

Abstract
1-(2-Chlorophenyl-N-methylpropyl)-3-isoquinolinecarboxamide (PK11195) is a proven enhancer of apoptotic cell death in a variety of cellular models. This effect is independent of its established cellular target, the mitochondrial benzodiazepine receptor (mBzR), since it is able to promote cell death also in mBzR knockout cells. Thus recently it was suggested that PK11195 might exert its effect by modulating the expression and function of the oncogene Bcl-2. We have previously demonstrated that Bcl-2 modulates cellular Ca2+ homeostasis as its overexpression reduces the Ca2+ concentration in the endoplasmic reticulum (ER) ([Ca2+]er), impairing mitochondrial and cytosolic Ca2+ overload during cellular stress and therefore inhibiting the induction of the apoptotic cascade. Here, using ER, mitochondria and cytosolic targeted aequorin probes, we show that cellular treatment with PK11195 induces opposite changes in cellular Ca2+ homeostasis, increasing the [Ca2+]er and amplifying IP3 induced Ca2+ transients in mitochondria ([Ca2+]m) and cytosol ([Ca2+]c). This work provides evidence for a novel pharmacological effect of PK11195 on Ca2+ signalling which may be linked to its effect on Bcl-2 and account for its role in apoptotic cell death.
Keywords: Apoptosis, PK11195, Bcl-2, Ca2+ signalling, Mitochondria, Endoplasmic reticulum
1. Introduction
1-(2-Chlorophenyl-N-methylpropyl)-3-isoquinolinecarboxamide (PK11195) [1,2] has been recently established as a chemosensitizer of tumour cells to a wide range of chemotherapeutic agents [3-7]. Although it is known as the ligand of the mitochondrial benzodiazepine receptor (mBzR) [8,9] (recently named also translocator protein, TSPO [10,11]), it has been repeatedly suggested that further targets are involved in its role of cell death enhancer. PK11195 efficiently facilitates the programmed cell death execution by a wide range of stimuli such as etoposide, doxorubicin, ceramide; besides improving the effect of the cytostatic drugs like doxorubicin and the Bcl-2 inhibitor HA14-1 [4,12,13]. Notably, PK11195 remains equally active even when the mBzR is knocked down [14,15] and thus different mechanisms for its action have been proposed [6]. It was shown to inhibit the multiple drug resistance (MDR) pumps facilitating the uptake of different therapeutics in human multidrug-resistant cells [3,16]. In parallel, also its ability to activate the p38 MAPK signaling pathway in esophageal cancer cells was demonstrated [5] as well as a specific effect on the intrinsic apoptotic pathway leading to a substantial release of cytochrome c from mitochondria was documented [17]. Still, the principal pathway targeted by the drug remains ill-defined.
Recently, it was proposed that PK11195 might reduce the expression levels and inhibit the function of the anti-apoptotic members of the Bcl-2 family. Indeed, it was shown to reverse the Bcl-2 mediated inhibition of apoptosis in specific cell types and to reduce the apoptosis threshold in others [12,18]. The presence of the compound facilitated the induction of apoptosis in EW36, a human B-cell lymphoma cell line that over-expresses Bcl-2 [19]. Moreover, in human cholangiocarcinoma cells, PK11195 reverted Bcl-2 mediated cytoprotection by promoting translocation of Bax to the mitochondrial outer membrane [12,13,20]. Moreover, in human hepatocellular carcinoma both Bcl-2 and Bcl-XL are downregulated by micromolar doses of PK11195, concomitantly with an upregulation of Bax levels [4]. Members of the Bcl-2 gene family are known to exert their pro- or anti-apoptotic effect determining the state of mitochondrial permeability, by promoting or inhibiting respectively the release of proapoptotic factors like cytochrome c (cyt-c) [21] and apoptosis inducing factor (AIF) from the intermembrane space [22-24]. To this aim, a tuned regulation of Ca2+ fluxes is essential and a role for this family of proteins in regulating ion fluxes has been long proposed [25]. We and other groups have recently demonstrated that anti- and pro-apoptotic Bcl-2 family members regulate intracellular Ca2+ homeostasis in opposite ways by targeting the endoplasmic reticulum (ER) [26,27]. Bcl-2/Bcl-XL over-expression reduces the state of filling of the ER Ca2+ store, impairs IP3 induced Ca2+ release, and consequently protects against treatment with various proapoptotic drugs acting through mitochondrial Ca2+ overload following Ca2+ release from the ER [28-36] (see also [37] and references therein). On the contrary, Bax was shown to chronically and acutely increase ER Ca2+ loading, leading to uploading of mitochondrial Ca2+ uptake hence manifestation of apoptotic cell death [32,38,39]. This led to the recognition of a Ca2+ mediated mitochondrial apoptotic pathway (see above and [40]), characterized also by Ca2+ dependent translocation of Bax to the mitochondria [41].
We have therefore hypothesized that cellular treatment with PK11195 might be able to modulate intracellular Ca2+ signaling due to its regulatory role on Bcl-2 and/or Bcl-2 family interactors (e.g. Bax, Bad, Bcl-XL). By the use of the Ca2+ sensitive photoprotein aequorin, we have discovered that in HeLa cells micromolar concentrations of PK11195 increase the steady-state [Ca2+] of the ER ([Ca2+]er) generating in turn higher IP3 generated mitochondrial ([Ca2+]m) and cytosolic ([Ca2+]c) Ca2+ transients. Furthermore, this effect is plausibly not due to mBzR binding since another prototypic PBR ligand, the 7-chloro-5-(4-chlorophenyl)-1,3-dihydro-1-methyl-2H-1,4-benzodiazepin-2-one (Ro5-4864) [42] has no effect on cellular Ca2+. Hence, we propose that the PK11195 induced alterations in Ca2+ signaling might be consequence of a direct blockade of Bcl-2 interaction with other Bcl-2 family members, relevant to cell death through mitochondrial membrane permeabilization.
2. Methods
2.1. Cell culture, plasmids, and transfection
For the aequorin measurements, cells were seeded onto 13 mm coverslips (BDH, Milan, Italy) and transfected with 4 μg mtAEQmut or cytAEQ using Ca2+-phosphate technique; experiments were performed 36 h after transfection as previously described [43,44]. HeLa cells were grown in DMEM (Celbio) supplemented with 10% FBS, 100 units penicillin per ml and 25 μg streptomycin per ml. Cells were grown at 37 °C in a 5% CO2 incubator.
