

Abstract
A major public health goal is to determine linkages between specific pollution sources and adverse health outcomes. This paper provides an integrative evaluation of the database examining effects of vehicular emissions, such as black carbon (BC), carbonaceous gasses, and ultrafine PM, on cardiovascular (CV) morbidity and mortality. Less than a decade ago, few epidemiological studies had examined effects of traffic emissions specifically on these health endpoints. In 2002, the first of many studies emerged finding significantly higher risks of CV morbidity and mortality for people living in close proximity to major roadways, vs. those living further away. Abundant epidemiological studies now link exposure to vehicular emissions, characterized in many different ways, with CV health endpoints such as cardiopulmonary and ischemic heart disease and circulatory-disease-associated mortality; incidence of coronary artery disease; acute myocardial infarction; survival after heart failure; emergency CV hospital admissions; and markers of atherosclerosis. We identify numerous in vitro, in vivo, and human panel studies elucidating mechanisms which could explain many of these cardiovascular morbidity and mortality associations. These include: oxidative stress, inflammation, lipoperoxidation and atherosclerosis, change in heart rate variability (HRV), arrhythmias, ST-segment depression, and changes in vascular function (such as brachial arterial caliber and blood pressure). Panel studies with accurate exposure information, examining effects of ambient components of vehicular emissions on susceptible human subjects, appear to confirm these mechanisms. Together, this body of evidence supports biological mechanisms which can explain the various CV epidemiological findings. Based upon these studies, the research base suggests that vehicular emissions are a major environmental cause of cardiovascular mortality and morbidity in the United States. As a means to reduce the public health consequences of such emissions, it may be desirable to promulgate a black carbon (BC) PM2.5 standard under the National Ambient Air Quality Standards, which would apply to both on and off-road diesels. Two specific critical research needs are identified. One is to continue research on health effects of vehicular emissions, gaseous as well as particulate. The second is to utilize identical or nearly identical research designs in studies using accurate exposure metrics to determine whether other major PM pollutant sources and types may also underlie the specific health effects found in this evaluation for vehicular emissions.
Keywords: Vehicular, Diesel, Cardiovascular, Exposure, Ambient, Epidemiology, Air pollution
Introduction
Method of assessing effects of vehicular emissions on cardiovascular endpoints
A considerable literature base is now available relating cardiovascular (CV) health effects from exposure to ambient particulate matter (PM) deriving from various sources. Specific components of PM from such sources—which are heterogeneous physically and chemically—will likely have differential health impacts.
Vehicular emissions consist of particulate and gaseous emissions, with biologically active carbonaceous products present in both phases. Black carbon, mainly from diesels, is found in ultrafine and fine size fractions, mainly less than 1 μm in size and predominantly below 0.18μm (Mauderly and Chow 2008). Such vehicular particulates are often coated with condensed organic and inorganic compounds (Mauderly 2001; Health Effects Institute 1995). Approximately 75% of diesel PM2.5 emissions consist of such carbon (Health Effects Institute 2003). While particulate vehicular emissions per se, notably in the ultrafine fraction, have been specifically associated with endpoints such as oxidative stress and mitochondrial damage (Li et al. 2003), lipid peroxidation (Pereira et al. 2007), upregulation of genes relevant to vascular inflammation (Gong et al. 2007), and early atherosclerosis and oxidative stress (Araujo et al. 2008), non-particulate emissions have also been specifically linked to a variety of health endpoints (Mauderly and Chow 2008).
Our evaluation begins with the considerable amount of epidemiological evidence which has become available mainly since 2002 linking vehicular emissions with a range of CV outcomes. We then examine in vitro, in vivo, and human panel studies to understand if biological mechanisms have been identified in such studies which would explain the epidemiological findings for each CV outcome. Many of these studies use diesel emissions or diesel emission particulates, or evaluate effects from ambient air collected near highways in major cities with associations found with black carbon, mainly a diesel emission. Thus, our assessment assesses the coherence of results across different methodologies for CV endpoints, related to vehicular emissions.
Interpretation of studies related to accurate exposure assessment
Before reviewing epidemiological evidence linking vehicular emissions with CV endpoints, it is important to understand how differences among studies in accuracy of subject exposure to spatially variable emissions such as vehicular emissions can affect the strength and biological significance of associations.
Many epidemiological and panel studies use data from central monitors to characterize exposure to pollutants which may have considerable local variability and thus do not accurately characterize subjects’ exposure to these emissions (Ito et al. 2004). Using central monitors or other less precise exposure estimation methods will result in underestimates of risks (Zeger et al. 2000), including those from vehicular emissions (Adar and Kaufman 2007). In multi-pollutant models, differential exposure error may cause risks to be transferred from variables having more exposure error to those having less (Goldberg and Burnett 2003; Hennekens and Buring 1987). Thus, it is essential to understand to what extent currently available studies include accurate assessment information for pollutants that might be particularly harmful, and to assess how these exposures relate to the magnitude and significance of risk estimates. This type of evaluation is necessary to determine whether emissions from specific sources rather than others may be more critical to regulate so as to preserve public health. Even with subject exposure misclassification, positive significant associations are not ruled out; however, in such a case, they are likely understated (Zeger et al. 2000).
This paper attempts to address these issues for vehicular emissions as they relate to CV health endpoints, with emphasis on epidemiological investigations incorporating reasonably accurate subject exposure information. We define “reasonably accurate exposure” as that exposure metric resulting from a methodology in which the measured exposure concentration varies with and, therefore, reflects reasonably closely the actual exposure for the population that is being assessed for adverse health outcomes. Methodologies which meet this criteria would include (1) personal monitors, (2) monitors which follow subjects closely as they go about their daily activities, (3) studies which use a combination of wind trajectories and pollution measurements to understand what sources were influencing exposure in the time period(s) of interest, and (4) studies in which the monitor was in close proximity to a roadway, and the subjects of the study also lived in close proximity to the same roadway not far distant from the monitor (Grahame 2009). On the other hand, an epidemiological study using a central monitored concentration and using this concentration as an exposure metric for subjects living over a wide area represents inaccurate exposure for traffic-related emissions, because such emissions have substantial variance across a city (Ito et al. 2004) and even within 100 m of a major highway vs. further away (Zhu et al. 2002a, b). A fuller discussion of these issues is found in Grahame (2009), which considers formally whether health effect associations vary consistently among studies which use reasonably accurate subject exposure information for spatially variable emissions such as BC, vs. studies using centrally monitored concentrations as a proxy for exposure to such local emissions.
The vehicle/traffic emissions category was selected since there is a large and growing evidence base suggesting that traffic-related pollution likely plays an important role in adverse health outcomes associated with ambient pollution such as urban PM, including black carbon (BC; White et al. 2005; Samet 2007; Adar and Kaufmann 2007; Li and Nel 2006; Delfino et al. 2008), the latter derived primarily from diesel engines.
We first consider epidemiological studies which utilize information relating proximity of residence to major roads, to determine the extent to which such proximity may be associated with health outcomes such as all-cause or cardiovascular mortality and morbidity. Some of these studies examined associations of specific vehicular emissions, e.g., BC as a marker of diesel emissions, with such outcomes. Similar studies use modeled exposure to vehicular emissions (BC, or mainly outside the U.S., NO2) at the residence, or use traffic density as a proxy for exposure to vehicular emissions (at county level or within 100 m of the residence).
We then consider studies which examine specific CV biological endpoints, e.g., oxidative stress, inflammation, change in EKG pattern (ST-segment depression), changes in heart rate variability (HRV), vasoconstriction and changes in blood pressure, arrhythmias, and lipoperoxidation/atherosclerosis. Such studies can vary considerably in accuracy of subject exposure; again, we emphasize findings from studies with more accurate exposure information. Toxicological evidence regarding specific biological mechanisms is also examined to understand if there might be common threads which extend from toxicological through panel studies to epidemiological evaluations.
Associations between vehicular emissions and lung cancer, and mutagenicity of vehicular emissions, have been reviewed elsewhere (Grahame and Schlesinger 2007) and will not be reviewed here. Similarly, the ample literature relating vehicular emissions and highway proximity to respiratory morbidity endpoints, such as asthma, will not be discussed.
Epidemiological evidence for association of vehicular emissions with adverse cardiovascular health outcomes
Investigating health effects specific to vehicular emissions in epidemiological studies has historically been problematic. Early studies did not monitor for pollutants most closely related to vehicular emissions (e.g., BC) and, thus, could not find associations with such emissions. Later reanalyses of the early studies were sometimes able to parse out such associations, despite the lack of vehicular pollution data (e.g., the Jerrett et al. 2005 reanalysis of the American Cancer Society cohort [Pope et al. 1995, 2002]). When studies began to monitor specifically for vehicular emissions such as BC, initial exposure assessment used central monitor data and associations were often not robust. NO2 is also seen as a marker of vehicular emissions, particularly in European studies. Although NO2 is emitted from sources other than vehicles, such as power plants and industry, vehicular NO2 emissions usually dominate in busy urban centers lacking major industry. In some cases, where monitors were located in close proximity to major highways, even SO2 and/or SO4 from different sources, including diesels before the 2007 changeover to ultra-low sulfur diesel fuel, may be intermixed (Grahame and Hidy 2007a, b).
