM-CSF), (c) CEA vaccine (108 pfu rV/F-CEA-TRICOM + 107 pfu rF-GM-CSF), (d) IFN-α (50,000 IU/mouse, 3× a week, alternating weeks), (e) IFN-α + non-CEA vaccine, and (f) IFN-α + CEA vaccine. Tumor volumes were measured twice per week with digital calipers. The experimental end point for survival was defined as the time point at which the tumor volume reached 2 cm3, whereupon the mice were euthanized.
Phenotypic analyses
Flow cytometry was used in four experiments to conduct phenotypic analyses. In the first experiment, inguinal lymph node cells were isolated from mice 24 h after the last injection of IFN-α (0-50,000 IU/mouse, three injections). The lymph node cells were stained with conjugated antibodies against CD3, CD4, CD8, CD25, CD11c, I-Ab, CD19, and NK1.1. In the second experiment, proliferating cultures of MC38.CEA and Panc02.CEA cells were treated with titrating doses of IFN-α (0-1,000 IU/mL) for 48 h. Following treatment, the cells were stained for H-2Kb, H-2Db, CD80 (B7-1), and CD66 (CEA). In the third experiment, the in vivo tumor cell surface phenotype was studied in mice following a 2-wk course of IFN-α (50,000 IU/mouse, 3× a week). The mice were sacrificed and their tumors were prepared and stained for expression of H-2Kb, H-2Db, CD80 (B7-1), and CD66 (CEA). In the fourth experiment, the spleens, inguinal lymph nodes, and tumor infiltrates of mice with Panc02.CEA tumors were analyzed for CD3, CD4, CD8, NK1.1, Gr-1, CD25, and I-Ab expression following a 2-wk course of IFN-α (50,000 IU/mouse, 3× a week). Intracellular expression of FoxP3, IFN-γ, and granzyme B was measured using a BD Cytofix/Cytoperm Fixation/Permeabilization kit following the manufacturer's recommendations (Becton Dickinson). In all in vivo experiments, the mice were euthanized 24 h after the last IFN-α treatment. All samples (106 cells per tube) were incubated with 1 μg of unlabeled antimouse CD16/CD32 to block nonspecific binding to Fcγ III/II receptors. Isotype control antibodies were matched for each fluorochrome and used to set gate cursors. All flow cytometry experiments were done using a BD LSR II (Becton Dickinson). Data are expressed as the percentage and number of cells positive for each marker and its mean fluorescence intensity.
Mixed lymphocyte culture
Draining inguinal lymph node cells from C57BL/6 mice were isolated 24 h after the last injection of IFN-α (50,000 IU/mouse, three injections). The lymph node cells were irradiated and cultured with splenic T cells isolated from naïve BALB/c mice at a ratio of 1:5 (1 BALB/c T cell:5 C57BL/6 lymph node cells) in 10 mL for 5 d at 37°C. Viable BALB/c lymphocytes were recovered by centrifugation over lymphocyte separation media following the manufacturer's recommendations (BioWhittaker) and used in alloreactive CTL assays.
β-Galactosidase production assay
In vitro, the cellular production of β-galactosidase (β-gal) protein was measured in MC38 cells following infection with rV-LacZ (0.01 multiplicity of infection) in the presence of increasing concentrations of IFN-α (0-100 IU/mL). After 24 h, MC-38 cells were lysed [lysis buffer contained 100 mmol/L Tris (pH 8.0), 100 mmol/L NaCl, and 0.5% NP40] and whole cell extracts were prepared. In vivo, C57BL/6 mice were vaccinated i.m. with rV-LacZ (108 pfu) in both quadriceps followed by administration of either vehicle or IFN-α (50,000 IU/mouse) for 3 consecutive days at the site of vaccination. On day 3, the mice were euthanized and whole cell extracts were prepared from the quadriceps muscles. In both experiments, the production of β-gal protein was measured by ELISA according to the manufacturer's recommendations (Roche Molecular Biochemicals).
Lymphoproliferation assays
To measure alloreactive lymphoproliferative responses, splenocytes from naïve BALB/c mice were enriched for T lymphocytes using magnetic murine pan B (B220) Dynabeads (Invitrogen Dynal AS) per manufacturer's recommendations. Naïve BALB/c T cells (2 × 105 per well) were incubated with irradiated lymph node cells (0-1 × 106 per well) from IFN-α–treated C57BL/6 mice for 4 d at 37°C in flat-bottomed 96-well plates. The cultures were pulsed on day 3 with 1 μCi/well of [3H]thymidine (Amersham Corp.).
In vaccinated mice, antigen-specific proliferation assays were initiated 2 to 3 wk following the booster vaccination and performed in a cell culture incubator at 37°C as described previously (25). Splenic CD4+ lymphocytes from each treatment group were stimulated for 5 d with CEA (3.13-50 μg/mL), β-gal (6.25-100 μg/mL), or ovalbumin (50 or 100 μg/mL) as a negative control. For positive controls, CD4+ lymphocytes (2 × 105 per well) were incubated in flat-bottomed 96-well plates with concanavalin A (0.25-2 μg/mL; Sigma Chemical Co.) for 3 d. All cultures were pulsed with 1 μCi/well of [3H]thymidine (Amersham Biosciences) for the last 18 h of incubation. Cultures were harvested onto glass fiber filter mats (Wallac, Inc.) using a multiple automated sample harvester (Tomtec), and radioactivity was determined by liquid scintillation on a Perkin-Elmer 1450 microbeta plate reader. Results from individual mice in triplicate wells were combined to yield a mean ± SE for each immunization group.
Cytotoxicity assays
All CTL responses were measured using either 4-h 51Cr or overnight 111In release assays as previously described (25, 34, 36). Allospecific CTL responses were evaluated by incubating BALB/c T cells, isolated following a 5-d mixed lymphocyte culture, with 111In-labeled MC38 (H-2b) or P815 (H-2d) targets. To measure antigen-specific CTL responses in vaccinated mice, splenocytes from each mouse (25 × 106) were cultured in an upright T-25 flask containing 10 μg/mL of either CEA526-533 (EAQNTTYL) peptide or β-gal96-103 (DAPIYTNV) peptide (CPC Scientific). After 1 wk, lymphocytes were collected on a Histopaque (Sigma-Aldrich) density gradient and quantified. Panc02.CEA target cells were pretreated with IFN-α (2 × 103 IU/mL, 48 h, 37°C) before the CTL assay. On the day of the assay, target cells (4 × 106) were incubated with 50 μCi of 111In-labeled oxine (GE Healthcare) for 30 min at 37°C. Target cells were washed twice and pulsed with 20 μg/mL of CEA526-533 peptide, β-gal96-103 peptide, or vesicular stomatitis virus nucleoprotein VSV-NP52-59 (RGYVYQGL) control peptide (CPC Scientific) for 30 min at 37°C. Tumor cells (2,500 per well) were coincubated with 3.13 × 104 to 5 × 105 lymphocytes for 18 h at 37°C. The final peptide concentrations in the CTL assay were 10 μg/mL. The amount of 111In released was measured using a gamma counter (Cobra II, Packard Instruments). The percentage of specific lysis was calculated as follows: % specific lysis = [(experimental cpm - spontaneous cpm) / (maximal cpm - spontaneous cpm)] × 100.