2.2. Aequorin measurements
mtAEQmut and cytAEQ transfected cells were used 36 h after transfection. The cells were incubated with 5 μM coelenterazine for 1-2 h in Krebs Ringer Buffer (KRB) supplemented with 1mM Ca2+ and then transferred to the perfusion chamber.
To test the effect of the PBR ligands, PK11195 or Ro5-4864, were added in concentration of 10 μM and 100 nM, respectively, in KRB supplemented with 1 mM CaCl2 for 2 h. PK11195 was dissolved in chloroform and Ro5-4864 in dimethyl sulfoxide (DMSO), utilized at a concentration of 1/1000, v/v and 1/10,000, v/v, respectively. Solvent only containing controls have been included throughout the experiments. All aequorin measurements were carried out in KRB, supplemented with either 1 mM Ca2+ or the indicated [Ca2+]. Agonists and other drugs were added to the same medium, as specified in the figure legends. The experiments were terminated by lysing cells with 100 μM digitonin in a hypotonic Ca2+-rich solution (10 mM CaCl2 in H2O), thus discharging the remaining aequorin pool. In brief, a 13 mm-round coverslip with the transfected cells was placed in a perfused, thermostatted chamber located in close proximity of a low-noise photomultiplier, with built-in amplifierdiscriminator. The output of the discriminator was captured by a Thorn-EMI photon counting board and stored in an IBM-compatible computer for further analyses. The aequorin luminescence data were calibrated off-line into [Ca2+] values, using a computer algorithm based on the Ca2+ response curve of wild-type and mutant aequorins, as previously described [43-47].
For the Er-AEQ measurements, as stated also in the results, before the reconstitution, it is necessary to reduce the Ca2+ content of the Golgi apparatus and the ER drastically. To this end, the cells were incubated for 1 h at 4 °C, in KRB (Krebs-Ringer modified buffer: 125 mM NaCl, 5 mM KCl, 1 mM Na3PO4, 1 mM MgSO4, 5.5 mM glucose, 20 mM HEPES, pH 7.4, 37 °C) supplemented with 5 mM coelenterazine, the Ca2+ ionophore ionomycin (2.5 μM) and 600 μM EGTA. After this incubation, the cells were washed extensively with KRB supplemented with 2% bovine serum albumin (BSA) and 1 mM EGTA. In the experiments, additions (1 mM SrCl2, 1 mM CaCl2, histamine, etc.) were made to the same medium, as specified in the figure legends. As for mtAEQ and cytAEQ, the experiments were terminated by lysing the cells with 100 mM digitonin in a hypotonic Ca2+-rich solution (10 mM CaCl2 in H2O), thus discharging the remaining aequorin pool [48].
2.3. Statistical analysis
All statistical analyses were performed using a two tailed Student's t-test assuming normal distributions with unequal variances. Error bars presented in graphs denote standard deviation (S.D.).
2.3.1. Reagents
Coelenterazin was purchased from molecular probes (Eugene, OR, USA). Histamine, ATP, ionomycin, EGTA, CaCl2 and all the reagents used for the Krebs Ringer Buffer were purchased from Sigma-Aldrich (ITALY). Krebs Ringer Buffer used has the following composition: 20 mM HEPES pH 7.4, 125 mM NaCl, 5 mM KCl, 1 mM MgSO4, 1mM Ha2HPO4, 20 mM NaHCO3, 5.5 mM glucose, 2 mM L-glutamine. PK11195 and Ro5-4864 and their respective solvents chloroform and dimethyl sulfoxide were purchased from Sigma-Aldrich (ITALY).
3. Results
3.1. Increased ER luminal Ca2+ concentrations in PK11195 treated HeLa cells
The ER is the major source of both physiological and pathological Ca2+ signals. To investigate the effect of PK11195 on ER Ca2+ handling, we used a low affinity aequorin construct, targeted to the ER [47]. HeLa cells were transfected with er-AEQ and treated with 10 μM PK11195 for 2 h or with an equivalent volume of the drug's solvent chloroform (1/1000, v/v) and the [Ca2+]er in the two cohorts were compared. To avoid consumption of the aequorin, the luminal [Ca2+] of these organelles must be reduced prior to the reconstitution of the probe with prostethic group coelenterazine. This was obtained by incubation of the cells in KRB supplemented with the low affinity coelenterazinen and 2.5 μMionomycin, a Ca2+ ionophore, in the absence of extracellular Ca2+ (see Section 2 and references therein). Aequorin luminescence signals were collected using a luminometer and calibrated into [Ca2+] values. Upon switching the perfusion medium to KRB buffer supplemented with 1 mM Ca2+, [Ca2+]er gradually increased, reaching plateau levels of ~390 μM Ca2+ in control cells whilst in PK11195 treated cells, an higher steady state level (~10%) was observed in the same compartment with values of ~430 μM Ca2+ (Fig. 1A). The consecutive addition of the IP3 generating agonist histamine resulted in a rapid decrease in [Ca2+]er in both cohorts, indicating that the sensitivity to agonists of the Ca2+ stores was retained. Taken together, these measurements indicate that micromolar doses of PK11195 induce an increase of the luminal [Ca2+] in the ER. Solvent treated cells did not show any modification as compared to those untreated (data not shown).
Fig. 1. PK11195 increases the Ca2+ content in the endoplasmic reticulum and IP3 generated Ca2+ transients in the cytosol and mitochondria of HeLa cells.