Studies using central monitoring for several different sources, including vehicles, did not always find daily mortality or morbidity associated with vehicular emissions. For example, Thurston et al. (2005) failed to find cardiovascular or non-accidental mortality associated with a traffic emissions factor in either Phoenix or Washington, DC, using different source apportionment models. However, the daily mortality study of Laden et al. (2000) found such associations, as did the Schwartz (2003) reanalysis of Laden et al. (2000), necessitated by problems in the original study with a statistical software package (which affected many other studies as well). Another multi-city study (Janssen et al. 2002) examined prevalence of air conditioning as an effect modifier, and found associations for daily morbidity (hospital admissions for CV disease) with diesel emissions, highway emissions, and vehicle miles traveled per square mile (traffic density). Two recent studies based in Atlanta (Sarnat et al. 2008; Tolbert et al. 2007) found significant associations between vehicular emissions and emergency department admissions for CV disease. However, Metzger et al. (2007), in a study also in the Atlanta area, failed to find associations between PM10, PM2.5, or vehicular emissions with daily morbidity (i.e., arrhythmias).
Four of these six multi-pollutant studies found CV health associations with vehicular emissions, but none of the studies utilized exposure measurements known to reflect subject exposure reasonably accurately, because all used pollution measurements from central monitors as a proxy for personal exposure. While these studies, taken as a whole, suggest the importance of vehicular emissions for the health endpoints examined, strength of effects may be underestimated (Ito et al. 2004), and results are not always consistent. Thus, the most credible studies linking spatially variable vehicular emissions to various health effect endpoints would be those which demonstrate that the ambient pollutant concentrations utilized relate reasonably well to actual exposure of populations examined for health effects.
Concentrations of vehicular emissions have been shown to drop by as much as an order of magnitude within 100 m of a major freeway. People living nearby major roadways will be exposed to greater amounts of vehicular emissions such as BC, ultrafine PM, and gaseous emissions, than will those living at a greater distance (Zhu et al. 2002a, b). As a result, the studies first to show large adverse cardiovascular effects clearly and consistently linked to vehicular emissions were “highway proximity studies.” These are studies designed to isolate the health risks of living near a major road from the traditional risk factors typically examined in cohort studies.
Conducted first in Western Europe and Canada, and later in the US, highway proximity studies found significantly elevated risks for cardiovascular death or morbidity outcomes and for all-cause mortality, for people living in close proximity (usually 100 m to major roadways or 50 m to a major urban road), compared to those living farther away. Several utilized data from existing cohort studies, adding only the highway proximity variables. In different ways and to different degrees, these studies accounted for socioeconomic and other variables which could confound pollution–health associations. These studies include the following (statistically significant associations are indicated):
Finkelstein et al. (2004, 2005; all-cause mortality [relative risk, RR = 1.18] and circulatory disease mortality [RR = 1.40], respectively);
Hoek et al. (2002; cardiopulmonary mortality [RR = 1.95]);
Gehring et al. (2006; cardiopulmonary mortality, RR = 1.70);
Tonne et al. (2007; acute myocardial infarction [MI], RR = 1.04 to 1.06);
Hoffmann et al. (2007; increased coronary artery calcification: RRs = 1.63 and 1.34 for distances of less than 50 m and 51 to 100 m from highways, vs. more than 200 m distant); and
Hoffman et al. (2006; incidence of coronary heart disease, RR of 1.85).
These RRs can be compared to RRs for all-cause and cardiopulmonary mortality of 1.04 and 1.06 (per 10 μg/m3 increase in PM2.5) found in the American Cancer Society (ACS) study (Pope et al. 2002). Finkelstein et al. (2004) found a significant mortality rate advancement period of 2.5 years associated with residence near a major roadway.
Another method of associating mortality risks with proximity to traffic and highways utilizes different measures of traffic density (e.g., annual vehicle miles per square kilometer in a county, or daily vehicle kilometer within 100 m of a residence) as a proxy for exposure to vehicular emissions. Relevant studies finding significant health associations with traffic density measures include:
Kan et al. 2008 (incidence of coronary heart disease [CHD]; significant increase in CHD incidence hazard ratio [HR] of 1.32 for those in highest quartile of traffic density vs. lowest quartile, HR of 1.38 for those in second highest quartile vs. lowest quartile);
Janssen et al. 2002 (hospital admissions for CVD; one interquartile range [IQR] increase in vehicle miles traveled per square mile significantly associated with a 21.2% increase in CVD hospital admissions);
Lipfert et al. 2006a, b (all-cause mortality; between these two studies, 30 of 33 tests of traffic density measures were significant, in single and multi-pollutant models, with two different ways of stating risks, with RRs centering around 1.16 in Lipfert et al. 2006a 1); and
Medina-Ramon et al. 2008 (survival after heart failure; an IQR increase in daily traffic within 100 m of home significantly associated with a mortality HR of 1.12 after adjustment for SES, and with a HR of 1.30 for those living within 50 m of a bus route).
The Medina-Ramon et al. (2008) study found that a measure of traffic density (vehicle km within 100 m of residence) was a stronger predictor of survival time after heart failure than were other measures, such as similar distance to a major roadway. This finding makes intuitive sense—ideally, exposure should be a product of both proximity and of the amount of pollution produced within that proximity. Just as proximity of a residence to a major highway is a better proxy for exposure to traffic emissions than is centrally monitored PM concentrations, traffic density within 100 m of a residence is likely a better proxy than distance of 100 m from a major road. Distance to a bus route was also an important predictor of survival in Medina-Ramon et al. (2008), suggesting the importance of diesel emissions.
Results of Kan et al. (2008) parallel findings of Hoffmann et al. (2006), in that both studies controlled for hypertension, a mechanism by which noise might cause stress and, thus, by which traffic might cause coronary heart disease via noise pollution rather than chemical pollution. As a result, findings associating mortality and morbidity risks to proximity to busy highways and traffic likely can be attributed primarily to chemical pollution rather than to noise.
Using geographic modeling systems to relate individual residences to either traffic emissions concentrations, distance to major roads, or traffic intensity, other studies also found elevated levels of vehicular pollutants to be associated with adverse cardiac-related health outcomes. Examples include:
Rosenlund et al. (2006; NO2 modeled to home significantly associated with out-of-hospital deaths, OR of 2.17 for a 5% to 95% increase in NO2 [in Stockholm, Sweden]);
Maynard et al. (2007; significant 2.3% increase in all-cause mortality per IQR increase in BC modeled to home);
Beelen et al. (2008; significant RR of 1.05 for all-cause mortality for a 5% to 95% increase in black smoke); and,
Nafstad et al. (2004; significant RR of 1.11 for ischemic heart disease mortality associated with a 10 μg/m3 increase in NO2 at home [in Oslo, Norway]).
Finally, Peters et al. (2004) found that a first MI was significantly associated with being in traffic 1 h prior to the event (odds ratio 2.92).
What all of these studies have in common is a demonstration that people exposed to the higher levels of traffic emissions found close to major urban roads or freeways, or modeled to their homes, have higher long-term risks for ischemic heart disease, acute MI morbidity and mortality, and all-cause mortality, or increased risks of a first-time MI with short-term exposure to traffic pollution. These studies, with reasonably accurate subject exposure information for traffic emissions, have results which often (but not always) contrast with studies using central monitors.
Several studies noted above also associated BC or its near equivalent, black smoke, with increased morbidity and mortality risks. Hoek et al. (2002) found risks of cardiopulmonary mortality significantly associated with black smoke (local plus background, RR = 1.71; background black smoke, RR = 1.34), suggesting the greater effects of fresh, local roadway black smoke. Maynard et al. (2007) found that an increase of one IQR (interquartile range) in previous days BC exposure was associated with a significant increase of 2.3% in all-cause mortality in single-pollutant models, and of 2.2% in two pollutant models. Beelen et al. (2008) found significant increases in all-cause and respiratory mortality associated with black smoke and/or NO2 but not with PM2.5 or SO2. Nafstad et al. (2004) found a 10 μg/m3 increase in nitrogen oxides at the home to be associated with significant increases in all-cause and ischemic heart disease mortality of 1.08 and 1.11, respectively. Relative risks of cardiopulmonary mortality of 1.57 and of all-cause mortality of 1.17 were related to near-highway NO2 exposure in Gehring et al. (2006). Rosenlund et al. (2006) found an odds ratio for out-of-hospital death of 2.17 related to NO2 exposure.
Mechanistic bases for adverse cardiovascular health outcomes from vehicular emissions-derived PM
In order to provide mechanistic plausibility for the epidemiological findings noted above, it would be helpful to have information about specific conditions or biological responses which could lead to these reported health outcomes. Such information can be derived from in vitro and in vivo toxicology studies, as well as from human panel studies using vehicular emissions or ambient air.
Personal monitors would provide better exposure assessment than would a proxy for personal exposure, such as distance of residence from a major road, or modeled exposure at one’s home. Although each of these proxies provides far better exposure differentiation than would a central monitor reading applied to all residents in a locality, there will still be differences in daily activity patterns and, thus, exposures, among similarly situated people. For obvious reasons, cohort studies of mortality and of many morbidity outcomes such as hospital admissions for CV disease could not utilize personal monitors. However, for certain short-term morbidity outcomes with fairly high frequencies, such as change in HRV, ST-segment depression, and arrhythmias, the use of personal pollution monitors or their equivalent may be possible.
Oxidative stress
Oxidative stress is a mechanism postulated to be involved in various PM-induced cardiovascular health effects, including chronic heart failure (McMurray et al. 1993), acute heart failure (especially when patients had atrial or ventricular arrhythmia [Charniot et al. 2008]), and atrial fibrillation (AF; Neuman et al. 2007). Kim et al. (2003) found in AF patients that gene expression profiles for reactive oxygen species were upregulated, while those for anti-oxidants were downregulated. Iravanian and Dudley (2006) suggest a unifying hypothesis that there are multiple triggers for oxidative stress and that oxidative stress, whatever the origin, causes AF. Furthermore, AF itself can result in further oxidative stress, creating a positive feedback loop.