NK cell cytotoxicity was assessed in standard 4-h chromium release assay as previously described (37). Experiments were done using 12.5:1 to 200:1 effector-to-target (E/T) ratios and 51Cr-labeled yeast artificial chromosome-1 target cells. All experiments were done in triplicate.
Antibody-based immune cell depletion studies
Antibody-based immune cell depletion studies were carried out in tumor-bearing mice to deplete CD4+ (rat antibody GK1.5), CD8+ (rat antibody 2.43), CD4+ and CD8+, or NK1.1+ (PK136) cells before receiving rV/F-CEA-TRICOM vaccination. Mice were challenged with Panc02.CEA tumors (106 cells per mouse) on day 0. Antibodies were injected i.p. on days 24 to 27, 34, and 41. Mice received a primary vaccination with rV-CEA-TRICOM and rF-GM-CSF on day 28 and a booster vaccination with rF-CEA-TRICOM and rF-GM-CSF on day 42 after tumor challenge. Tumor growth and survival were monitored for up to 100 d following tumor challenge.
Serum cytokine assays
In tumor-bearing mice treated with either vehicle or IFN-α, serum was collected at sacrifice. ELISA assays were done to measure the serum concentrations of vascular endothelial growth factor and transforming growth factor β1 per manufacturer's recommendations (R&D Systems).
Measurement of effector cytokine production
Intracellular cytokine production of IFN-γ+ and granzyme B+, as a percentage of CD8+ T cells or NK1.1+ cells, was assessed in mice with established Panc02.CEA tumors receiving either no treatment or IFN-α (50,000 IU, 3× a week for 2 wk; n = 4 mice per group). Following 2 wk of immunotherapy with IFN-α, the mice were sacrificed and single-cell suspensions were prepared from the tumor mass of each animal. Following the manufacturer's recommendations, tumor samples (106 cells/mL) were stimulated for 5 h with a leukocyte activation cocktail containing phorbol 12-myristate 13-acetate, ionomycin, and brefeldin A (Becton Dickinson).
Statistical analyses
Differences between treatment groups were determined using ANOVA and Tukey's multiple comparisons test. Survival estimates were generated using Kaplan-Meier plots and the log-rank test. All analyses were conducted using the GraphPad Prism software package (Prism 4 for Windows, version 4.03, GraphPad Software, Inc.) and statistical significance was accepted at the P < 0.05 level.
Results
IFN-α induces phenotypic and functional changes in draining lymph node cells
Titrating doses of IFN-α (0-50,000 IU/mouse × 3 injections) resulted in significant phenotypic and functional changes in the draining inguinal lymph node cells. IFN-α induced a dose-dependent cellular expansion in the total lymph node cells (Fig. 1A; P < 0.0001). The highest dose of IFN-α (50,000 IU/mouse, three injections) significantly elevated antigen presentation function as measured by an allospecific lymphoproliferation assay (Fig. 1B; P < 0.0001). Moreover, IFN-α treatment dose-dependently improved T-cell (Fig. 1C; P < 0.0001) and NK cell (Fig. 1D; P = 0.0002) cytotoxicity. The cellular expansion induced by IFN-α was locoregional because the draining inguinal lymph nodes were expanded by 60% over the contralateral inguinal lymph nodes of the same mice (Supplementary Fig. S2A; P = 0.03). Finally, this IFN-induced cellular expansion was composed of many cell populations including CD19+ B lymphocytes (Supplementary Fig. S2B; P < 0.0001), CD3+ T lymphocytes (Supplementary Fig. S2C; P < 0.0001), CD11c+/I-Ab+ dendritic cells (Supplementary Fig. S2D; P < 0.0001), and NK1.1+ cells (Supplementary Fig. S2E; P < 0.0001).
Fig. 1.
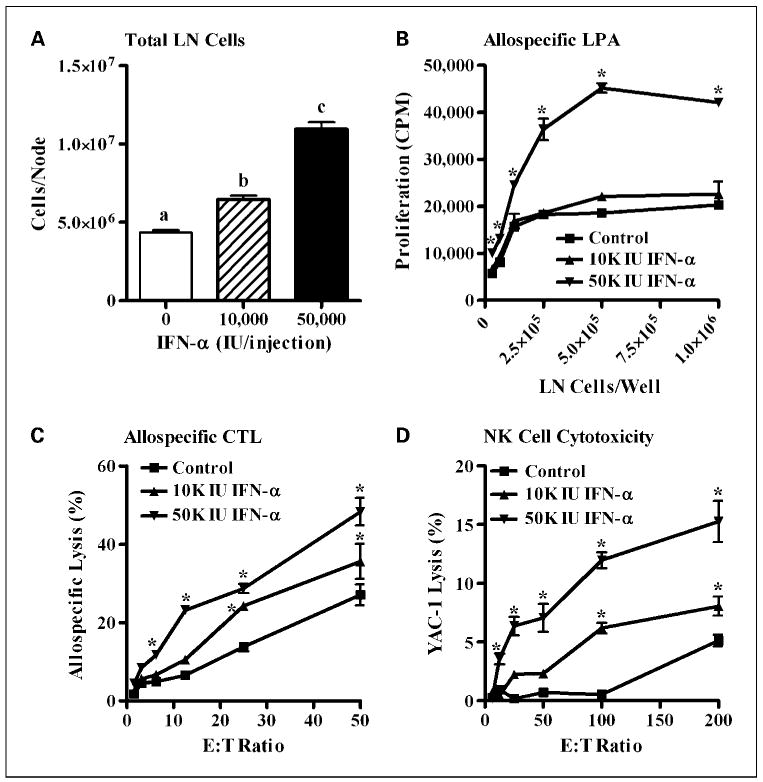
Effect of IFN-α administration on lymph node phenotype, antigen presentation, and cytotoxicity. Twenty-four hours following the last of three injections of IFN-α (0, 10,000, or 50,000 IU), phenotypic and functional changes in the inguinal lymph node cells were evaluated by flow cytometry and in vitro T lymphocyte and NK cell cytotoxicity assays (n = 3 mice per group). A, dose-dependent phenotypic responses in the draining inguinal lymph node (LN) following administration of IFN-α at 0 IU (open columns), 10,000 IU (hatched columns), and 50,000 IU (solid columns) are shown as the average number of lymph node cells per mouse. B, lymph node cells isolated from mice treated with 50,000 IU of IFN-α showed functional improvements in antigen presentation in an allospecific lymphoproliferation assay (LPA). C, IFN-α treatment induced dose-dependent improvements in T-cell–mediated lysis of 111In-labeled MC38 tumor cells in an alloreactive CTL assay using draining inguinal lymph node cells. D, we observed dose-dependent improvements in NK cell – mediated lysis of 51Cr-labeled yeast artificial chromosome-1 (YAC-1) target cells in a 4-h chromium release assay.
To evaluate the efficacy of IFN-α as an adjuvant to protein-based vaccination, we measured immune responses in mice vaccinated with β-gal protein (100 μg, s.c.) in the presence or absence of IFN-α (50,000 IU/mouse, three injections) at the site of vaccination (Supplementary Fig. S3). Coadministration of β-gal and IFN-α increased CD4+ proliferative responses ∼3-fold (1,500 versus 4,500 cpm/2 × 105 CD4+ cells; P < 0.0001). Total IgG serum antibody titers were also increased 3- to 4-fold in mice vaccinated with β-gal and IFN-α, compared with those vaccinated with β-gal alone (P < 0.001).