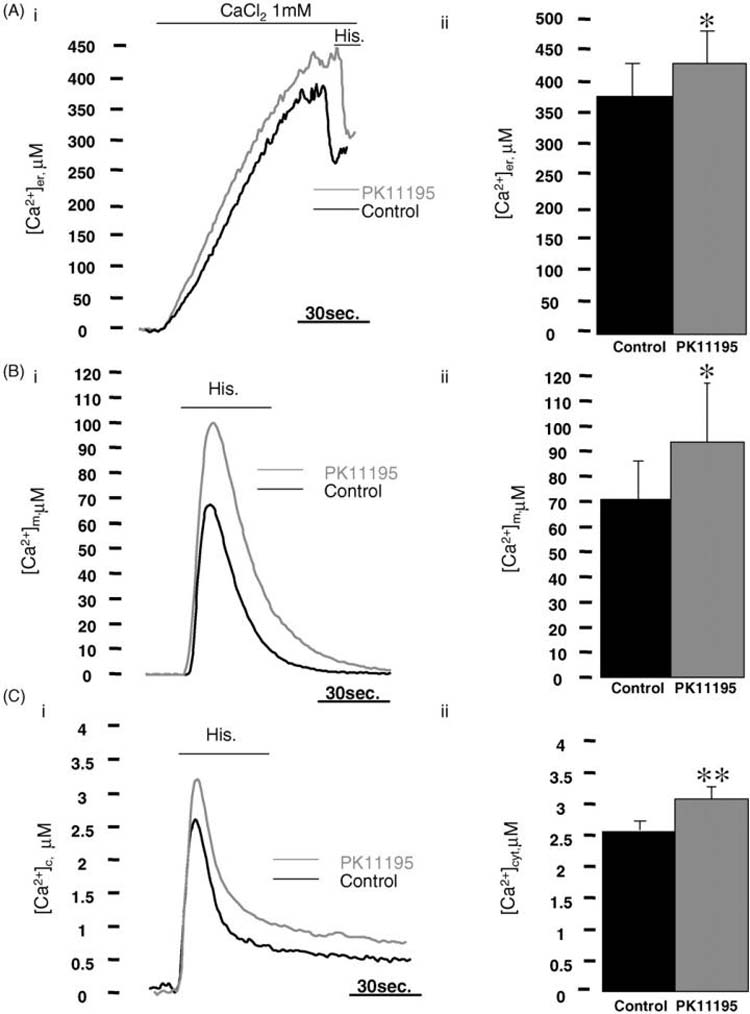
(Ai) Shows representative traces of the ER Ca2+ refilling in HeLa cells expressing endoplasmic reticulum targeted Aequorin (er-AEQ) treated for 2 h with 10 μM PK11195 (grey trace) or kept in PK11195 solvent as control (black trace). (Aii) Summarizes the average [Ca2+]er values of 14 individual experiments (control: 384.93 ± 78.52 μM Ca2+, PK11195: 426.55 ± 55 μM Ca2+, n =24 *p < 0.05). (Bi) Shows representative records of histamine (100 μM) induced Ca2+ rise in mitochondria of HeLa cells expressing mitochondrially targeted mutated aequorin (mt-AEQMut). The degree of Ca2+ accumulation recorded in untreated (black trace) cells is statistically lower than in cells treated with 10 μM PK11195 (grey trace). Peak [Ca2+]m values corresponding to 32 individual experiments are plotted on (Bii) (control: 68.17 ± 23.81 μM Ca2+, PK11195: 84.82 ± 29.25 μM Ca2+, n =33 *p < 0.05). (Ci) Shows histamine (100 μM) induced cytosolic Ca2+ transients in control (black trace) and PK11195 treated cells (grey trace) expressing cytosolic based aequorin (cyt-AEQ). As for the mitochondrial environment, PK11195 treatment promotes a statistically higher cytosolic Ca2+ peak also in this cellular compartment and values corresponding to 15 individual experiments are shown in (Cii) (control: 2.43 ± 0.28 μM Ca2+, PK11195: 3.1 ± 0.33 μM Ca2+, n = 27, **p < 0.01).
3.2. Stimulus-induced increases in [Ca2+]m and [Ca2+]c in PK11195 treated HeLa cells
Mitochondria play an important role in intracellular Ca2+ homeostasis as they lie in close proximity of inositol 1,4,5-trisphosphate (IP3)-gated channels and are capable of taking up the Ca2+ released by IP3-generating agonists (e.g. histamine H1 and ATP), thereby buffering the [Ca2+]c [45,49]. We hypothesized that the PK11195-induced increase in the steady state [Ca2+]er level and the ensuing increase in the driving force for IP3-induced Ca2+ release should also increase the mitochondrial Ca2+ uptake. To test this hypothesis, HeLa cells were transfected with a mitochondrial targeted low affinity aequorin construct (mt-AEQmut), exposed to PK11195 or only the solvent (control) and then challenged with histamine in the presence of extracellular Ca2+. Fig. 1B shows that the peak mitochondrial response was markedly increased ~25% in PK11195 treated cells than solvent treated cells showing average values of ~85 μM Ca2+ in PK11195 and ~70 μM Ca2+ in control. The effect of PK11195 on mitochondrial Ca2+ transient was also measured with equal outcome in single cells transfected with the fluorescent Ca2+ probe 2 mt-YC 2.1 (data not shown) [50] confirming that PK11195-induced increment of [Ca2+]er leads to a consequent increase in the stimulus-induced mitochondrial Ca2+ uptake.
Consequently, we expected to see similar result also for the [Ca2+]c following agonist stimulation. To this purpose, HeLa cells were transfected with cytosolic high affinity aequorin (cyt-AEQ) [43] and treated with PK11195 or with its solvent. Also in this cellular compartment, the amplitude of the histamine induced [Ca2+]c peak was increased by PK11195 treatment (Fig. 1C) resulting in a peak value higher (~25%) than controls.
3.3. Mitochondrial and cytosolic Ca2+ handling in Ro5-4864 treated HeLa cells
To ascertain that PK11195 mediated effect on Ca2+ signalling was not due to a modulation of the mBzR we tested mitochondrial and cytosolic Ca2+ transients in HeLa cells treated with another high affinity ligand of the mBzR, Ro5-4864. HeLa cells expressing mt-AEQmut and cyt-AEQ respectively were incubated with 100 nM Ro5-4864 or with equal volume of DMSO (1/10,000, v/v) as control for 2 h. Fig. 2 shows that Ro5-4864 treated cells did not show any statistically significant modification of the cytosolic and mitochondrial Ca2+ peak values. No alterations were observed either after the application of higher concentrations of Ro5-4864 (1-10 μM, data not shown). In this way, we could postulate that the PK11195 induced alterations of the intracellular Ca2+ signalling are most probably not mediated by the mBzR.
Fig. 2. The prototypical PBR ligand Ro5-4864 does not affect the [Ca2+]m and [Ca2+]c in HeLa cells.