Chahine et al. (2007) found that individuals lacking genes protective against oxidative stress (GSTM1 and the short repeat variant of HMOX-1), but not those with such genes, are vulnerable to HRV changes due to pollution exposure. Instillation of urban air particles and inhalation of concentrated ambient air particles caused oxidative stress in the heart in vivo, as well as reduction in HRV (Rhoden et al. 2005); increases in heart oxidant levels were demonstrated by increases in chemiluminescence or TBARS. Oxidant effects were abolished by the anti-oxidant, NAC. When oxidative stress was abolished, HRV returned to normal (Rhoden et al. 2005). Thus, findings of both Chahine et al. (2007) and Rhoden et al. (2005) suggest that reductions in HRV appear to be due to increased oxidative stress. If this is true, then lack of HRV changes may indicate lack of oxidative stress in some to many cases.
Given the evidence that oxidative stress can be mechanistically linked to cardiac pathophysiology, we next review studies which find that exposure to vehicular emissions generally, and to diesel emissions specifically, are linked to oxidative stress.
Pereira et al. (2007) found that in vivo exposure to ambient emissions taken adjacent to a busy road in Porto Alegre, Brazil caused oxidative stress and lipid peroxidation in rat lungs. Similarly, Huang et al. (2003) found that PM1.0 was more likely to cause lipid peroxidation in human bronchial epithelial cells than either PM1.0–2.5 or PM2.5–10, and that the components of PM1.0 associated with increased lipid peroxidation were organic carbon (OC) and elemental carbon (EC), but not various ions.
A number of studies suggest that diesel emissions, most specifically ultrafine PAH, appear to be associated with increased levels of cellular oxidative stress. As reviewed in Grahame and Schlesinger (2007), a series of studies demonstrated the ability of both diesel emissions and of ambient air in Los Angeles to cause oxidative stress in vitro in bronchial epithelial cells. These effects were highly correlated with organic carbon and PAH content. Briefly, Li et al. (2002a) found that organic diesel emission particles (DEP) caused oxidative stress in vitro (marked by increases in heme oxigenase-1, HO-1), and Li et al. (2002b) found that concentrated emissions from Los Angeles air, collected from near a major freeway, also caused increases in oxidative stress, e.g., in HO-1. Ultrafine (UF) fractions of Los Angeles air stimulated higher production of HO-1 than did larger PM fractions (Li et al. 2003). Production of HO-1 was correlated with the organic and PAH content of the ultrafine PM. Electron microscopy showed these UF particles penetrated into subcellular structures more easily than did larger particles, and damaged mitochondria. Taken together, these in vitro studies of Los Angeles air suggest that the ultrafine fraction of diesel emissions in Los Angeles air, likely including ultrafine BC coated with a mix of organic compounds, appear to be causally related to the increases in oxidative stress also found to be caused by organic DEP.
Findings of oxidative stress were confirmed in vivo, in a study where HO-1 levels in mice exposed to the exhaust of a normally running newer diesel increased significantly vs. filtered air control (McDonald et al. 2004). Furthermore, when a new catalytic trap was retrofitted on the diesel, most of the carbonaceous emissions were reduced by large percentages, BC was entirely oxidized, and the HO-1 levels were no longer elevated.
The panel study of Delfino et al. (2008) used indoor and outdoor monitors at the residences of 29 non-smoking elderly subjects with coronary artery disease living in Los Angeles. Decreased levels of an anti-oxidant enzyme were significantly associated with increased concentrations of BC, primary OC of outdoor origin, NO2, and ultrafine PM.
Mills et al. (2005) exposed healthy human volunteers to diluted diesel exhaust (300 μg/m3) or to filtered air for 1 h in a double-blind, randomized, crossover study. The authors found that inhalation of diesel exhaust impaired the regulation of vascular tone and endogenous fibrinolysis. Net release of “clotbusting” t-PA antigen was significantly reduced 6 h after exposure. The authors postulated that effects might be caused by reduced availability of nitric oxide (NO) in the vasculature due to oxidative stress induced by the ultrafine particle fraction of diesel exhaust, providing a mechanistic link for associations between PM and acute MIs. Consistent with this supposition, previous work has found that the ultrafine fraction of diesel emissions (likely with adsorbed carbonaceous species) causes greater levels of oxidative stress than larger fractions (Li et al. 2002a, b, 2003; Araujo et al. 2008). The results of Mills et al. (2005) suggest one possible explanation for the significant finding of an initial myocardial infarction in 1 h after being in traffic (as a driver or on public transit) noted by Peters et al. (2004), and for increases in cardiovascular mortality linked to traffic emissions on high pollution days (Schwartz 2003).
Because of its potential to be involved in various specific aspects of CV pathophysiology, further discussion of the role of oxidative stress in pollutant-related health outcomes is provided in discussions of individual CV endpoints below.
Alteration in heart rate variability
Many recent studies assess HRV, which refers to alterations in the beat to beat heart rate and is regulated by the autonomic nervous system. While normal sinus rhythm is characterized by regular R–R intervals in the EKG, the heart does normally show some variability from beat to beat, which can be measured by examination of these intervals. HRV changes appear to be predictive of MI for those who have had a previous MI (Tapanainen et al. 2002), or who have chronic congestive heart failure (Bilchick et al. 2002). Schwartz et al. (2005a) found that in those lacking the allele for glutathione-S-transferase M1 (GSTM1), a component of the cellular defense against oxidative stress, PM2.5 effects on HRV (a decrease in high-frequency component, HF) is mediated by reactive oxidant species. Use of statins, which have anti-oxidant effects, eliminated the effects of PM2.5 on HRV. Thus, while changes in HRV may be a cause of cardiac mortality, in particular for those who have had a previous MI, it also appears to be another possible sequela of oxidative stress in the heart (Rhoden et al. 2005; Schwartz et al. 2005a), which has broader health implications than change in HRV alone.
Several recent studies with accurate exposure information suggest that either vehicular emissions specifically, or urban emissions generally, are associated with changes in HRV. A literature search was performed to find studies of HRV which used ambient air and human subjects in the US, and which monitored for at least two of the three pollutants (PM2.5, BC, sulfate).
Adar et al. (2007) used a mobile monitor, which followed 44 non-smoking elderly residents of a seniors’ home who were going about daily activities, and found significant associations between increased BC exposure for the entire period and changes in six measures of HRV (see Table 1), for two different time periods, a total of 12 significant associations in 12 tests. When the subject boarded a bus and BC concentrations rose by about an order of magnitude, changes in most HRV measures were similar.
Table 1.
Vehicular emissions and heart rate variability changes
| Study | Subject exposure method | Characterization of HRV changes | BC levels |
|---|---|---|---|
| A. In vivo animal study | |||
| 1. Anselme et al. (2007) | Healthy and CHF rats exposed to diesel emissions | Immediate decrease in RMSSD in both CHF and healthy rats immediately after exposure, returning to baseline after 2.5 h | BC not measured |
| B. Human studies with accurate exposure | |||
| 1. Schwartz et al. (2005b) | Subjects lived adjacent to same urban road to which monitor was adjacent, less than 1 km distant | Monotonic decrease in SDNN with increase in BC exposure; significant BC associations in seven of eight tests (SDNN, RMSSD, PNN50, LF/HF, 1 and 24-h averages); no significant associations in eight tests for PM2.5 without BC (“non-traffic secondary particles”) | BC mean = 1.2 μg/m3 |
| 2. Adar et al. (2007) | Monitor followed subjects during activities, in residence at night | For change of one IQR, BC significantly associated with changes in all six measures of HRV, for both 5-min and 24-h means; sharply increased exposure to BC when subjects on buses associated with changes of similar magnitude in all six HRV measures (decreases in SDNN, PNN50 + 1, RMSSD, LF, and HF; increase in LF/HF), similar to Schwartz et al. (2005b) | BC mean not given; BC IQR for all periods was 330 ng/m3; for bus periods, IQR was 2911 ng/m3 |
| 3. Riediker et al. (2004b) | Presence of young patrol officers in vehicle for 9 h before tests | Significant increases in SDNN, PNN50 associated with “speed change” source factor, (braking and diesel emissions), but not “crustal,” “steel wear” or gasoline factors | BC not measured |
| 4. Ebelt et al. (2005) | Personal monitors in panel study in Vancouver | Estimated non-sulfate urban PM2.5 associated with decreased RMMSD, sulfate not associated | BC not measured |
| C. Human studies using central monitors not far from street level (horizontal exposure misclassification) | |||
| 1. Wheeler et al. (2006) | Central monitor for greater Atlanta area subjects | EC associated with SDNN changes in only one of four tests, NO2 in only 4 of 13 tests; authors discuss exposure error due to spatial variability of NO2, note “this greater exposure error is consistent with the fact that traffic, which varies spatially over short distances, is a significant source of outdoor NO2.” | EC mean = 1.6 μg/m3 |
| 2. Park et al. (2005) | Central monitor for subjects living within 40 km of monitor | BC associated with one of four measures of HRV changes; exposure discussed in context of PM2.5 (little exposure error) but not discussed for BC | BC mean = 0.92 μg/m3 |
| D. Studies using highly elevated central monitors (horizontal and vertical exposure misclassification) | |||
| 1. Luttmann-Gibson et al. (2006) | Central monitor elevated 400 feet above town where subjects lived, a mile from monitor | For IQR change in PM2.5 or sulfate, significant reductions in SDNN, RMSSD, HF, and LF (sulfate borderline for LF), no associations for BC; exposure error not discussed | BC mean = 1.0 μg/m3 |
SDNN standard deviation of normal-to-normal intervals, RMSSD square root of mean squared difference between adjacent normal-to-normal intervals, PNN50 percentage of adjacent normal-to-normal intervals differing by more than 50 ms, HF high-frequency power, LF low frequency power, LF/HF ratio LF to HF, RR risk ratio, OR odds ratio, IQR interquartile range increase, SD standard deviation
Schwartz et al. (2005b) examined HRV in 27 seniors living in close proximity to a busy urban road in Boston. The monitor, located about 0.5 km distant from the living quarters and also adjacent to the same road, recorded BC and PM concentrations which would mirror concentrations at the residences reasonably well (because of similar close proximity to the major urban road). The investigators found in six of eight tests (four HRV measures, two time frames), that increased BC was significantly associated with changes in different measures of HRV (a seventh test was borderline significant). PM2.5 was significantly associated in two of eight tests. When an algorithm was used to remove BC from PM2.5 on an hourly basis and thus obtain a measure of what the authors viewed as regional PM2.5 without fresh BC and correlated emissions, no associations were found between PM2.5 and HRV measures (Fig. 1). These findings appear to show that previous studies which did not monitor for BC and found associations with PM2.5, may have been detecting unmeasured BC (i.e., vehicular) effects, and that PM2.5 was associated with various changes in HRV only when PM2.5 was highly correlated with BC, another finding of Schwartz et al. (2005b).