IFN-α differentially modulates poxvirus-based vaccines
Given the inherent role of type I IFNs in early antiviral defense (38, 39), we were intrigued about whether the observed enrichment of the regional lymph nodes draining the site of IFN-α injection could be exploited to improve host immune responses to poxvirus-based vaccines. A series of experiments was designed to optimize the administration of IFN-α with recombinant vaccinia and fowlpox vaccines. Antigen-specific proliferative and cytotoxic responses were measured in mice vaccinated with either rV-LacZ-TRICOM or rF-LacZ-TRICOM. In rF-LacZ-TRICOM–vaccinated mice, the administration of IFN-α (50,000 IU/mouse, three injections), whether local or distal to the site of vaccination, significantly improved β-gal–specific CD4+ T-cell proliferative responses (Fig. 2A; P < 0.001). The local administration of IFN-α, at the site of rF-LacZ-TRICOM vaccination, significantly improved β-gal–specific CD8+-mediated tumor cell lysis (P < 0.001), whereas distal administration of IFN-α did not alter CTL activity in this system (Fig. 2B). When administered at the site of rV-LacZ-TRICOM vaccination, IFN-α significantly inhibited β-gal–specific proliferative responses (Fig. 2C; P < 0.001) and CTL activity (Fig. 2D; P < 0.001). Distal administration of IFN-α in rV-LacZ-TRICOM–vaccinated mice significantly improved β-gal–specific proliferation (Fig. 2C; P < 0.001) but did not alter CTL activity (Fig. 2D). All controls for these experiments, concanavalin A–induced T-cell proliferation or VSV-NP–specific CTL activity, were normal (Supplementary Fig. S4A-D).
Fig. 2.
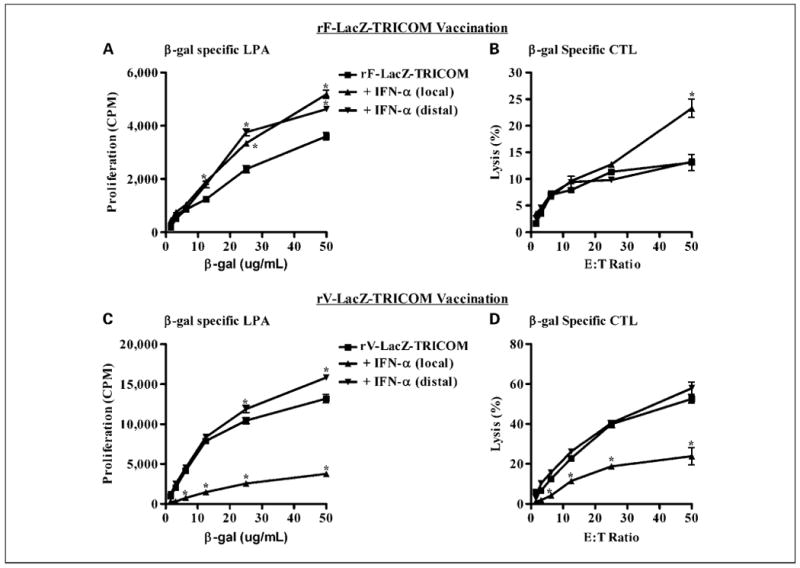
Effect of IFN-α on recombinant vaccinia and fowlpox vaccinations. The route of IFN-α administration and its effect on poxvirus vaccine responses were evaluated in mice receiving vaccine alone (■) or vaccine in combination with IFN-α given either locally (▲; right ventral surface) or distally (▼; left dorsal surface) to the site of vaccination. In rF-LacZ-TRICOM–vaccinated mice, the local administration of IFN-α significantly improved β-gal – specific CD4+ T-cell proliferative responses (A) and CD8+-mediated lysis of MC38 tumor cells pulsed with β-gal96-103 (DAPIYTNV) peptide (B). In rV-LacZ-TRICOM–vaccinated mice, the local but not distal administration of IFN-α significantly inhibited β-gal – specific CD4+ T-cell proliferation (C) and CTL responses (D). Concanavalin A – induced T-cell proliferation was normal in all experiments (Supplementary Fig. S4A and C). Negligible CTL activity was noted in MC38 tumor cells pulsed with the VSV-NP52-59 (RGYVYQGL) control peptide (Supplementary Fig. S4B and D). In each experiment, a 1-wk course of IFN-α (50,000 IU/mouse, 3× a week, MWF) was initiated on the day of vaccination with either vaccinia or fowlpox (Monday). The immune data presented here were generated from the pooled splenocytes of three mice per group and the experiments were conducted thrice with consistent findings.
The role of IFN-α–mediated inhibition of replication and/or infectivity by the vaccinia virus was evaluated in vitro and in vivo. Recombinant IFN-α (0-100 IU) was shown to dose-dependently inhibit the production of β-gal protein in cultured MC38 cells infected with 0.01 multiplicity of infection of the rV-based LacZ vaccine (Supplementary Fig. S5A; P < 0.01). Likewise, the i.m. coadministration of rV-based LacZ vaccine (108 pfu) and IFN-α (50,000 IU, 3×) in C57BL/6 mice resulted in significant inhibition of β-gal protein production at the site of vaccination (Supplementary Fig. S5B; P < 0.009). No such inhibition was observed when the fowlpox-based LacZ construct was coinjected with IFN-α (data not shown).
IFN-α alters the tumor cell surface phenotype
Although the baseline cell surface phenotype of MHC class I (H-2Kb and H-2Db) and CD80 (B7-1) expression on MC38.CEA and Panc02.CEA cells are quite different, in vitro titration of IFN-α (0-1,000 IU/mL, 48 hours) dose-dependently up-regulated MHC class I expression to a similar degree (Table 1A). In vivo, the mean fluorescence intensity for MHC class I on MC38.CEA tumor cells increased substantially in mice treated with IFN-α (50,000 IU/mouse, 3× a week for 2 wk; Table 1B). The same dose and schedule of IFN-α, in vivo, up-regulated the percentage of MHC class I–positive cells on the surface of Panc02.CEA tumors (Table 1B).
Table 1.