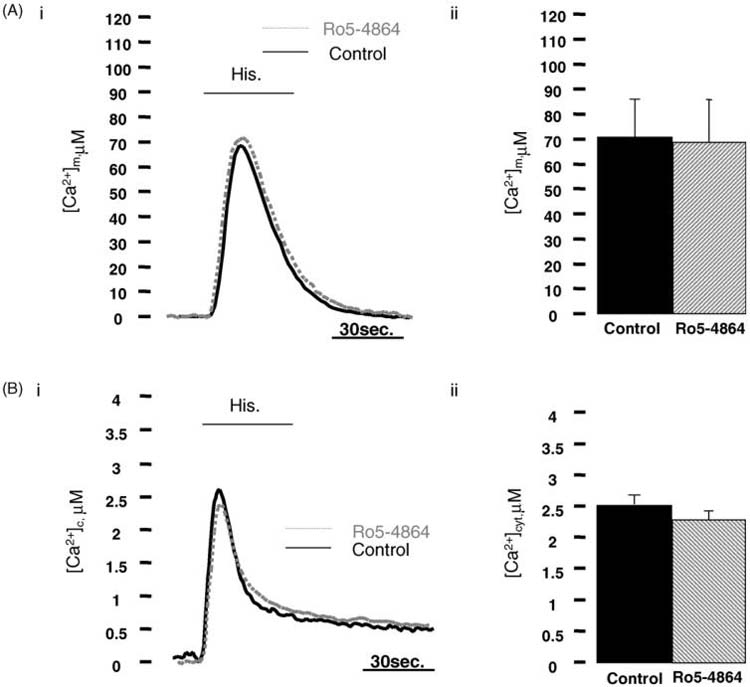
(Ai) Shows Ca2+ traces of HeLa cells transfected with mitochondrially-targeted aequorin (mt-AEQMut) challenged with 100 μM histamine (Hist.) Ro5-4864 (dot grey trace) and control cells (black trace) present identical amplitude of the Ca2+ response as reported by the statistical analysis in (Aii) (control: 67.41 ± 22.68 μM Ca2+, Ro5-4864: 69.41 ± 92 μM Ca2+, mean ± S.D. of 28 individual experiments, P = 0.61). (Bi) As for the mitochondrial Ca2+ uptake, neither in the bulk cytosol, Ro5-4864 affects Ca2+ rise after histamine stimulation. Representative traces of HeLa cells expressing cyt-AEQ are pictured in the panel. Control (black trace) and Ro5-4864 (dot grey trace) cells have similar Ca2+ peak values as the graph plotted in (Bii) underlines (control: 2.23 ± 0.27 μM Ca2+, Ro5-4864: 2.33 ± 0.35 μM Ca2+, average ± S.D. of 12 individual experiments, P = 0.79).
3.4. Capacitative Ca2+ influx in control and PK11195 treated cells
Previously, we have demonstrated that in Bcl-2 overexpressing cells a reduction in the steady-state of [Ca2+]er was strikingly paralleled with a long term depression of capacitive Ca2+ influx [28,30]. We assumed therefore that if PK11195 modulates global Ca2+ signaling in the opposite way of Bcl-2, it should also increase the capacitative Ca2+ influx, following agonist induced emptying of the ER Ca2+ stores. For this purpose, PK11195 treated and control HeLa cells expressing cyt-AEQ were first challenged with histamine in Ca2+-free extracellular medium containing 100 μM EGTA. This maneuver ensures that increases of the [Ca2+]c will be due only to the release of Ca2+ from the intracellular stores. Moreover, leaving the cells in this medium leads to the partial depletion of the ER Ca2+ store, which we benefited to even out the steady state ER luminal [Ca2+] differences in control and PK11195 treated cells, and thus allowing to assess the specific effects of the drug on the Ca2+ influx following Ca2+ re-addition to the extracellular medium (Fig. 3Ai). Indeed, this procedure, observed similar Ca2+ release in the control and PK11195 treated groups, while PK11195 induced a markedly higher [Ca2+]c increase (~28%) via the capacitative Ca2+ influx pathway in PK11195 treated cells (see graphs in Fig. 3Aii). This indicates a specific activation of the Ca2+ influx pathway in PK11195 treated cells, leading to a higher filling state of ER as observed in the presence of extracellular Ca2+. This effect on the cellular Ca2+ homeostasis is the contrary of what have been observed in Bcl-2 overexpressing cells.
Fig. 3. Capacitative Ca2+ influx in PK11195 treated HeLa cells.
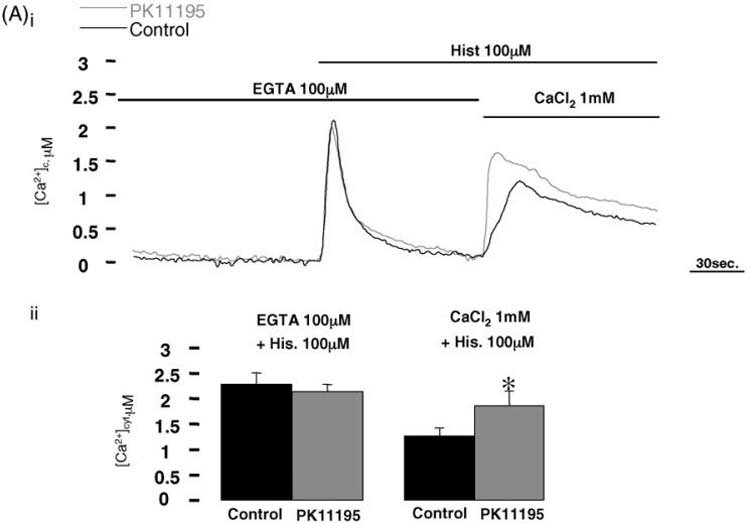
(Ai) Cyt-AEQ expressing control and PK11195 treated HeLa cells were kept in Ca2+-free medium containing 100 μM EGTA for about 5 min and then challenged with 100 μM histamine. Ca2+-containing medium was then replaced still in presence of histamine in order to estimate the Ca2+ influx from extracellular medium. Panel (Aii) shows mean ± S.D. values of 12 individual experiments (control: 2.2 ± 0.14 μM Ca2+, PK11195: 2.22 ± 0.21 μM Ca2+, P = 0.20; control: 1.31 ± 0.33 μM Ca2+, PK11195: 1.81 ± 0.66 μM Ca2+,*p < 0.05).
3.5. Bcl-2 as target for PK11195 induced modulation of Ca2+ homeostasis: a hypothesis
By these means, we suggest that a direct effect of PK11195 on Bcl-2 is a plausible mechanism by which steady-state levels and agonist dependent fluxes of Ca2+ ions occur (Fig. 4Ai). Thus PK11195 incubation affects key mechanisms of intracellular Ca2+ homeostasis in a Bcl-2 opposite manner: the state of refilling of the ER, the influx trough the plasma membrane (PM) and consequent mitochondrial Ca2+ accumulation and cytosolic Ca2+ transients in response to IP3 generation (Fig. 4Aii).
Fig. 4. Model for the PK11195 proposed mechanism of action on intracellular Ca2+ signalling in HeLa cells.