Fig. 1.
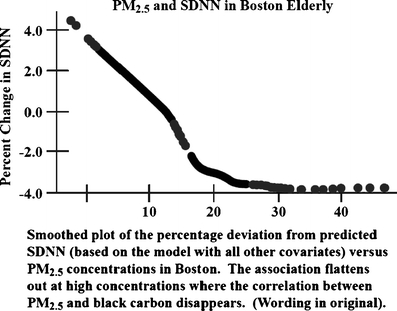
SDNN monotonically decreases with increased PM2.5 when PM2.5 is highly correlated with BC, but is not affected by rising levels of PM2.5 when PM2.5 is higher and correlated with regional PM, but not BC [from Schwartz et al. (2005b), reproduced with permission]
Creason et al. (2001), in a study of 56 non-smoking seniors with a mean age of 82, used wind trajectories to determine which air masses (containing different types of pollution) were associated with changes in HRV. The authors initially found a “U-shaped” association between increasing PM2.5 levels and decreasing HRV—as PM concentrations increased from the lowest levels, HRV reductions occurred with increasing PM2.5, but toward the highest PM2.5 concentrations, HRV reductions reversed, and the HRV measure returned to where it was at the lowest PM2.5 levels (null effect). Inclusion of a 2-day air mass with high PM2.5 (highest and third highest of 24 days) had caused the reversion. On these 2 days, the investigators found, wind trajectories showed that the air masses had come from rural north-central Pennsylvania. When these 2 days were removed from the analysis, there was a monotonic decrease in HRV with increasing PM, similar to the monotonic decrease in PM found in Schwartz et al. (2005b) when the PM was highly correlated with urban BC. The authors noted that HRV reductions were found when the emissions reflected urban or industrial activity, but that there was no toxicity in the 2 days with high PM from rural areas.
Similar effects were noted by Park et al. (2007), a study of 487 male veterans. The authors found that trajectories reflecting urban emissions (either a stagnant local air mass in Boston or an air mass transported from the Washington, DC through New York City corridor) were associated with changes in several HRV measures. Two other trajectories reflecting mostly rural air masses were not so associated, even though monitored levels of sulfate, BC, and PM2.5 were very similar among the air masses. Finally, Ebelt et al. (2005) used personal monitors in a panel study of 16 non-smoking COPD patients, and found that local urban particles, but not sulfates, were associated with HRV changes.
All these above-studies are characterized by reasonably accurate exposure information, whether for BC (Adar et al. 2007; Schwartz et al. 2005b), for PM in urban/industrial air masses vs. rural air masses (Creason et al. 2001; Park et al. 2007) or for urban emissions vs. sulfate (Ebelt et al. 2005). The two studies which monitored for BC (Adar et al. 2007; Schwartz et al. 2005b) showed consistently significant associations between changes in HRV measures and BC concentrations, with virtually no insignificant associations. Further, HRV changes were essentially monotonic with increasing BC levels in these studies.
Since the main contributor to “urban excess” PM2.5 is vehicular emissions marked by carbonaceous species (Rao et al. 2002), the urban vs. rural air mass results are consistent with results of exposure to higher BC and vehicular emissions in an urban area. Furthermore, Anselme et al. (2007) exposed Wistar rats (ten healthy, ten with chronic ischemic heart failure, CHF) to diluted diesel emissions. Immediate decreases in a HRV measure (RMSSD) were observed in both healthy and CHF rats following exposure, a finding which parallels the reductions in HRV found in Schwartz et al. (2005b) and Adar et al. (2007) in association with increased BC exposure.
Other extant studies lacking accurate exposure information for locally variable emissions because they used central monitor readings for subjects living over a wide area, generally showed weak (Wheeler et al. 2006; Park et al. 2005) or non-existent (Luttmann-Gibson et al. 2006) associations for locally variable BC (Table 1), while some studies with central monitor readings still found associations with vehicular emissions (de Hartog et al. 2009). The exposure assessment for Luttmann-Gibson et al. in particular, uses a monitor several hundred feet in the air, adding substantial vertical exposure misclassification (Restrepo et al. 2004) to the horizontal exposure misclassification (Ito et al. 2004) found in Wheeler et al. (2006) and Park et al. (2005). We are unaware of any study with reasonably accurate exposure information for BC or for urban emissions which fails to find associations between such emissions and HRV alteration when the latter was examined. A detailed review of US HRV studies utilizing ambient air and human subjects concludes that results of these studies appear to vary consistently with accuracy of exposure to vehicular emissions (Grahame 2009), with consistently strong BC associations with changes in various measures of HRV, almost always significant, only when BC exposure measures are reasonably accurate.
In a study of 643 healthy men and women aged 55 to 75 and with no prior history of heart disease or stroke, Sajadieh et al. (2004) suggested the possibility of interactions between HRV changes (imbalance in sympathetic system) and subclinical inflammatory processes in promoting atherosclerosis. Thus, there are likely linkages between various aspects of cardiopulmonary pathophysiology related to pollutant exposure.
ST-segment changes
The ST interval in the EKG represents the period during which the ventricles depolarize. A depression in the ST-segment is associated with increased risk for future cardiac events; those with undetected ST-segment depression have significantly greater risk of death from cardiovascular disease and stroke (Kurl et al. 2003). Pekkanen et al. (2002) found elevated levels of PM2.5 mass in urban areas to be associated with ST-segment depression, but could not determine which specific components of PM2.5 resulted in such a response. On the other hand, Gold et al. (2005), in a study with the same reasonably accurate exposure protocol as its companion study (Schwartz et al. 2005b), found BC strongly associated with ST-segment depression. Total PM2.5, which would include a mixture of both regional and local pollution, was not associated with ST-segment depression in this study.2 Lanki et al. (2006) found that of several local and regional pollutants, only a traffic factor marked by absorbance coefficient (ABS, similar to black carbon) was associated with ST-segment depression. Subject exposure in Lanki et al. (2006), however, was based on central monitor readings.
Although Lanki et al. (2006) did not have as accurate exposure characterization as did Gold et al. (2005), both of these studies came to the same conclusion, namely, that vehicular emissions marked by BC or ABS were associated with ST-segment depression. This endpoint, as well as HRV/oxidative stress in the heart and arrhythmias, is among the likely causes for a first-time MI 1 h after being in traffic (Peters et al. 2004).
In a double-blind, random crossover study (Mills et al. 2007), subjects with coronary heart disease were exposed for 1 h to diluted diesel exhaust or filtered air (alternating 15 min of mild exercise with 15 min of rest). ST-segment depression occurred in both sets of subjects, but the average change in ST-segment depression was twice as great in subjects exposed to diluted diesel exhaust. This result with diesel emissions parallels results of the two epidemiological studies of this endpoint reviewed above, which found associations with BC or its equivalent, ABS.
Yan et al. (2008) examined left ventricular function in healthy rats exposed to diesel exhaust particles (NIST standard) and in those subjected to isoproterenol-induced injury as a model for congestive heart failure. Diesel exhaust particles impaired left ventricular functioning, such as end diastolic diameter, in healthy mice, and this was further impaired in the myocardial injured animals. This suggests that acute exposure to these particles may result in exacerbation of congestive heart failure, which supports epidemiological findings of an association between PM components, fine PM, OC, and EC, and hospital admissions for congestive heart failure (e.g., Metzger et al. 2004).
Cardiac arrhythmia
Although some studies have associated measures of air pollution with arrhythmias monitored by implantable cardioverter-defibrillators (ICDs), none to date have utilized either personal pollution monitors or ambient monitors which follow residents of a seniors’ home, or the specific exposure protocol of Schwartz et al. (2005b) and Gold et al. (2005). Thus, the available database having good spatial exposure information, and which would also allow evaluation of those pollutants that may be important for triggering arrhythmias, is limited.