Up-regulation of tumor cell surface MHC class I expression by IFN-α
| (A) In vitro cell surface antigen expression | |||||
|---|---|---|---|---|---|
| Cell Line | IFN-α (IU/mL)* | % Positive (MFI) † | |||
| MHC class I (H-2Kb) | MHC class I (H-2Db) | CD80 (B7-1) | CD66 (CEA) | ||
| MC38.CEA | 0 | 57.5 (36.5) | 40.5 (33.0) | Neg | 94.6 (139.8) |
| 1 | 99.8 (168.6) | 99.8 (161.8) | Neg | 93.9 (131.0) | |
| 10 | 99.8 (200.5) | 99.7 (179.8) | Neg | 95.0 (145.1) | |
| 100 | 99.8 (205.5) | 99.4 (183.1) | Neg | 94.7 (177.4) | |
| Panc02.CEA | 0 | Neg | Neg | 40.3 (1,687) | 80.6 (5,093) |
| 1 | Neg | Neg | 40.9 (975) | 82.0 (4,764) | |
| 10 | 42.8 (527) | 54.7 (715) | 45.4 (952) | 80.9 (6,413) | |
| 100 | 70.1 (516) | 76.5 (896) | 48.1 (1,224) | 80.0 (6,806) | |
| 1,000 | 89.6 (918) | 89.9 (1,306) | 41.7 (1,391) | 77.9 (7,299) | |
| (B) In vivo cell surface antigen expression | |||||
| Tumor | IFN-α (IU) ‡ | % Positive (MFI) | |||
| MHC class I (H-2Kb) | MHC class I (H-2Db) | ||||
| MC38.CEA | 0 | 94.2 (133.5) | 94.5 (110.4) | ||
| 50,000 | 96.4 (335.2) | 96.0 (270.3) | |||
| Panc02.CEA | 0 | 23.1 (1,010) | 18.7 (270) | ||
| 50,000 | 47.7 (1,058) | 43.4 (267) | |||
NOTE: The effect of IFN-α on cell surface MHC class I expression was evaluated in murine pancreatic and colorectal cancer cells. In vitro titration of IFN-α (0-1,000 IU/mL) was shown to dose-dependently up-regulate the percentage of cells positive for MHC class I (H-2Kb and H-2Db) in MC38.CEA and Panc02.CEA tumor cells (A). As shown in A, Panc02.CEA, but not MC38.CEA, tumor cells constitutively express the T-cell costimulatory molecule CD80 (B7-1). In mice with established subcutaneous tumors, IFN-α treatment (50,000 IU/injection, 3× a week, 2 wk) up-regulated MHC class I (H-2Kb and H-2Db) mean fluorescence intensity on MC38.CEA tumors and the percentage of MHC class I–positive cells in Panc02.CEA tumors (B).
Abbreviations: MFI, mean fluorescence intensity; Neg, negative.
Each cell line was pretreated at the indicated dose of murine IFN-α for 48 h.
I-Ab, CD54 (intercellular adhesion molecule-1), CD48 (muLFA-3), and CD40 were evaluated but did not respond to titrating doses of IFN-α in the cell lines tested.
Mice received six injections (every other day) of either PBS or the indicated dose of IFN-α. The last injections were given 24 h before analysis.
The combination of IFN-α and rV/F-CEA-TRICOM yields significant antitumor activity
Following 21 days of treatment, the growth of MC38.CEA tumors was significantly delayed only in mice receiving the combination of IFN-α and rV/F-CEA-TRICOM (Fig. 3B; P < 0.001). This treatment combination conferred a significant survival benefit over all other treatment groups (Fig. 3C; P = 0.01). The antitumor efficacy of IFN-α in combination with rV/F-CEA-TRICOM vaccination was CEA dependent because the combination of IFN-α and rV/F-TRICOM (non-CEA vaccine) did not alter the growth of MC38.CEA tumors (Fig. 3A) or improve survival outcomes relative to untreated controls (Fig. 3C). In a subset analysis (n = 4 mice per group) of CTL activity in tumor-bearing mice, only mice vaccinated with rV/F-CEA-TRICOM or the combination of IFN-α and rV/F-CEA-TRICOM generated significant CEA526-533–specific CTL activity against MC38.CEA targets (P < 0.05; Fig. 3D).
Fig. 3.
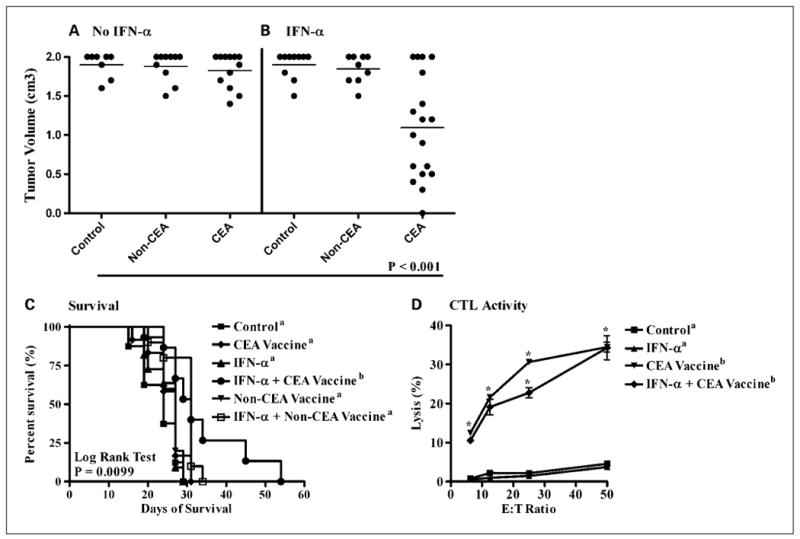
The antitumor efficacy of IFN-α combined with rV/F-CEA-TRICOM vaccination against subcutaneous murine colorectal adenocarcinomas. Mice with established MC38.CEA tumors were randomized into one of six treatment groups (n = 10 mice per group): untreated controls (■); rV/F-CEA-TRICOM + rF-GM-CSF (◆, CEA vaccine); IFN-α (▲, 50,000 IU/mouse, 3× a week, alternating weeks); IFN-α and rV/F-CEA-TRICOM + rF-GM-CSF (●, IFN-α + CEA vaccine); rV/F-TRICOM + rF-GM-CSF (▼, non-CEA vaccine); and IFN-α and rV/F-TRICOM + rF-GM-CSF (□, IFN-α + non-CEA vaccine). A and B, the individual and average tumor volumes in each group are shown following 21 d of treatment. The growth of MC38.CEA tumors was significantly delayed only in mice receiving the combination of IFN-α and the CEA vaccine (B). The combination conferred a significant survival benefit over all other treatment groups (C). In a subset analysis (n = 4 mice per group) of CTL activity in tumor-bearing mice, only mice vaccinated with rV/F-CEA-TRICOM or the combination of IFN-α and rV/F-CEA-TRICOM generated significant CEA-specific CTL activity against MC38.CEA targets (D). Statistically significant differences in the mean tumor volume and percent lysis of each treatment group were compared using ANOVA and Tukey's multiple comparison test. Survival estimates were generated using Kaplan-Meier plots and the log-rank test. Statistical significance was accepted at P < 0.05 and is indicated by superscript letters.