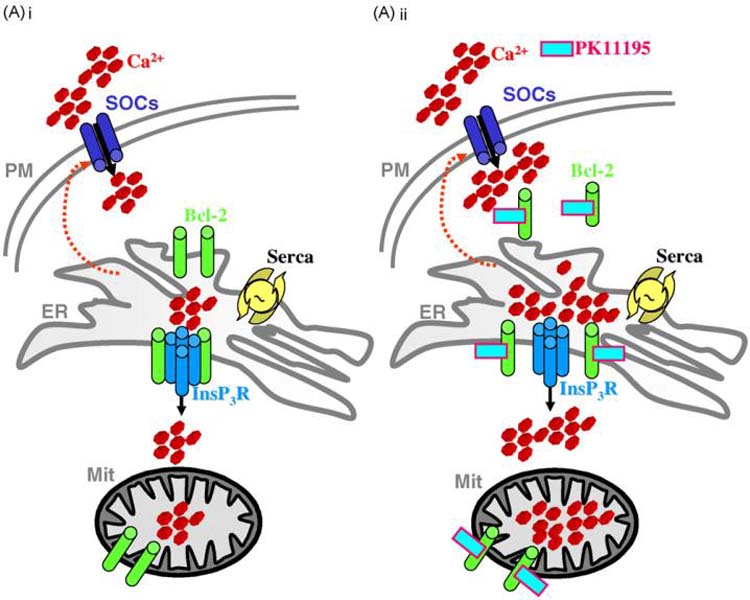
(Ai) shows the principal effects of the anti-apoptotic protein Bcl-2 on Ca2+ homeostasis. The oncogene reduces the Ca2+ content of the ER increasing the leak of Ca2+ from the lumen and downregulating the influx of the ion trough the plasma membrane (PM). This ultimately modulates the steady-state levels and the agonist dependent fluxes of intracellular Ca2+ in the mitochondria and cytosol. We suggest that incubation with PK11195 counteracts this Bcl-2 mediated effect reducing the leak from the ER, re-enforcing the Ca2+ influx and increasing the amplitude of mitochondrial and cytosolic Ca2+ transients following stimulation with Ca2+ mobilizing agonists (Aii).
4. Discussion
Using genetically encoded Ca2+ indicators, we have demonstrated that pharmacological treatment with PK11195 modulates intracellular Ca2+ signaling in HeLa cells independently of mBzR binding and consistently with a direct inhibition of Bcl-2 effect on cellular Ca2+ homeostasis.
In detail, 2 h treatment with 10 μM PK11195 led to: (i) a substantial increase (~10%) of the steady-state [Ca2+] level in the ER, (ii) higher histamine evoked mitochondrial and cytosolic Ca2+ transients (~25%) and (iii) increased capacitative Ca2+ influx (~28%).
Conversely, treatment with another prototypical mBzR ligand, the diazepam Ro5-4864 did not affect Ca2+ signalling. Interestingly, previous reports showed opposite effects on [Ca2+]i for both mBzR ligands. Frigo and co-workers thus reported a Ro5-4864 mediated rise of [Ca2+]c trough extracellular Ca2+ influx in human neutrophils and a parallel inhibitory effect for PK11195 [51]. Similarly, McLarnon and co-workers [52] proposed a PK11195 mediated inhibition of the Ca2+ store-operated channels (SOCs) activity in human microglia; hence in apparent contradiction with our findings. Whilst, more recently, the group of Lacapere demonstrated in human colon cancer cells HT-29 that PK11195 was able to promote a rapid and transient dose-dependent rise in intracellular [Ca2+]c, unaffected by extracellular Ca2+ [53]. However, the differences of the experimental protocol, the cell type, methods of Ca2+ measurements, and the way of drugs administration-tested in acute and not in incubation as in our model- do not allow a neat comparison. Nevertheless, in the cited papers, the authors aimed to characterize the mBzR contribution to intracellular Ca2+ signalling via its acute pharmacological modulation whilst in our case, as the role of PK11195 in apoptotic cell death was proven to be mBzR independent, we enrolled a long term pharmacological approach to define the functional results of the drug interaction with target genes in apoptotic response modulation.
Despite that, as mitochondria should amplify the differences recorded in the cytosol (Fig. 1B and C), an inhibitory role for mBzR itself on mitochondrial Ca2+ uptake in PK11195 treated HeLa cells cannot be excluded.
It is now widely appreciated that alteration of cellular Ca2+ handling is a common strategy used by different proteins in order to modulate the apoptotic response: Bcl-2 exerts some of its antiapoptotic effects by reducing [Ca2+]er and down-regulating Ca2+ fluxes between the ER and mitochondria and the capacitative Ca2+ influx [26,28].
A similar effect was observed with the coxsackievirus 2B protein [54] and by reducing the mitochondrial Ca2+ uptake directly [55] (see also [56]). In the same way, experimental conditions that lowered [Ca2+]er protect HeLa cells from ceramide-induced apoptosis [30] whilst maneuvers that increase [Ca2+]er have the opposite effect and this is the case of the proapoptotic protein Bax [36]. This accounts for a signaling mechanism between the ER and the mitochondria in which alterations in Ca2+ uptake modulate the apoptotic response [57].
Key events occurring in the mitochondrial matrix are all sensitive to increases in [Ca2+]m such as (i) ATP production, whose concentration is crucial for apoptosis vs. necrosis choice [58], (ii) reactive oxygen species production [59], (iii) the opening of the permeability transition pore (mPTP) followed by the release of proapoptotic proteins [60,61]. Accordingly, down-regulation of Ca2+ fluxes protect mitochondria from cytotoxic rises in [Ca2+]m.
Our findings show that PK11195 up regulates Ca2+ fluxes and this can be considered one of the mechanisms utilized by the compound to exert its role in promoting cell death, in line with the previously established proapoptotic role of ER-mitochondrial Ca2+ transfer [62,63]. In addition, we hypothesize that the combined PK11195 mechanism to increase either the IP3-dependent release of Ca2+ from the ER and the capacitative Ca2+ entry support also a direct inhibitory effect on Bcl-2. Bcl-2 selectively modulates two hubs of Ca2+ signaling, the [Ca2+]er and the capacitative Ca2+ entry, acting via a “double hit” strategy to avoid the Ca2+ mediated apoptosis [30]. PK11195 appears to act in an identical, but opposite manner thus it will potentiate this pathway, leading to cell death induction.