Albert et al. (2007) examined risks of an ICD shock during and after driving, finding increased risk in the hour after driving (significant RR = 2.24). The risks were specific for ventricular tachycardia or ventricular fibrillation (VT/VF), which occurred primarily in the half hour after driving (RR = 4.46, CI = 2.92 to 6.82). These risks are similar to but higher than the risks for a first MI found by Peters et al (2004) in the first hour after being in traffic (odds ratio 2.92; 95% CI = 2.22 to 3.83). Charniot et al. (2008) found that oxidative stress in the heart was linked to acute heart failure when a subject had a ventricular arrhythmia. Thus, ventricular arrhythmias and oxidative stress, both of which are linked to exposure to vehicular emissions, together appear to contribute to acute heart failure. However, the role of oxidative stress as a specific cause of such arrhythmias is not yet fully understood. If oxidative stress were to be the cause of VT/VF, then the studies linking BC to HRV/oxidative stress (e.g., Schwartz et al. 2005b; Adar et al. 2007) would point to BC as a primary cause of arrhythmias as well. The actual cause of the oxidative stress could be BC itself, emissions adsorbed onto the BC, and/or emissions highly correlated with BC.
Riediker et al. (2004a, b) examined 11 different health endpoints, including changes in supraventricular ectopic (SVE) beats, an arrhythmia variable, in healthy young patrol officers. Measurements for this endpoint took place 10 h after a 9-h duty shift on roads and highways. Using the suite of emissions measured, the authors established three different factors of emissions, reflecting PM on road surfaces, gasoline emissions, and a “speed change” factor reflecting accelerating diesels and brake wear. Increases in SVE beats were associated only with the speed-change factor.
Ebelt et al. (2005) also found, in a study using personal monitors, increases in SVE beats associated with ambient urban PM and ambient non-sulfate PM, but not with sulfate, in a panel study in the Vancouver area. Table 2 shows results of studies of vehicular emissions and arrhythmias, stratified as in Table 1 by accuracy of exposure.
Table 2.
Vehicular emissions and arrhythmia risks
| Study | Subject exposure method | Risks of arrhythmia |
|---|---|---|
| A. In vivo animal study | ||
| 1. Anselme et al. (2007) | CHF rats exposed to diesel emissions; no effect in healthy rats | 200% to 500% increase in ventricular premature beats, persisting up to 5 h after exposure |
| B. Human studies with accurate exposure | ||
| 1. Albert et al. (2007) | Presence in vehicle before ICD events | RR of ICD shock in hour after driving =2.24; RR of ventricular tachycardia or ventricular fibrillation in half hour after driving =4.46 |
| 2. Riediker et al. (2004b) | Presence of young patrol officers in vehicle for 9 h before tests | ∼ 40% increase in SVE beats per change of one SD in “speed change” source factor (braking and diesel emissions), but not in “crustal,” “steel wear” or “gasoline” factors |
| 3. Ebelt et al. (2005) | Personal monitors in panel study in Vancouver area | Ambient and estimated PM2.5, non-sulfate PM2.5 each associated with ∼ln 0.2 SVE effect estimate; sulfate not associateda |
| C. Human studies using central monitors not far from street level (horizontal exposure misclassification) | ||
| 1. Peters et al. (2000) | Central monitor for eastern Massachusetts area subjects | Risks highest for NO2 and BC, then PM2.5; results seen by authors as related predominantly to traffic emissions; OR of ICD shock 2 days later =1.8 for 26 ppb increase in NO2; due to single monitor, authors’ expectation would have been to bias estimates of gaseous emissions toward null |
| 2. Dockery et al. (2005) | Central monitor for subjects living within 40 km of monitor in Boston | Ventricular tachyarrhythmias associated with BC, NO2, CO, and PM2.5, for those with an arrhythmia in previous 3 days, authors see these as indicative of vehicular emissions; for BC, OR = 1.74 for increase of 0.74 μg/m3 IQR exposure, for NO2, OR = 1.34 for 7.7 ppb IQR increase; exposure misclassification discussed, thought to weaken associations observed |
| 3. Metzger et al. (2007) | Central monitor data for patients living in metro Atlanta area | No associations with ICD events with PM2.5, NO2, CO, EC, OC, SO4; exposure misclassification discussed, study “does not contribute evidence regarding whether personal exposure may be a determinant of ventricular tachyarrhythmia” |
| 4. Rich et al. (2006) | Central monitor for subjects living within 40 km of monitor in Boston | No risk associations for paroxysmal atrial fibrillation episodes with PM2.5, NO2, BC, CO, or SO2; associations only with ozone; small number of episodes, thus reduced statistical power discussed, but exposure misclassification not discussed |
| D. Studies using highly elevated central monitors (horizontal and vertical exposure misclassification) | ||
| 1. Sarnat et al. (2006) | Central monitor elevated 400 feet above town where subjects lived, a mile from monitor | For 5-day moving average in pollution concentration, OR for having an SVE = 1.42 for PM2.5; 1.70 for sulfate; no associations for BC |
RR risk ratio; OR odds ratio; IQR interquartile range increase; ICD implantable cardioverter-defibrillators; SVE supraventricular ectopy; SD standard deviation
aAssociation taken from Fig. 2 in Ebelt et al. (2005). Association in Ebelt et al. also reported [in Sarnat et al. (2006)] as a 22% increase in rate of SVE for subjects whose mean rate of SVE was 33 bph
Peters et al. (2000) examined the incidence of arrhythmias among 100 patients in eastern Massachusetts in relation to various air pollutants (PM10, PM2.5, BC, NO2, SO2, CO, O3) measured in South Boston. Associations were found with PM2.5, BC, CO, and NO2 with a 2-day lag. The strongest associations were found for BC and NO2, which the authors took as evidence of effects of local traffic. However, because of exposure misclassification, BC and NO2 effects were likely underestimated, a possibility suggested by others (Zeger et al. 2000; Ito et al. 2004) and discussed by authors of several studies with exposure error (Tables 1 and 2). As Peters et al. (2000) noted, “…we would expect any exposure misclassification [for a centrally monitored emission] to…bias the estimates toward the null.”
Dockery et al. (2005) followed 203 cardiac patients with implanted ICDs living in metropolitan Boston for an average of 3.1 years each between 1993 and 2002. Pollution (PM2.5, BC, SO4, particle number, NO2, CO, SO2, O3) was monitored at several sites in the metropolitan area. Average concentrations were derived for pollutants with multiple measurements. No significant associations for ventricular tachyarrhythmias were found for an interquartile range increase in any pollutant, for a 2-day pollution mean. However, when stratifying by a recent arrhythmia, significant associations were found for PM2.5, BC, NO2, CO and SO2, and a borderline significant association for SO4, for an interquartile increase in the 2-day pollution mean; this finding suggests that the most at-risk patients in general may also be most at risk for a pollution-related ventricular tachyarrhythmia. The authors took the associations with PM2.5, CO, NO2, and BC as indicative of a link with motor vehicle emissions, and the sulfate association as evidence of a link with regional fossil fuel emissions. SO2 was highly correlated with vehicular emissions and was seen as local in origin. In light of the findings of Albert et al. (2007), the lack of an association in this study with particle number, which is considerably higher in the 100 m nearest a major road (Zhu et al. 2002a, b), suggests that exposure misclassification may also have occurred in this study, perhaps understating effects of local vehicular emissions and transferring health associations to other emissions (Goldberg and Burnett 2003). The authors note that “…improved estimate of subject specific air pollution exposures would be expected to find stronger, more statistically significant associations.”
Another study having exposure misclassification is that of Metzger et al. (2007), conducted in the Atlanta area and involving 518 patients with ICDs; there were found to be no associations with tachyarrhythmias for any of the emissions monitored (PM10, ozone, NO2, CO, and SO2 for 10 years, PM2.5 and oxygenated hydrocarbons for 4 years). Metzger et al. also discussed exposure misclassification, stating that the study “does not contribute evidence regarding whether personal exposure may be a determinant of ventricular tachyarrhythmia.”
Sarnat et al. (2006) examined pollutant associations with supraventricular and ventricular arrhythmias. This was a companion study to that of Luttmann-Gibson et al. (2006), who examined pollution associations with reduced HRV in Steubenville, Ohio during summer and fall 2000. Both studies used a monitor several hundred feet higher than the living quarters of the subjects, and about a mile distant. Like Luttmann-Gibson et al., Sarnat et al. found associations with sulfate, but not with BC, although the companion study also found associations with PM2.5. Exposure misclassification for locally variable emissions such as BC is particularly severe, as with the companion study, due to the monitor’s height.
Anselme et al. (2007) found an immediate 200% to 500% increase in ventricular premature beats in CHF rats compared to healthy rats exposed to diluted diesel emissions. This finding is similar that of Dockery et al. (2005), in which the significant pollution associations for ventricular arrhythmias were only for those who had had a previous arrhythmia in the previous 3 days vs. more healthy people. The increase in arrhythmias following exposure to diesel exhaust in Anselme et al. (2007) appears to have its strongest parallels in the findings of Albert et al. (2007), e.g., increases in VT/VF in subjects half an hour after being in traffic, and of Riediker et al. (2004a, b), which found that the “speed change” factor, marked by diesel emissions and brake wear, was associated with significant increases of about 40% in SVE beats. Of the four epidemiological studies, none of which had good exposure information for spatially variable local vehicular emissions, two nevertheless suggest that traffic emissions are associated with arrhythmias, and three discussed exposure misclassification as a possible reason for results being smaller and weaker than had exposure information been more accurate.