In mice with Panc02.CEA tumors, significant antitumor activity and improved survival were observed with rV/F-CEA-TRICOM vaccination (Fig. 4A and C; P < 0.001), IFN-α treatment (Fig. 4B and C; P < 0.001), and the combination of IFN-α and rV/F-CEA-TRICOM vaccination (Fig. 4B and C; P < 0.001). The antitumor efficacy of the vaccine was CEA specific because rV/F-TRICOM (non-CEA) vaccination had no effect on Panc02.CEA tumor growth (Fig. 4A) or survival (Fig. 4C) relative to untreated controls. In antibody-based immune cell depletion studies, nonsignificant trends for reduced survival were observed in mice depleted of CD4+/CD8+ T cells or NK1.1+ cells (Supplementary Fig. S6A). The median survival of mice receiving the CEA vaccine was 52 days after tumor challenge whereas the median survival in vaccinated mice depleted of CD4+/CD8+ T cells or NK1.1+ cells was 42 days (P = 0.07) and 45.5 days (P = 0.27), respectively. Actuarial survival at 60 days after tumor challenge was 43% (vaccine only), 33% (vaccine + CD4-), 33% (vaccine + CD8-), 0% (vaccine + CD4-/CD8-), and 0% (vaccine + NK1.1-). Significant CEA526-533–specific CTL lysis of Panc02.CEA targets was observed in mice receiving IFN-α, rV/F-CEA-TRICOM, and the combination of IFN-α and rV/F-CEA-TRICOM (Fig. 4D; P < 0.05). Antibody blockade of B7-1 resulted in a statistically significant decline in Panc02.CEA tumor cell lysis by a CEA526-533–specific CD8+ T-cell line (Supplementary Fig. S6B).
Fig. 4.
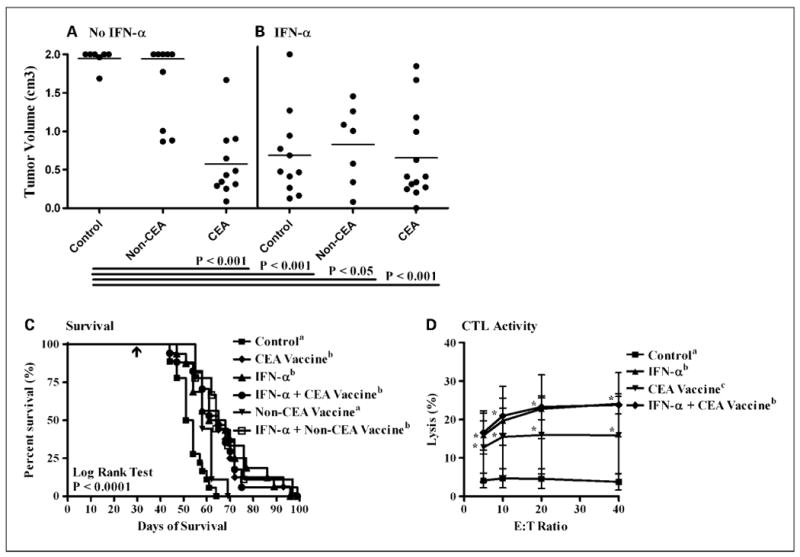
The antitumor efficacy of IFN-α combined with rV/F-CEA-TRICOM vaccination against subcutaneous murine pancreatic adenocarcinomas. Mice with established Panc02.CEA tumors were randomized into one of six treatment groups (n = 10 mice per group): untreated controls (■); rV/F-CEA-TRICOM + rF-GM-CSF (◆, CEA vaccine); IFN-α (▲, 50,000 IU/mouse, 3× a week, alternating weeks); IFN-α and rV/F-CEA-TRICOM + rF-GM-CSF (●, IFN-α + CEA vaccine); rV/F-TRICOM + rF-GM-CSF (▼, non-CEA vaccine); and IFN-α and rV/F-TRICOM + rF-GM-CSF (□, IFN-α + non-CEA vaccine). In mice with Panc02.CEA tumors, significant antitumor efficacy and improved survival were observed with rV/F-CEA-TRICOM vaccination (A and C), IFN-α monotherapy (B and C), and the combination of IFN-α and rV/F-CEA-TRICOM vaccination (B and C). C, arrow, initiation of treatment. Significant CEA-specific CTL lysis of Panc02.CEA targets was observed in mice receiving IFN-α, rV/F-CEA-TRICOM, and the combination of IFN-α and rV/F-CEA-TRICOM (D). Statistically significant differences in the mean tumor volume and percent lysis of each treatment group were compared using ANOVA and Tukey's multiple comparison test. Survival estimates were generated using Kaplan-Meier plots and the log-rank test. Statistical significance was accepted at P < 0.05 and is indicated by superscript letters.
To better understand the mechanism(s) by which IFN-α therapy inhibited Panc02.CEA tumor growth, we treated mice (n = 6 mice per group) for 2 wk with IFN-α (50,000 IU, 3× a week) and then examined their tumor infiltrates (Fig. 5A-D) and lymphoid organs (Supplementary Fig. S7A and B) using six-color flow cytometry and measured serum cytokine levels by ELISA (Supplementary Fig. S7C and D). When compared with untreated controls, IFN-α treatment significantly suppressed tumor growth (Fig. 5A; P = 0.03) and increased the percentage of CD8+ (Fig. 5B, 6.7% versus 11.2%; P = 0.03), NK1.1+ (Fig. 5C, 1.8% versus 3.2%; P = 0.03), and Gr-1+ (Fig. 5D, 2.4% versus 4.5%; P = 0.03) tumor infiltrates. IFN-α treatment did not alter the percentage of CD4+ tumor infiltrates (data not shown). Nonsignificant trends were noted for normalization of the spleen size (Supplementary Fig. S7A; P = 0.22) and expansion of the draining inguinal lymph nodes (Supplementary Fig. S7B; P = 0.25) in these tumor-bearing mice. Treatment with IFN-α significantly reduced serum vascular endothelial growth factor levels (Supplementary Fig. S7C; P = 0.03) whereas serum transforming growth factor-β1 concentrations were unchanged (Supplementary Fig. S7D; P = 0.44). To assess effector cytokine production by tumor infiltrates, tumor samples from control and IFN-α–treated mice were stimulated in vitro with a leukocyte activation cocktail. Intracellular IFN-γ and granzyme B expression was higher in the tumor-infiltrating CD8+ T cells (46.1% versus 55.7%; P < 0.05) and NK1.1+ cells (14.7% versus 25.8%; P < 0.05) of IFN-α–treated mice.
Fig. 5.
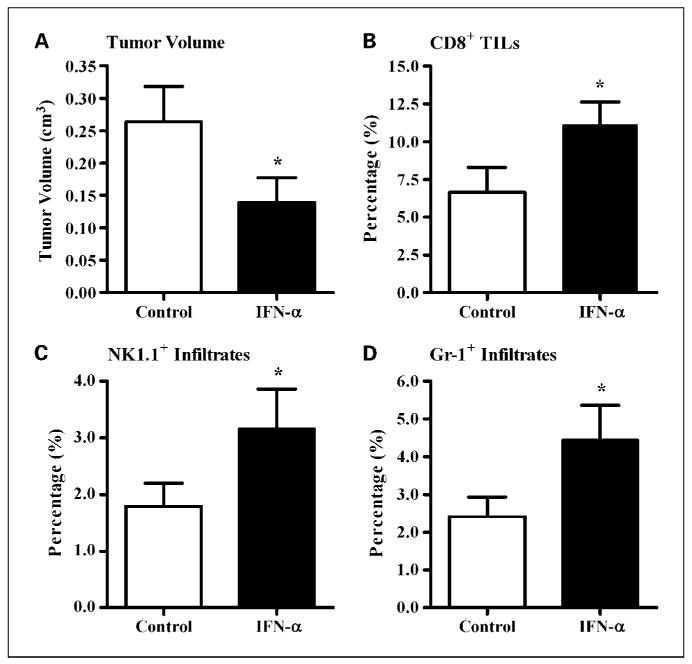
The antitumor activity of IFN-α in mice with established murine pancreatic adenocarcinomas. Mice with established Panc02.CEA tumors (n = 6 mice per group) were treated for 2 wk with either vehicle (open columns) or IFN-α (50,000 IU/mouse, 3× a week; solid columns). The mice were sacrificed 24 h after the last injection of IFN-α. The spleens, tumor-draining inguinal lymph nodes, tumors, and sera were isolated from each mouse. Single-cell suspensions of each tissue were prepared and six-color flow cytometry was used for the phenotypic analysis. IFN-α therapy significantly slowed the growth of Panc02.CEA tumors (A) and increased the percentage of CD8+ tumor-infiltrating lymphocytes (TILs; B), NK cells (NK1.1+; C), and granulocytes (Gr-1+; D). *, P < 0.05, versus the control group.