Although direct experimental evidences will be needed to confirm that this effect is mediated by a direct inhibition of the oncoprotein Bcl-2, it appears as a plausible target to justify the observed effects. An intriguing possibility might be also that PK11195 modifies the phosphorylation state of Bcl-2, as previously demonstrated for specific mitochondrial proteins [64], thus changing its interaction with other Bcl-2 family members and components of the Ca2+ signaling machinery on the ER surface [65,66]. Further studies will be then necessary to explore these prospects in depth thus paving the way to identify new molecular targets for modifying the apoptotic sensitivity of cancer cells.
Acknowledgments
This work was supported by Telethon grant GGP05284, the Italian Association for Cancer Research (AIRC), the local funds from Ferrara University, the Italian University Ministry, the PRRIITT program of the Emilia Romagna Region, the Italian Space Agency (ASI) and NIH (Grant: a mitochondrial longevity pathway: p66shc mechanisms); Central Research Fund, University of London, UK (Grant: a bioluminescent approach to investigate the function role of the mitochondrial benzodiazepine receptor, mBzR); internal funds of the Royal Veterinary College. We would also like to thank Prof. Michael R Duchen for support.
REFERENCES
- [1].Le FG, Perrier ML, Vaucher N, Imbault F, Flamier A, Benavides J, et al. Peripheral benzodiazepine binding sites: effect of PK 11195, 1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinolinecarboxamide. I. In vitro studies. Life Sci. 1983;32:1839–47. doi: 10.1016/0024-3205(83)90062-0. [DOI] [PubMed] [Google Scholar]
- [2].Le FG, Guilloux F, Rufat P, Benavides J, Uzan A, Renault C, et al. Peripheral benzodiazepine binding sites: effect of PK 11195, 1-(2-chlorophenyl)-N-methyl-(1-methylpropyl)-3 isoquinolinecarboxamide. II. In vivo studies. Life Sci. 1983;32:1849–56. doi: 10.1016/0024-3205(83)90063-2. [DOI] [PubMed] [Google Scholar]
- [3].Walter RB, Pirga JL, Cronk MR, Mayer S, Appelbaum FR, Banker DE. PK11195, a peripheral benzodiazepine receptor (pBR) ligand, broadly blocks drug efflux to chemosensitize leukemia and myeloma cells by a pBR-independent, direct transporter-modulating mechanism. Blood. 2005;106:3584–93. doi: 10.1182/blood-2005-02-0711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sutter AP, Maaser K, Grabowski P, Bradacs G, Vormbrock K, Hopfner M, et al. Peripheral benzodiazepine receptor ligands induce apoptosis and cell cycle arrest in human hepatocellular carcinoma cells and enhance chemosensitivity to paclitaxel, docetaxel, doxorubicin and the Bcl-2 inhibitor HA14-1. J Hepatol. 2004;41:799–807. doi: 10.1016/j.jhep.2004.07.015. [DOI] [PubMed] [Google Scholar]
- [5].Sutter AP, Maaser K, Gerst B, Krahn A, Zeitz M, Scherubl H. Enhancement of peripheral benzodiazepine receptor ligand-induced apoptosis and cell cycle arrest of esophageal cancer cells by simultaneous inhibition of MAPK/ERK kinase. Biochem Pharmacol. 2004;67:1701–10. doi: 10.1016/j.bcp.2004.01.009. [DOI] [PubMed] [Google Scholar]
- [6].Banker DE, Cooper JJ, Fennell DA, Willman CL, Appelbaum FR, Cotter FE. PK11195, a peripheral benzodiazepine receptor ligand, chemosensitizes acute myeloid leukemia cells to relevant therapeutic agents by more than one mechanism. Leuk Res. 2002;26:91–106. doi: 10.1016/s0145-2126(01)00112-6. [DOI] [PubMed] [Google Scholar]
- [7].Santidrian AF, Cosialls AM, Coll-Mulet L, Iglesias-Serret D, de FM, Gonzalez-Girones DM, et al. The potential anticancer agent PK11195 induces apoptosis irrespective of p53 and ATM status in chronic lymphocytic leukemia cells. Haematologica. 2007;92:1631–8. doi: 10.3324/haematol.11194. [DOI] [PubMed] [Google Scholar]
- [8].Casellas P, Galiegue S, Basile AS. Peripheral benzodiazepine receptors and mitochondrial function. Neurochem Int. 2002;40:475–86. doi: 10.1016/s0197-0186(01)00118-8. [DOI] [PubMed] [Google Scholar]
- [9].Weizman R, Gavish M. Molecular cellular and behavioral aspects of peripheral-type benzodiazepine receptors. Clin Neuropharmacol. 1993;16:401–17. doi: 10.1097/00002826-199310000-00003. [DOI] [PubMed] [Google Scholar]
- [10].Veenman L, Papadopoulos V, Gavish M. Channel-like functions of the 18-kDa translocator protein (TSPO): regulation of apoptosis and steroidogenesis as part of the host-defense response. Curr Pharm Des. 2007;13:2385–405. doi: 10.2174/138161207781368710. [DOI] [PubMed] [Google Scholar]
- [11].Papadopoulos V, Baraldi M, Guilarte TR, Knudsen TB, Lacapere JJ, Lindemann P, et al. Translocator protein (18kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci. 2006;27:402–9. doi: 10.1016/j.tips.2006.06.005. [DOI] [PubMed] [Google Scholar]
- [12].Hirsch T, Decaudin D, Susin SA, Marchetti P, Larochette N, Resche-Rigon M, et al. PK11195, a ligand of the mitochondrial benzodiazepine receptor, facilitates the induction of apoptosis and reverses Bcl-2-mediated cytoprotection. Exp Cell Res. 1998;241:426–34. doi: 10.1006/excr.1998.4084. [DOI] [PubMed] [Google Scholar]
- [13].Chen J, Freeman A, Liu J, Dai Q, Lee RM. The apoptotic effect of HA14-1, a Bcl-2-interacting small molecular compound, requires Bax translocation and is enhanced by PK11195. Mol Cancer Ther. 2002;1:961–7. [PubMed] [Google Scholar]
- [14].Gonzalez-Polo RA, Carvalho G, Braun T, Decaudin D, Fabre C, Larochette N, et al. PK11195 potently sensitizes to apoptosis induction independently from the peripheral benzodiazepin receptor. Oncogene. 2005;24:7503–13. doi: 10.1038/sj.onc.1208907. [DOI] [PubMed] [Google Scholar]
- [15].Hans G, Wislet-Gendebien S, Lallemend F, Robe P, Rogister B, Belachew S, et al. Peripheral benzodiazepine receptor (PBR) ligand cytotoxicity unrelated to PBR expression. Biochem Pharmacol. 2005;69:819–30. doi: 10.1016/j.bcp.2004.11.029. [DOI] [PubMed] [Google Scholar]