The rapid initiation of arrhythmias after exposure to diesel emissions may suggest a direct effect on the myocardium. Anselme et al. (2007) state that such a quick response supports the idea that agents such as ultrafines, gasses, or soluble PM do cross the pulmonary epithelium into the circulation. However, rapid initiation might instead support the findings of Rhoden et al. (2005), who found that antagonists of both the sympathetic and parasympathetic nervous systems prevented oxidative stress in the heart caused by instillation of urban PM or inhalation of CAPs. In this second case, it might not be necessary for pollution to enter the bloodstream; oxidative stress in the heart due to exposure to PM (Rhoden et al.) or diesel emissions (Anselme et al.) might cause arrhythmias via direct modulation of the vagal nerve.
Inflammation
Initiation of an inflammatory response is another potential mechanism underlying PM-induced cardiovascular effects. Ridker et al. (2008) demonstrated that reducing systemic inflammation, initially marked by levels of high-sensitivity C-reactive protein (CRP), resulted in significant reduction of cardiovascular events (MI; revascularization or unstable angina; stroke; combined end point of MI, stroke, and death from any cardiovascular cause; and death from any cause) among those with low LDL cholesterol levels (<130 mg/dL) but initially high CRP (>2.0 mg/L). CRP itself is not the cause of progression of disease, however, but rather an indicator of inflammation (Schunkert and Samani 2008). Thus, elevated chronic systemic inflammation appears to be a cause of increased cardiovascular mortality and morbidity, even among apparently healthy people without hyperlipidemia. Systemic inflammation as marked by interleukin-6 (IL-6) is also associated with higher risks of mortality among older female CVD patients (Volpato et al. 2001) and with higher risks of a future MI among apparently healthy men (Ridker at al. 2000).
Bonvallot et al. (2001) exposed human bronchial epithelial cells to DEP and to DEP organic extracts, both of which induced activation of pro-inflammatory NF-κB, but the stripped carbonaceous core induced less intense responses. NF-κB is involved in inducement of inflammatory cytokines such as TNF-α, IL-6, and IL-8. Bonvallot et al. showed that DEP induced expression of a cytochrome P-450 specifically involved in PAH metabolism. This finding suggests the importance of PAH organic compounds in diesel emissions in causing inflammatory responses.
McDonald et al. (2004) found that diesel emissions from a relatively new diesel engine operating in steady state caused significant increases in several biological endpoints in mice, including inflammatory cytokines IL-6, TNF-α, and INF-γ (as well as oxidative stress, as marked by HO-1), with exposures of 200 μg/m3 (6 h/day, 7 days). Use of a catalyzing trap eliminated virtually all the black carbon and a large percentage of many carbonaceous emissions, and also abolished the significant increases in cytokines and HO-1.
Tornquist et al. (2007) exposed 15 healthy men to diesel exhaust or filtered air for 1 h. In those exposed to diesel emissions, there were significant increases in TNF-α and IL-6, paralleling in vitro and in vivo findings just reviewed.
Seaton et al. (1999) estimated exposure to PM10 in 108 elderly subjects in Belfast and Edinburgh, modeling estimated personal exposures based upon activity diaries and multiple monitoring sites. Increased CRP was significantly associated with city center measurements of PM10, but IL-6 was not. Another European multi-city study (Ruckerl et al. 2007) found in pooled results a significant increase in IL-6 per interquartile range increase (IQR) of particle number count 12–17 h before blood withdrawal, and per same day IQR increase in NO2 (primarily a marker for vehicular emissions in European cities); no such association was found with CRP.
Riediker et al. (2004a), Riediker (2007), and Delfino et al. (2008) are examples of studies with excellent exposure assessments. Both of the Riediker et al. studies examined effects of 9 h of exposure to in-vehicle pollution on various cardiovascular parameters in healthy young male patrol officers. In Riediker et al. (2004a), elevated CRP was significantly associated with in-vehicle PM2.5, but not with roadside PM2.5. In Riediker (2007), IL-6 levels were significantly increased by about 20% for a one-standard-deviation increase in in-vehicle PM2.5. Although IL-6 was not measured in the 2004 study, and CRP was not measured in the 2007 study, we can reasonably conclude that both CRP and IL-6 are elevated when healthy young men are exposed to several hours of vehicular emissions at levels found inside on-road vehicles.
Delfino et al. (2008) examined biomarkers of inflammation, anti-oxidant activity, and platelet activation in a panel study of 29 non-smoking elderly with CHD in retirement homes in Los Angeles. Using monitors just outside and inside the residences, the authors modeled concentrations of outdoor emissions inside the homes, where subjects spent most of their time. Delfino et al. (2008) found that several biomarkers of inflammation (CRP, IL-6, and soluble receptor-II for TNF-α) increased significantly with increasing exposure to various vehicular emissions (current day and multi-day averages). Reduced anti-oxidant activity was found for all but three subjects with several different traffic emissions. Among PM size fractions, only the smallest particles (“quasi-ultrafines,” PM0.25) were significantly associated with these biomarkers, paralleling findings regarding particles in Los Angeles taken close to freeways with regard to oxidative stress, as reviewed above. Particle number, BC, EC, CO, and primary OC were also associated with one or more of the biomarkers. Interestingly, secondary organic carbon showed no associations.
Thus, three studies of humans exposed to ambient air with accurate exposure monitoring found elevated levels of inflammatory indicators CRP and IL-6 associated with increased in-vehicle PM2.5, or with several vehicular emissions. Of the other two studies (Seaton et al. 1999 and Ruckerl et al. 2007), one found elevated CRP in subjects in relation to modeled central city PM10, while the other found IL-6 was elevated relative to centrally monitored particle number and NO2 concentrations. Tornquist et al. (2007) found in healthy human volunteers significant increases in IL-6, TNF-a, and soluble P-selectin 24 h after inhalation (1 h) of 300 μg/m3 diesel exhaust, suggesting the role of diesel emissions in the epidemiological findings noted above. Findings of these studies are in concert with the in vitro (Bonvallot et al. 2001) and in vivo (McDonald et al. 2004) studies reviewed above as well. Bonvallot et al. establishes a possible biological mechanism of inflammation involving PAHs.
Zeka et al. (2006) studied 710 members of the VA Normative Aging Study cohort living in the greater Boston area, with air pollution measured at a central monitor. Despite the potential exposure misclassification, the authors found an association for elevated CRP with an increase in one standard deviation in BC (for those with a BMI over 30, e.g., obese). Risks of elevated CRP were approximately four times higher for obese than for non-obese. Significant associations were not found for CRP with other types of PM. Those lacking a measure of genetic protection against oxidative stress, e.g., GSTM1-null subjects, were significantly more likely to have increased levels of CRP associated with increased BC concentrations. The authors discuss exposure misclassification, noting that a limitation of the study was the inability to measure personal exposure to different types of PM, and that they “would expect an underestimation of the effects of air pollution observed in the present study.”
Atherosclerosis
Atherosclerosis refers to a thickening of the luminal wall of arteries that, depending upon the specific type of condition, may or may not result in reduction in blood flow. Atherosclerosis may be considered an inflammatory disease which progresses in concert with oxidation of LDL lipids (Steinberg 2002). Thus, initiation or exacerbation of inflammation, associated with pollutant exposure, may be a mechanism for some of the health outcomes noted herein.
Studies reviewed above show links between vehicular emissions and acute HRV reductions/oxidative stress reactions. A study of 1,133 non-smokers in a Swiss cohort (Probst-Hensch et al. 2008) found that those with glutathione-S-transferase polymorphism deletions (which reduce anti-oxidant defenses) have altered autonomic control, as marked by reduced HRV. The authors conclude that their results are consistent with an important pathological role for systemic, chronic oxidative stress among the general population. Although unexplored in this study, these findings may suggest as well that vehicular emissions which cause acute oxidative stress and thus change in HRV may be causally related to increases in arterial plaque formation via oxidation of LDL cholesterol. In this regard, Romieu et al. (2008) examined residents of a Mexico City nursing home for 6.5 months. Mexico City’s elevated pollution levels contain high levels of vehicular emissions, likely resulting in increased oxidative stress. Subjects were treated with anti-oxidants (fish oil and soy oil supplements). Both supplements increased antioxidant activity, as measured by biomarkers of response to oxidative stimuli [glutathione (GSH) and Cu/Zn superoxide dismutase (SOD) activity]. Those treated with fish oil supplements, but not soy oil, also had 72% less lipoperoxidation. These results suggest that chronic oxidative stress caused by urban pollution is linked to increased lipoperoxidation, likely enhancing development of atherosclerotic plaques.
In the panel study of elderly non-smoking subjects reviewed above, Delfino et al. (2008) also examined associations of platelet activation, marked by soluble P-selectin, with various emissions. P-selectin is an adhesion molecule which plays an important role in atherosclerosis via leukocyte recruitment (Woollard and Chin-Dusting 2007). Delfino et al. (2008) found increased levels of soluble P-selectin significantly associated with increased levels of EC of outdoor origin and primary OC, suggesting that these emissions can produce platelet activation and, thus, advance atherosclerosis.
Araujo et al. (2008) showed that inhalation of concentrated ultrafine PM from close to a Los Angeles freeway, enriched in PAHs, caused significantly larger early atherosclerotic lesions in genetically susceptible (apolipoprotein E-deficient) mice inhaling concentrated PM2.5 or filtered air. Exposure to ultrafine PM also resulted in inhibition of anti-inflammatory capacity of plasma HDL and greater systemic oxidative stress, in part as evidenced by upregulation of Nrf2-regulated anti-oxidant genes. The atherosclerotic lesions were likely caused by the combination of oxidative stress and inflammation, as both appear to be necessary for development of atherosclerosis (Steinberg 2002).