Discussion
Previous studies have enumerated the pleiotropic adjuvant actions of IFN-α including cellular enrichment of the draining regional lymph nodes as well as the ability to up-regulate antigen expression on tumor targets (9, 10, 40). The locoregional changes brought on by IFN-α can be argued to improve priming of the cellular host immune response, whereas the systemic distribution of IFN-α may improve the effector arm of host immunity by improving immune cell target recognition. In this study, we identified a dose and schedule of IFN-α (50,000 IU, 3× a week) that induced a locoregional expansion of the draining lymph nodes and functional improvements in antigen presentation and cellular cytotoxicity. These findings are consistent with previous reports about the adjuvant characteristics of IFN-α on dendritic cell (16, 41) and NK cell (42) functions.
Our studies show that inhibition of infectivity and/or replication of the vaccinia virus observed with coadministration of IFN-α can be avoided by injecting IFN-α at a site distal to the site of vaccination. These data suggest that the systemic effects of IFN-α allow for its continued use as a biological response modifier in antitumor studies (i.e., to up-regulate tumor MHC class I expression) by anatomically separating the administration of IFN-α from the site of primary vaccination with recombinant vaccinia. Fowlpox vectors, being replication defective and resistant to IFN-induced suppression of viral replication, yielded improved lymphoproliferative and CTL responses when coadministered with IFN-α at the site of booster vaccinations.
Based on these observations, we designed antitumor studies to evaluate the combination of poxvirus vaccines with the optimal dose, schedule, and site of IFN-α administration. In mice with MC38.CEA tumors, vaccination alone generated CEA526-533–specific CD8+ T-cell responses but was not sufficient to yield therapeutic antitumor responses. One interpretation of these findings is that IFN-α potentiated the action of the vaccine regimen in mice with MC38.CEA tumors through one or more of its immunomodulatory effects (activation of dendritic cells and NK cells or MHC up-regulation).
Our immune cell depletion studies in mice with Panc02.CEA tumors suggest that CD4+ and CD8+ T cells and NK1.1+ cells are important mediators of vaccine efficacy. These findings are consistent with the work of Sivinski et al. (43) who reported a significant role for these cell populations in generating antitumor immunity against parental Panc02 tumors. The absence of synergy between IFN-α and CEA vaccine in mice with Panc02.CEA tumors was surprising considering the efficacy of the individual treatments and the synergistic effects of this combination in mice with MC38.CEA tumors. One possible explanation is that in mice with Panc02.CEA tumors, IFN-α and CEA vaccine may operate through shared immunologic pathways such as the activation of CEA526-533–specific CTL activity.
The phenotypic differences in Panc02.CEA (MHC-I-/CD80+) and MC38.CEA (MHC-I+/CD80-) tumor cells allowed us to ask fundamental questions about immunosurveillance and response to immunotherapy in these two model systems. The functional consequences of these phenotypic differences may explain why IFN-α seems to activate adaptive immune responses in mice with Panc02.CEA but not MC38.CEA tumors. One might infer from our studies that the treatment of Panc02.CEA tumor–bearing mice with IFN-α induces CEA526-533–specific CTL activity and the accumulation of CD8+ tumor-infiltrating lymphocytes by up-regulating MHC class I expression (signal 1) in a tumor cell with constitutive CD80 expression (signal 2). The Panc02 model recapitulates some of the conditions reported by Hiroishi et al. (11, 12) in which MC38 cells transduced with either IFN-α or CD80 generated MC38-specific CTLs and long-lasting antitumor immunity. Our antibody blockade experiments support a role of CD80 expression in mediating antitumor immunity. Costimulation through CD80 is likely an important mediator of adaptive immunity in the Panc02 system because CD28-/- mice show markedly reduced survival following a challenge with the parental Panc02 cell line (43). The role of CD80 in mediating the interaction between tumor and the immune system is complex, however, because low CD80 expression is reported to contribute to tumor escape through preferential engagement of CD152 (CTLA-4) on T cells (44). The findings by our laboratory and others would suggest that a threshold may exist above which CD80 expression induces positive costimulation resulting in therapeutic responses to IFN-α therapy.
Our data also support a role for immune surveillance of Panc02 tumors by NK cells. Panc02.CEA tumors remained 50% to 60% MHC class I negative following IFN-α therapy and, thus, may also be more susceptible than MC38.CEA cells to recognition and lysis by NK cells. The activation of NK1.1+ cytotoxicity in tumor-free mice and the accumulation of NK1.1+ infiltrates in tumor-bearing mice would suggest that activation of the innate immune system is another mechanism by which IFN-α regulated Panc02.CEA tumor growth in our experiments. To summarize our findings in mice with Panc02.CEA tumors, IFN-α therapy was shown to stimulate CEA-specific CTL activity, increase CD8+ and NK1.1+ tumor infiltrates and their effector cytokine production (IFN-γ and granzyme B), slow tumor growth, and improve survival.
Clinically, IFN-α is well documented to enhance the expression of CEA and MHC antigens on freshly isolated human adenocarcinomas (40) and improve monoclonal antibody-targeting of carcinoma lesions following systemic administration in patients (45, 46). In patients with CEA-expressing carcinomas, therefore, we would expect the full repertoire of immunomodulatory properties of IFN-α to improve the clinical efficacy of our vaccine strategy. In a phase I study of patients with advanced CEA-expressing carcinomas (primarily colorectal), Marshall et al. (26) used recombinant poxvirus vaccines to elicit CEA-specific T-cell responses without significant toxicities. In a recent phase I study of advanced pancreatic cancer, recombinant poxvirus vaccines expressing CEA and MUC-1 (termed PANVAC-TRICOM) were successful in generating antigen-specific immune responses in 62.5% of evaluable patients (27). Overall survival was significantly better in those patients who developed antigen-specific immune responses to vaccine (15.1 versus 3.9 months, respectively; P = 0.002). In pancreatic cancer patients who undergo pancreaticoduodenectomy, an adjuvant IFN-α–based chemoradiation protocol has led to 5-year survival rates of 55% despite a high incidence of nodal involvement and advanced tumor stage before surgery (47). These studies suggest that recombinant poxvirus vaccines and IFN-α can be potentially used as effective forms of immunotherapy in patients with gastrointestinal malignancies. Our research provides both a rationale and mechanistic insights to support a clinical trial evaluating the combination of IFN-α and CEA-directed poxvirus vaccines in patients with CEA-expressing carcinomas.