- [16].Jakubikova J, Duraj J, Hunakova L, Chorvath B, Sedlak J. PK11195, an isoquinoline carboxamide ligand of the mitochondrial benzodiazepine receptor, increased drug uptake and facilitated drug-induced apoptosis in human multidrug-resistant leukemia cells in vitro. Neoplasma. 2002;49:231–6. [PubMed] [Google Scholar]
- [17].Li J, Wang J, Zeng Y. Peripheral benzodiazepine receptor ligand, PK11195 induces mitochondria cytochrome c release and dissipation of mitochondria potential via induction of mitochondria permeability transition. Eur J Pharmacol. 2007;560:117–22. doi: 10.1016/j.ejphar.2006.12.027. [DOI] [PubMed] [Google Scholar]
- [18].Decaudin D, Castedo M, Nemati F, Beurdeley-Thomas A, De PG, Caron A, et al. Peripheral benzodiazepine receptor ligands reverse apoptosis resistance of cancer cells in vitro and in vivo. Cancer Res. 2002;62:1388–93. [PubMed] [Google Scholar]
- [19].Muscarella DE, O'Brien KA, Lemley AT, Bloom SE. Reversal of Bcl-2-mediated resistance of the EW36 human B-cell lymphoma cell line to arsenite- and pesticide-induced apoptosis by PK11195, a ligand of the mitochondrial benzodiazepine receptor. Toxicol Sci. 2003;74:66–73. doi: 10.1093/toxsci/kfg052. [DOI] [PubMed] [Google Scholar]
- [20].Okaro AC, Fennell DA, Corbo M, Davidson BR, Cotter FE. Pk11195, a mitochondrial benzodiazepine receptor antagonist, reduces apoptosis threshold in Bcl-X(L) and Mcl-1 expressing human cholangiocarcinoma cells. Gut. 2002;51:556–61. doi: 10.1136/gut.51.4.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–32. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- [22].Kroemer G, Zamzami N, Susin SA. Mitochondrial control of apoptosis. Immunol Today. 1997;18:44–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- [23].Chao DT, Korsmeyer SJ. BCL-2 family: regulators of cell death. Annu Rev Immunol. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- [24].Zamzami N, Brenner C, Marzo I, Susin SA, Kroemer G. Subcellular and submitochondrial mode of action of Bcl-2-like oncoproteins. Oncogene. 1998;16:2265–82. doi: 10.1038/sj.onc.1201989. [DOI] [PubMed] [Google Scholar]
- [25].Minn AJ, Velez P, Schendel SL, Liang H, Muchmore SW, Fesik SW, et al. Bcl-x(L) forms an ion channel in synthetic lipid membranes. Nature. 1997;385:353–7. doi: 10.1038/385353a0. [DOI] [PubMed] [Google Scholar]
- [26].Pinton P, Ferrari D, Magalhaes P, Schulze-Osthoff K, Di VF, Pozzan T, et al. Reduced loading of intracellular Ca(2+) stores and downregulation of capacitative Ca(2+) influx in Bcl-2-overexpressing cells. J Cell Biol. 2000;148:857–62. doi: 10.1083/jcb.148.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hanson CJ, Bootman MD, Roderick HL. Cell signalling: IP3 receptors channel calcium into cell death. Curr Biol. 2004;14:R933–5. doi: 10.1016/j.cub.2004.10.019. [DOI] [PubMed] [Google Scholar]
- [28].Foyouzi-Youssefi R, Arnaudeau S, Borner C, Kelley WL, Tschopp J, Lew DP, et al. Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc Natl Acad Sci USA. 2000;97:5723–8. doi: 10.1073/pnas.97.11.5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Szalai G, Krishnamurthy R, Hajnoczky G. Apoptosis driven by IP(3)-linked mitochondrial calcium signals. EMBO J. 1999;18:6349–61. doi: 10.1093/emboj/18.22.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Pinton P, Ferrari D, Rapizzi E, Di VF, Pozzan T, Rizzuto R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J. 2001;20:2690–701. doi: 10.1093/emboj/20.11.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Palmer AE, Jin C, Reed JC, Tsien RY. Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc Natl Acad Sci USA. 2004;101:17404–9. doi: 10.1073/pnas.0408030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, et al. The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat Cell Biol. 2005;7:1021–8. doi: 10.1038/ncb1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hanson CJ, Bootman MD, Distelhorst CW, Wojcikiewicz RJ, Roderick HL. Bcl-2 suppresses Ca(2+) release through inositol 1,4,5-trisphosphate receptors and inhibits Ca(2+) uptake by mitochondria without affecting ER calcium store content. Cell Calcium. 2008 doi: 10.1016/j.ceca.2008.01.003. [DOI] [PubMed] [Google Scholar]
- [34].Distelhorst CW, Shore GC. Bcl-2 and calcium: controversy beneath the surface. Oncogene. 2004;23:2875–80. doi: 10.1038/sj.onc.1207519. [DOI] [PubMed] [Google Scholar]
- [35].Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–9. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- [36].Chami M, Prandini A, Campanella M, Pinton P, Szabadkai G, Reed JC, et al. Bcl-2 and Bax exert opposing effects on Ca2+ signaling, which do not depend on their putative pore-forming region. J Biol Chem. 2004;279:54581–9. doi: 10.1074/jbc.M409663200. [DOI] [PubMed] [Google Scholar]
- [37].Mathai JP, Germain M, Shore GC. BH3-only BIK regulates BAX, BAK-dependent release of Ca2+ from endoplasmic reticulum stores and mitochondrial apoptosis during stress-induced cell death. J Biol Chem. 2005;280:23829–36. doi: 10.1074/jbc.M500800200. [DOI] [PubMed] [Google Scholar]
- [38].Vanoverberghe K, Vanden AF, Mariot P, Lepage G, Roudbaraki M, Bonnal JL, et al. Ca2+ homeostasis and apoptotic resistance of neuroendocrine-differentiated prostate cancer cells. Cell Death Differ. 2004;11:321–30. doi: 10.1038/sj.cdd.4401375. [DOI] [PubMed] [Google Scholar]
- [39].Jones RG, Bui T, White C, Madesh M, Krawczyk CM, Lindsten T, et al. The proapoptotic factors Bax and Bak regulate T Cell proliferation through control of endoplasmic reticulum Ca(2+) homeostasis. Immunity. 2007;27:268–80. doi: 10.1016/j.immuni.2007.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Benli-Furet NL, Chami M, Houel L, De GF, Vernejoul F, Lagorce D, et al. Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene. 2005;24:4921–33. doi: 10.1038/sj.onc.1208673. [DOI] [PubMed] [Google Scholar]