Studies reviewed noted above found ambient levels of traffic emissions PM to be associated with both oxidative stress and inflammation. Findings of Araujo et al. (2008), with a sensitive murine model, are in concert with both the findings of Delfino et al. (2008) and the epidemiological results of Kuenzli et al. (2005), i.e., that people exposed to higher annual levels of ambient air in Los Angeles had higher prevalence of atherosclerotic plaque or of important precursors of such plaque. The results of Araujo et al. (2008) are also consistent with the findings of Romieu et al. (2008), who found that lipoperoxidation in residents of a highly polluted city was reduced by use of a common anti-oxidant. Findings of these studies are also consistent with findings of studies in the sections on oxidative stress and inflammation, with regard to exposure to diesel emissions, urban pollution, and specific emissions such as BC.
Gong et al. (2007) examined exposure to ambient ultrafine particles that were highly enriched in redox cycling organic chemicals in terms of promotion of atherosclerosis in mice. The investigators found that an interaction between diesel exhaust particles and oxidized LDL lipids synergistically affected gene expression corresponding to pathways relevant to vascular inflammatory processes such as atherosclerosis. This study suggests how the lipid peroxidation found in the Huang et al. (2003) and Pereira et al. (2007) studies could lead to atherosclerosis such as found by Kuenzli et al. (2005) and lipoperoxidation as noted by Romieu et al. (2008). Similarly, Sharman et al. (2002) found that people regularly occupationally exposed to vehicular emissions (auto mechanics) had greater plasma susceptibility to oxidation, and, thus, a greater risk of developing atherosclerosis than did matched controls.
Kuenzli et al. (2005) and Jerrett et al. (2005) utilized a modeled PM2.5 “surface” created for Los Angeles by interpolating data from 23 fixed-site monitors, creating within-city gradients for exposure to PM2.5. Motor vehicles are the major source of emissions in Los Angeles, since other potentially major stationary sources of pollution (e.g., coal-fired power plants, steel mills, and coke industries) are absent. Thus, PM2.5 in the Los Angeles basin as a whole is related primarily to vehicular emissions, although near ports shipping emissions may also be important.
Using the ACS data base for Los Angeles, Jerrett et al. (2005) found the relative risks for all-cause and cardiovascular mortality three to four times larger than in the original ACS study, which examined inter-city differences in PM2.5, vs. intracity differences. Kuenzli et al. (2005) found higher levels of PM2.5 within the Los Angeles airshed to be associated with higher levels of atherosclerotic plaques in the carotid artery (associations were strongest for women 60 years old or older.). These results, suggesting that mainly vehicular-derived PM2.5 in Los Angeles may be causally related to the development of atherosclerosis, parallel those of Hoffmann et al. (2007), noted above, who found that people living close to major roads had significantly higher levels of coronary atherosclerosis as measured by coronary artery calcification.
Vascular function and blood pressure
Another potential mechanism by which vehicular-derived PM could cause cardiovascular injury involves changes in vascular tone. Urch et al. (2004, 2005) exposed healthy human volunteers to ozone plus CAPs (concentrated ambient particles) obtained in close proximity to a major Toronto freeway. The resultant increase in vasoconstriction of the brachial artery (2004 study) was associated with EC and OC, but not with any of the other 23 components of PM2.5 examined, suggesting traffic emissions as a likely causal source. Diastolic blood pressure (2005 study), possibly related to the vasoconstriction findings of the first study, increased with increased levels of organic carbon and was attributed to traffic emissions.
Miller et al. (2009) exposed rat aortic rings in vitro to diesel exhaust particles (DEP) to explore mechanisms of effects on vascular function. The authors found that oxidative stress caused by DEP reduced the bioavailability of endothelium-derived nitric oxide “without prior interaction with the lung or vascular tissue.”
Bartoli et al. (2009) examined the effects of CAPs collected adjacent to a major urban road on blood pressure of dogs. Increases in systolic blood pressure (SBP), diastolic BP (DBP), mean BP, heart rate, and rate times pressure were all associated with an IQR increase in PM2.5, BC, and particle count, with the size of the effect increasing in that order (BC marginally significant for systolic pressure). As with the studies of Urch et al. the CAPs were taken from next to a major road, exposure was accurately known, and the BC, carbonaceous and/or ultrafine PM (emitted from traffic) were highly associated with the increases in BP.
Subjects in the multi-city study of Auchincloss et al. (2008) were aged 45–84 and clinically free of CVD. The authors found several associations between PM2.5 and pulse pressure (PP), with only one model (which included traffic emissions) finding associations with SBP. Further analysis showed that when PM2.5 exposure for subjects was stratified by either of three traffic variables (NO2 levels above median; residence within 300 m of highway; or high density of roads nearby residence), a 10-μg/m3 increase in PM2.5 was associated with increased PP (all three cases) and SPB (two or three cases) when the traffic variable was “positive,” but not when the traffic variable was “negative.” Thus, PM2.5 exposure was not associated with either endpoint unless exposure level to traffic emissions was stratified as “high.” Using different methods of assessing exposure to vehicular emissions in a multi-city study, these investigators found similar SBP results to the studies above using CAPs taken from nearby major roads.
Findings have been inconsistent with regard to elevated blood pressure associations among studies lacking reasonably accurate information for exposure to locally variable emissions, such as those from vehicles. Ibald-Mulli et al. (2004) examined 131 subjects with coronary heart disease in three European cities, used centrally monitored PM2.5, accumulation mode, and ultrafine particle mass concentrations. Results were controlled for temperature, barometric pressure, and relative humidity. Very small negative, but significant, associations were found for both SBP and DBP with particles of different sizes. However, the authors cautioned against inferring clinical relevance from these findings. Zanobetti et al. (2004), in a study of residents of greater Boston, found positive and significant PM2.5 associations (mean level for 5 days before physician visits) with resting SBP, DBP, and mean arterial BP (MAP). In addition, for those with resting heart rate >70 bpm, mean PM2.5 level for the 2 days preceding the visit were associated with increases in DBP and MAP during exercise. Temperature, dew-point temperature, and barometric pressure, as well as standard socioeconomic variables, were controlled for. Pollutants were measured at central sites. Associations were found with SO2, O3, BC, but not with NO2 or CO in single-pollutant models, but only PM2.5 remained associated with elevated DBP in multi-pollutant models.
Thus, it appears that when exposure to vehicular emissions are reasonably well characterized—as when ambient air is from near a major road, or when results are stratified by whether someone lives near major roads or has a high density of roads near their residence—increased blood pressure effects are consistently found associated with vehicular emissions. However, in the absence of reasonably well-characterized exposure information for vehicular emissions, associations become inconsistent, and vehicle-specific emissions are less likely to be associated with change in blood pressure.
Lai et al. (2005) found that toll workers exposed to traffic exhausts had significantly higher levels of plasma NO, an agent affecting vascular tone, than similar workers not so exposed, suggesting another pathway by which vehicular emissions could adversely influence vascular tone.
Peretz et al. (2008) exposed 27 adult volunteers (ten healthy, 17 with metabolic syndrome) to diluted diesel exhaust (100 or 200 μg/m3 PM2.5) or filtered air. The authors examined brachial arterial diameter change, and collected systemic blood samples for endothelin-1 (ET-1), a vasoconstrictor. Reduction in brachial artery diameter was linearly related to increasing exposure concentration of the exhaust, paralleling the findings of Urch et al. (2004), which used CAPs from nearby a Toronto freeway, and suggesting the importance of diesel emissions specifically for this endpoint. Plasma levels of ET-1 were increased only at the highest concentration of exposure.
Campen et al. (2005) found that fresh diesel emissions can cause vasoconstriction in the blood vessels of mice ex vivo, but that filtering diesel exhaust to remove particles did not change the vasoconstrictive properties of the emissions. Further analysis suggested that two specific gaseous emissions, aldehydes and alkanes, appeared to be responsible for these effects, suggesting a potential biological mechanism for the findings of Urch et al. (2004, 2005).
Changes in vascular homeostasis may be due to oxidative stress on endothelial cells or to systemic inflammation that affects the endothelium. Peretz et al. (2007) exposed healthy adults to diluted diesel exhaust and used microarray techniques to assess effects in peripheral blood leukocytes, since these cells are involved in inflammation and control of vascular homeostasis, including development of atherogenesis (Kristovich et al. 2004; Libby et al. 2002). They noted that the diesel exhaust exposure preferentially modulated genes involved in oxidative stress, inflammation, leukocyte activation, and vascular homeostasis, mechanisms by which adverse health effects may be modulated.
Research recommendations
Different types of particles have different biological effects, and some are likely to be more harmful than others; for example, some might cause more oxidative stress. Additional effort is needed to move closer to the goal of regulating those specific types of particles and emissions which may have the greatest health relevance. Research recommendations are made with this goal in mind.
First, although we suggest, based upon the data base discussed, that creation of a black carbon PM standard under the National Ambient Air Quality Standards may serve to protect public health, there is still much research to be done with regard to the different components of vehicular emissions. Non-PM carbonaceous components of vehicular emissions have adverse health effects (Mauderly and Chow 2008), but relatively little research has been done to date on them. Even if a black carbon standard were regulated under the NAAQS, and even if as a consequence of such a standard, many VOC and SVOC emissions would be controlled simultaneously, gasoline engines emit many of the same VOCs as diesels, and many different ones as well.