Supplementary Material
Acknowledgments
We thank Eileen Thompson and Garland Davis for their technical assistance in the laboratory and Debra Weingarten for her editorial assistance in the preparation of the manuscript.
Grant support: Intramural Research Program of the Center for Cancer Research, National Cancer Institute, NIH.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Kageshita T, Hirai S, Ono T, Hicklin DJ, Ferrone S. Down-regulation of HLA class I antigen-processing molecules in malignant melanoma: association with disease progression. Am J Pathol. 1999;154:745–54. doi: 10.1016/S0002-9440(10)65321-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Natali PG, Nicotra MR, Bigotti A, et al. Selective changes in expression of HLA class I polymorphic determinants in human solid tumors. Proc Natl Acad Sci U S A. 1989;86:6719–23. doi: 10.1073/pnas.86.17.6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Imanishi T, Kamigaki T, Nakamura T, et al. Correlation between expression of major histocompatibility complex class I and that of antigen presenting machineries in carcinoma cell lines of the pancreas, biliary tract and colon. Kobe J Med Sci. 2006;52:85–95. [PubMed] [Google Scholar]
- 4.Levin I, Klein T, Kuperman O, et al. The expression of HLA class-I antigen in prostate-cancer in relation to tumor differentiation and patient survival. Cancer Detect Prev. 1994;18:443–45. [PubMed] [Google Scholar]
- 5.Grandis JR, Falkner DM, Melhem MF, Gooding WE, Drenning SD, Morel PA. Human leukocyte antigen class I allelic and haplotype loss in squamous cell carcinoma of the head and neck: clinical and immunogenetic consequences. Clin Cancer Res. 2000;6:2794–802. [PubMed] [Google Scholar]
- 6.Ryschich E, Notzel T, Hinz U, et al. Control of T-cell-mediated immune response by HLA class I in human pancreatic carcinoma. Clin Cancer Res. 2005;11:498–504. [PubMed] [Google Scholar]
- 7.Romero JM, Aptsiauri N, Vazquez F, et al. Analysis of the expression of HLA class I, proinflammatory cytokines and chemokines in primary tumors from patients with localized and metastatic renal cell carcinoma. Tissue Antigens. 2006;68:303–10. doi: 10.1111/j.1399-0039.2006.00673.x. [DOI] [PubMed] [Google Scholar]
- 8.Isaacs A, Lindenmann J. Virus interference. 1. The interferon. Proc R Soc Lond B Biol Sci. 1957;147:258–67. [PubMed] [Google Scholar]
- 9.Gresser I. The antitumor effects of interferon: a personal history. Biochimie. 2007;89:723–28. doi: 10.1016/j.biochi.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 10.Ferrantini M, Capone I, Belardelli F. Interferon-α and cancer: mechanisms of action and new perspectives of clinical use. Biochimie. 2007;89:884–93. doi: 10.1016/j.biochi.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 11.Hiroishi K, Tuting T, Tahara H, Lotze MT. Interferon-α gene therapy in combination with CD80 transduction reduces tumorigenicity and growth of established tumor in poorly immunogenic tumor models. Gene Ther. 1999;6:1988–94. doi: 10.1038/sj.gt.3301034. [DOI] [PubMed] [Google Scholar]
- 12.Hiroishi K, Tuting T, Lotze MT. IFN-α-expressing tumor cells enhance generation and promote survival of tumor-specific CTLs. J Immunol. 2000;164:567–72. doi: 10.4049/jimmunol.164.2.567. [DOI] [PubMed] [Google Scholar]
- 13.Santodonato L, Ferrantini M, Palombo F, et al. Antitumor activity of recombinant adenoviral vectors expressing murine IFN-α in mice injected with metastatic IFN-resistant tumor cells. Cancer Gene Ther. 2001;8:63–72. doi: 10.1038/sj.cgt.7700274. [DOI] [PubMed] [Google Scholar]
- 14.Kuwashima N, Nishimura F, Eguchi J, et al. Delivery of dendritic cells engineered to secrete IFN-α into central nervous system tumors enhances the efficacy of peripheral tumor cell vaccines: dependence on apoptotic pathways. J Immunol. 2005;175:2730–40. doi: 10.4049/jimmunol.175.4.2730. [DOI] [PubMed] [Google Scholar]
- 15.Prell RA, Li B, Lin JM, VanRoey M, Jooss K. Administration of IFN-α enhances the efficacy of a granulocyte macrophage colony stimulating factor-secreting tumor cell vaccine. Cancer Res. 2005;65:2449–56. doi: 10.1158/0008-5472.CAN-04-1975. [DOI] [PubMed] [Google Scholar]
- 16.Le Bon A, Schiavoni G, D'Agostino G, Gresser I, Belardelli F, Tough DF. Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity. 2001;14:461–70. doi: 10.1016/s1074-7613(01)00126-1. [DOI] [PubMed] [Google Scholar]
- 17.Di Pucchio T, Pilla L, Capone I, et al. Immunization of stage IV melanoma patients with Melan-A/MART-1 and gp100 peptides plus IFN-α results in the activation of specific CD8+ T cells and monocyte/dendritic cell precursors. Cancer Res. 2006;66:4943–51. doi: 10.1158/0008-5472.CAN-05-3396. [DOI] [PubMed] [Google Scholar]
- 18.James CM, Abdad MY, Mansfield JP, et al. Differential activities of α/β IFN subtypes against influenza virus in vivo and enhancement of specific immune responses in DNA vaccinated mice expressing haemagglutinin and nucleoprotein. Vaccine. 2007;25:1856–67. doi: 10.1016/j.vaccine.2006.10.038. [DOI] [PubMed] [Google Scholar]
- 19.Mitchell MS, Abrams J, Thompson JA, et al. Randomized trial of an allogeneic melanoma lysate vaccine with low-dose interferon α2b compared with high-dose interferon α2b for Resected stage III cutaneous melanoma. J Clin Oncol. 2007;25:2078–85. doi: 10.1200/JCO.2006.10.1709. [DOI] [PubMed] [Google Scholar]
- 20.Potebnya GP, Kudryavets YY, Lisovenko GS, et al. Experimental study of the efficacy of combined use of cancer vaccine and interferon. Exp Oncol. 2007;29:102–05. [PubMed] [Google Scholar]
- 21.Hodge JW, McLaughlin JP, Kantor JA, Schlom J. Diversified prime and boost protocols using recombinant vaccinia virus and recombinant non-replicating avian pox virus to enhance T-cell immunity and antitumor responses. Vaccine. 1997;15:759–68. doi: 10.1016/s0264-410x(96)00238-1. [DOI] [PubMed] [Google Scholar]
- 22.Hodge JW, Sabzevari H, Yafal AG, Gritz L, Lorenz MG, Schlom J. A triad of costimulatory molecules synergize to amplify T-cell activation. Cancer Res. 1999;59:5800–07. [PubMed] [Google Scholar]