- [41].Deniaud A, Sharaf el DO, Maillier E, Poncet D, Kroemer G, Lemaire C, et al. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene. 2008;27:285–99. doi: 10.1038/sj.onc.1210638. [DOI] [PubMed] [Google Scholar]
- [42].File SE, Pellow S. RO5-4864, a ligand for benzodiazepine micromolar and peripheral binding sites: antagonism and enhancement of behavioural effects. Psychopharmacology (Berl) 1983;80:166–70. doi: 10.1007/BF00427962. [DOI] [PubMed] [Google Scholar]
- [43].Brini M, Marsault R, Bastianutto C, Alvarez J, Pozzan T, Rizzuto R. Transfected aequorin in the measurement of cytosolic Ca2+ concentration ([Ca2+]c). A critical evaluation. J Biol Chem. 1995;270:9896–903. doi: 10.1074/jbc.270.17.9896. [DOI] [PubMed] [Google Scholar]
- [44].Rizzuto R, Brini M, Bastianutto C, Marsault R, Pozzan T. Photoprotein-mediated measurement of calcium ion concentration in mitochondria of living cells. Methods Enzymol. 1995;260:417–28. doi: 10.1016/0076-6879(95)60155-4. [DOI] [PubMed] [Google Scholar]
- [45].Rizzuto R, Simpson AW, Brini M, Pozzan T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature. 1992;358:325–7. doi: 10.1038/358325a0. [DOI] [PubMed] [Google Scholar]
- [46].Rizzuto R, Brini M, Pozzan T. Intracellular targeting of the photoprotein aequorin: a new approach for measuring, in living cells, Ca2+ concentrations in defined cellular compartments. Cytotechnology. 1993;11(Suppl 1):S44–6. [PubMed] [Google Scholar]
- [47].Montero M, Alvarez J, Scheenen WJ, Rizzuto R, Meldolesi J, Pozzan T. Ca2+ homeostasis in the endoplasmic reticulum: coexistence of high and low [Ca2+] subcompartments in intact HeLa cells. J Cell Biol. 1997;139:601–11. doi: 10.1083/jcb.139.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Pinton P, Pozzan T, Rizzuto R. The Golgi apparatus is an inositol 1,4,5-trisphosphate-sensitive Ca2+ store, with functional properties distinct from those of the endoplasmic reticulum. EMBO J. 1998;17:5298–308. doi: 10.1093/emboj/17.18.5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–6. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- [50].Filippin L, Magalhaes PJ, Di BG, Colella M, Pozzan T. Stable interactions between mitochondria and endoplasmic reticulum allow rapid accumulation of calcium in a subpopulation of mitochondria. J Biol Chem. 2003;278:39224–3. doi: 10.1074/jbc.M302301200. [DOI] [PubMed] [Google Scholar]
- [51].Cosentino M, Marino F, Cattaneo S, Di Grazia L, Francioli C, Fietta AM, et al. Diazepam-binding inhibitor-derived peptides induce intracellular calcium changes and modulate human neutrophil function. J Leukoc Biol. 2000;67:637–43. doi: 10.1002/jlb.67.5.637. [DOI] [PubMed] [Google Scholar]
- [52].Hong SH, Choi HB, Kim SU, McLarnon JG. Mitochondrial ligand inhibits store-operated calcium influx and COX-2 production in human microglia. J Neurosci Res. 2006;83:1293–8. doi: 10.1002/jnr.20829. [DOI] [PubMed] [Google Scholar]
- [53].Ostuni MA, Ducroc R, Peranzi G, Tonon MC, Papadopoulos V, Lacapere JJ. Translocator protein (18 kDa) ligand PK 11195 induces transient mitochondrial Ca2+ release leading to transepithelial Cl- secretion in HT-29 human colon cancer cells. Biol Cell. 2007;99:639–47. doi: 10.1042/BC20070048. [DOI] [PubMed] [Google Scholar]
- [54].Campanella M, de Jong AS, Lanke KW, Melchers WJ, Willems PH, Pinton P, et al. The coxsackievirus 2B protein suppresses apoptotic host cell responses by manipulating intracellular Ca2+ homeostasis. J Biol Chem. 2004;279:18440–5. doi: 10.1074/jbc.M309494200. [DOI] [PubMed] [Google Scholar]
- [55].Szabadkai G, Simoni AM, Chami M, Wieckowski MR, Youle RJ, Rizzuto R. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell. 2004;16:59–68. doi: 10.1016/j.molcel.2004.09.026. [DOI] [PubMed] [Google Scholar]
- [56].Campanella M, Pinton P, Rizzuto R. Mitochondrial Ca2+ homeostasis in health and disease. Biol Res. 2004;37:653–60. doi: 10.4067/s0716-97602004000400022. [DOI] [PubMed] [Google Scholar]
- [57].Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–65. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- [58].Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Natl Acad Sci USA. 1999;96:13807–12. doi: 10.1073/pnas.96.24.13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Camello-Almaraz C, Gomez-Pinilla PJ, Pozo MJ, Camello PJ. Mitochondrial reactive oxygen species and Ca2+ signaling. Am J Physiol Cell Physiol. 2006;291:C1082–8. doi: 10.1152/ajpcell.00217.2006. [DOI] [PubMed] [Google Scholar]
- [60].Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans. 2006;34:232–7. doi: 10.1042/BST20060232. [DOI] [PubMed] [Google Scholar]
- [61].Duchen MR. Mitochondria and calcium: from cell signalling to cell death. J Physiol. 2000;529(Pt 1):57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–24. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- [63].Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, et al. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J Cell Biol. 2002;159:613–24. doi: 10.1083/jcb.200205091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Azarashvili T, Krestinina O, Yurkov I, Evtodienko Y, Reiser G. High-affinity peripheral benzodiazepine receptor ligand, PK11195, regulates protein phosphorylation in rat brain mitochondria under control of Ca(2+) J Neurochem. 2005;94:1054–62. doi: 10.1111/j.1471-4159.2005.03260.x. [DOI] [PubMed] [Google Scholar]
- [65].Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T, et al. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci USA. 2005;102:105–10. doi: 10.1073/pnas.0408352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Bassik MC, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ. Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 2004;23:1207–16. doi: 10.1038/sj.emboj.7600104. [DOI] [PMC free article] [PubMed] [Google Scholar]