- 23.Hodge JW, Grosenbach DW, Aarts WM, Poole DJ, Schlom J. Vaccine therapy of established tumors in the absence of autoimmunity. Clin Cancer Res. 2003;9:1837–49. [PubMed] [Google Scholar]
- 24.Reali E, Canter D, Zeytin H, Schlom J, Greiner JW. Comparative studies of Avipox-GM-CSF versus recombinant GM-CSF protein as immune adjuvants with different vaccine platforms. Vaccine. 2005;23:2909–21. doi: 10.1016/j.vaccine.2004.11.060. [DOI] [PubMed] [Google Scholar]
- 25.Kass E, Panicali DL, Mazzara G, Schlom J, Greiner JW. Granulocyte/macrophage-colony stimulating factor produced by recombinant avian poxviruses enriches the regional lymph nodes with antigen-presenting cells and acts as an immunoadjuvant. Cancer Res. 2001;61:206–14. [PubMed] [Google Scholar]
- 26.Marshall JL, Gulley JL, Arlen PM, et al. Phase I study of sequential vaccinations with fowlpox-CEA(6D)-TRICOM alone and sequentially with vaccinia-CEA(6D)-TRICOM, with and without granulocyte-macrophage colony-stimulating factor, in patients with carcinoembryonic antigen-expressing carcinomas. J Clin Oncol. 2005;23:720–31. doi: 10.1200/JCO.2005.10.206. [DOI] [PubMed] [Google Scholar]
- 27.Kaufman HL, Kim-Schulze S, Manson K, et al. Poxvirus-based vaccine therapy for patients with advanced pancreatic cancer. J Transl Med. 2007;5:60. doi: 10.1186/1479-5876-5-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsang KY, Zhu M, Nieroda CA, et al. Phenotypic stability of a cytotoxic T-cell line directed against an immunodominant epitope of human carcinoembryonic antigen. Clin Cancer Res. 1997;3:2439–49. [PubMed] [Google Scholar]
- 29.Zhu MZ, Marshall J, Cole D, Schlom J, Tsang KY. Specific cytolyticT-cell responses to human CEA from patients immunized with recombinant avipox-CEA vaccine. Clin Cancer Res. 2000;6:24–33. [PubMed] [Google Scholar]
- 30.Liu G, Zhai Q, Schaffner DJ, et al. Prevention of lethal respiratory vaccinia infections in mice with interferon-α and interferon-γ. FEMS Immunol Med Microbiol. 2004;40:201–06. doi: 10.1016/S0928-8244(03)00358-4. [DOI] [PubMed] [Google Scholar]
- 31.Corbett TH, Roberts BJ, Leopold WR, et al. Induction and chemotherapeutic response of two transplantable ductal adenocarcinomas of the pancreas in C57BL/6 mice. Cancer Res. 1984;44:717–26. [PubMed] [Google Scholar]
- 32.Robbins PF, Kantor JA, Salgaller M, Hand PH, Fernsten PD, Schlom J. Transduction and expression of the human carcinoembryonic antigen gene in a murine colon carcinoma cell line. Cancer Res. 1991;51:3657–62. [PubMed] [Google Scholar]
- 33.Clarke P, Mann J, Simpson JF, Rickard-Dickson K, Primus FJ. Mice transgenic for human carcinoembryonic antigen as a model for immunotherapy. Cancer Res. 1998;58:1469–77. [PubMed] [Google Scholar]
- 34.Schmitz J, Reali E, Hodge JW, et al. Identification of an interferon-γ-inducible carcinoembryonic antigen (CEA) CD8+ T-cell epitope, which mediates tumor killing in CEA transgenic mice. Cancer Res. 2002;62:5058–64. [PubMed] [Google Scholar]
- 35.Hodge JW, Poole DJ, Aarts WM, Gomez YA, Gritz L, Schlom J. Modified vaccinia virus ankara recombinants are as potent as vaccinia recombinants in diversified prime and boost vaccine regimens to elicit therapeutic antitumor responses. Cancer Res. 2003;63:7942–9. [PubMed] [Google Scholar]
- 36.Kass E, Schlom J, Thompson J, Guadagni F, Graziano P, Greiner JW. Induction of protective host immunity to carcinoembryonic antigen (CEA), a self-antigen in CEA transgenic mice, by immunizing with a recombinant vaccinia-CEA virus. Cancer Res. 1999;59:676–83. [PubMed] [Google Scholar]
- 37.Rogers CJ, Berrigan D, Zaharoff DA, et al. Energy restriction and exercise differentially enhance components of systemic and mucosal immunity in mice. J Nutr. 2008;138:115–22. doi: 10.1093/jn/138.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kimura T, Nakayama K, Penninger J, et al. Involvement of the Irf-1 transcription factor in antiviral responses to interferons. Science. 1994;264:1921–4. doi: 10.1126/science.8009222. [DOI] [PubMed] [Google Scholar]
- 39.Muller U, Steinhoff U, Reis LFL, et al. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–21. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 40.Guadagni F, Schlom J, Johnston WW, et al. Selective interferon-induced enhancement of tumor-associated antigens on a spectrum of freshly isolated human adenocarcinoma cells. J Natl Cancer Inst. 1989;81:502–12. doi: 10.1093/jnci/81.7.502. [DOI] [PubMed] [Google Scholar]
- 41.Luft T, Pang KC, Thomas E, et al. Type IIFNs enhance the terminal differentiation of dendritic cells. J Immunol. 1998;161:1947–53. [PubMed] [Google Scholar]
- 42.Herberman RR, Ortaldo JR, Bonnard GD. Augmentation by interferon of human natural and antibody-dependent cell-mediated cytotoxicity. Nature. 1979;277:221–3. doi: 10.1038/277221a0. [DOI] [PubMed] [Google Scholar]
- 43.Sivinski CL, Kohlgraf KG, VanLith ML, Morikane K, Tempero RM, Hollingsworth MA. Molecular requirements for CD8-mediated rejection of a MUC1-expressing pancreatic carcinoma: implications for tumor vaccines. Cancer Immunol Immunother. 2002;51:327–40. doi: 10.1007/s00262-002-0277-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tirapu I, Huarte E, Guiducci C, et al. Low surface expression of B7-1 (CD80) is an immunoescape mechanism of colon carcinoma. Cancer Res. 2006;66:2442–50. doi: 10.1158/0008-5472.CAN-05-1681. [DOI] [PubMed] [Google Scholar]
- 45.Roselli M, Guadagni F, Buonomo O, et al. Systemic administration of recombinant interferon α in carcinoma patients up-regulates the expression of the carcinoma-associated antigens tumor-associated glycoprotein-72 and carcinoembryonic antigen. J Clin Oncol. 1996;14:2031–42. doi: 10.1200/JCO.1996.14.7.2031. [DOI] [PubMed] [Google Scholar]
- 46.Milenic DE, Roselli M, Brechbiel MW, et al. In vivo evaluation of a lead-labeled monoclonal antibody using the DOTA ligand. Eur J Nucl Med. 1998;25:471–80. doi: 10.1007/s002590050246. [DOI] [PubMed] [Google Scholar]
- 47.Picozzi VJ, Kozarek RA, Traverso LW. Interferon-based adjuvant chemoradiation therapy after pancreaticoduodenectomy for pancreatic adenocarcinoma. Am J Surg. 2003;185:476–80. doi: 10.1016/s0002-9610(03)00051-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.