Several toxicology studies of gasoline emissions, using genetically engineered mice, have generated hypotheses about their effects. Lund et al. (2007), in a study using atherosclerosis-prone genetically modified ApoE−/− mice, showed that the gaseous fraction of gasoline emissions were associated with increased markers of vascular oxidative stress and transcriptional upregulation of factors associated with vascular remodeling important to the development of atherosclerosis. Campen et al. (2006), using the same mouse model, found that fresh gasoline emissions, but not paved road dust, altered cardiac repolarization. Should research establish that these emissions also cause serious harm to public health, technology solutions will have to be found for these as well.
More health endpoints appear to have been examined for diesel than for gasoline engine exhaust at this point, especially when using ambient air or human subjects or both; thus, further research is needed. To establish a comprehensive and consistent basis of comparison of gasoline with diesel emissions, ways should be found to test gasoline emissions in protocols using human subjects parallel to those of Mills et al. (2005, 2007), for example.
A second research recommendation is also quite important to determining which emissions need to be controlled to protect public health. It may be reasonable to think that today, there are two widespread types of air pollution of public health concern: those from vehicles and those from power plants, especially those using coal. Two to three decades ago, use of residual oil use was much more widespread than now, and the oil also contained higher amounts of metals (V, Ni) and sulfur (Thurston and Spengler 1985). With less than 3% of electric generation from residual oil today vs. 17% three decades ago, today’s residual oil emissions are lower and more localized. Similarly, there were formerly more coking and non-electric arc steel plants, and these plants did not face the emission regulations they face today. Thus, epidemiological studies of the 1970s and 1980s would have had to be attentive to specific emissions from these sources in more locations and on a more regional scale than more recent studies.
Many early studies of PM did not recognize, as we now do, either the importance of monitoring specifically for emissions from vehicles, or the need to have reasonably accurate exposure information for such emissions. Now that methodologies rectifying these deficiencies are available, we recommend they be used to examine whether cardiovascular effects found to be associated with vehicular emissions, as in the studies above, are also found with emissions representing coal emissions (secondary sulfates and coal fly ash) as well as associated products of atmospheric chemistry involving such emissions.
Thus, one recommendation would be to perform tests utilizing ambient air masses, as with the work of Schwartz et al. (2005b), Gold et al. (2005), and Creason et al. (2001), in regions such as north-central Pennsylvania or in central New York State, where on most days emissions would reflect little industrial or vehicular emissions relative to urban locations. To be consistent with many of the studies reviewed above, recruitment of those living in a retirement center would be recommended. Would days with higher total PM2.5, and/or with higher levels of sulfate, exhibit similar changes in inflammatory indicators (CRP, IL-6), levels of anti-oxidant enzyme activity, or adhesion molecules (soluble P-selectin), as in Delfino et al. (2008)? Such proposed studies should also examine blood pressure and vasoconstriction, as in Auchincloss et al. (2008) and Urch et al. (2004, 2005). ST-segment depression, oxidative stress, and arrhythmias should also be examined in studies paralleling those reviewed above.
Wind trajectory analysis, such as used in Park et al. (2007) and Creason et al. (2001), and which Lippmann et al. (2006) used to demonstrate associations with Ni from Canadian nickel smelters on reduced HRV in mice housed in a rural location in New York State, should also be used to see if on days with elevated measures of health effects the air masses might come from an unsuspected source, as in Lippmann et al. (2006).
These studies would allow a direct comparison with the studies examined in this assessment, and thus would enable researchers to see if ambient coal emissions, including reaction products, would cause the same cardiovascular health effects as diesel and/or vehicular emissions. Recommendation of research of this type, however, is not to recommend that more innovative research is any less important. For instance, the “highway gradient” studies are an example of the kind of innovation that caused researchers to focus on biological mechanisms of vehicular emissions.
More generally, the use of new personal monitoring tools, such as vests being developed by EPA which are easy to wear and monitor many different emissions, may broaden the endpoints which can be examined with regard to pollution associations. Arrhythmias would be one primary endpoint, since up to now, central monitors have been used to provide pollution data in studies of arrhythmias. Since this health endpoint has not yet apparently been examined in studies using accurate exposure information for vehicular emissions, arrhythmias would be an excellent candidate for use in studies with better monitoring, perhaps using the protocols of Schwartz et al. (2005b) and Gold et al. (2005). Furthermore, although V and Ni now tend to be relatively local emissions (e.g., near major ports as shipping fuel and in a few Northeastern locales), it is still important to separate effects of metals from carbonaceous materials.
Conclusions
Epidemiologic studies with good exposure information for locally variable levels of particulate emissions from motor vehicles consistently find associations between such exposure and cardiopulmonary disease mortality, circulatory disease mortality, ischemic heart disease mortality, and all-cause mortality, and with many CV morbidity endpoints, such as cardiovascular hospital admissions, markers of atherosclerosis, survival after heart failure, incidence of coronary heart disease, initial myocardial infarction, and acute myocardial infarction. For each of the cardiovascular health endpoints reviewed herein—oxidative stress, HRV changes, ST-segment depression, inflammation, arrhythmia, vascular function and blood pressure, and atherosclerosis—there are mechanistic studies supporting a pathophysiological basis for how diesel and/or vehicular emissions could cause such outcomes. The mechanistic studies for each endpoint are briefly summarized in Table 3. These cardiovascular health endpoints, in turn, provide multiple biological mechanisms with explanatory value for the mortality and morbidity findings in the epidemiology studies.
Table 3.
Summary of effects of vehicular emissions and black carbon on CVD health endpoints
| Health endpoint | In vitro studies | In vivo studies | Human panel studies |
|---|---|---|---|
| 1. Oxidative stress | Li et al. 2002a (increases in HO-1, diesel PM) | McDonald et al. 2004 (diesel emissions, increased HO-1 in normal mice; oxidative stress abolished with use of catalyzing trap which totally eliminated black carbon, largely eliminated most organics, including many PAHs) | Mills et al. 2005 (diesel emissions, healthy human volunteers; reduction in t-PA, impairment of vascular tone, postulated by authors to be related to oxidative stress; also see HRV Alteration [HRV changes often caused by oxidative stress]) |
| Li et al. 2002b (increases in HO-1, Los Angeles air) | Delfino et al. 2008 (decreased levels of anti-oxidant enzyme activity, in panel of 29 non-smoking elderly subjects with history of coronary artery disease associated with BC, NO2, primary OC of outdoor origin, and ultrafine PM, for current day and multi-day averages, in study with excellent exposure characterization, using both indoor and outdoor monitors at Los Angeles residences) | ||
| Li et al. 2003 (increases in HO-1, most harm caused by ultrafines in Los Angeles air, correlated with organics and PAHs) | |||
| 2. HRV alteration | NA | Anselme et al. 2007 (diesel emissions, HRV decreases in healthy and CHF rats immediately after exposure) | Adar et al. 2007 (changes in six different types of HRV associated with BC exposure; when subjects on bus with high BC levels, larger HRV changes roughly correspond with larger changes in BC; monitor followed subjects wherever they went) |
| Schwartz et al. 2005b (changes in 4 types of HRV associated with BC concentrations, but not with concentrations of non-BC regional PM2.5; subjects live on same road as monitor is located, both in close proximity to road, 0.5 miles apart) | |||
| Creason et al. 2001 (HRV changes monotonically associated with increasing PM2.5, after two days with high PM2.5 from only rural sources eliminated from regression) | |||
| Ebelt et al. 2005 (HRV associations found for ambient urban PM, not found for sulfate; personal monitors used) | |||
| In studies using central monitors, Wheeler et al. (2006) and Park et al. (2005) show associations with BC in only one fourth of tests; Luttmann-Gibson et al. (2006) find no BC associations. | |||
| Park et al. (2007) is same study as Park et al. (2005), but uses wind trajectories to determine sources, thus has better exposure information than Park et al. (2005); HRV associations found for urban air masses, not for rural air masses | |||
| 3. ST-segment Depression | NA | Yan et al. 2008 (diesel exhaust particles impaired left ventricular functioning in healthy rats, with further impairment in rats with myocardial injury) | Mills et al. 2007 (ST-segment depression in subjects with CHD exposed to diesel emissions twice as great as for subjects without CHD, suggesting how diesel emissions could harm susceptible subjects) |
| Gold et al. 2005 (in parallel study to Schwartz et al. 2005b HRV study, e.g., with accurate exposure information, ST-segment depression associated with BC but not with PM2.5) | |||
| Lanki et al. 2006 (in study of several local and regional pollutants lacking good exposure information, ST-segment depression associated with ABS [EU equivalent of BC] but not with sulfate or other pollutants) | |||
| 4. Cardiac Arrhythmia | NA | Anselme et al. 2007 (diesel emissions, 200% to 500% increase in ventricular premature beats in CHF rats, but not in normal rats) | Albert et al. 2007 (risks of ICD shock elevated in hour after driving, RR = 2.24; risks for ventricular fibrillation or tachycardia elevated in half hour after driving, RR = 4.46) |
| Riediker et al. 2004a, b (∼40% increase in SVE beats for change of one SD in “speed change” factor reflecting diesel emissions and brake wear) | |||
| Ebelt et al. (2005) (SVE associations found for ambient urban PM, non-sulfate ambient urban PM, not found for sulfate; personal monitors used) | |||
| Peters et al. (2000), Dockery et al. (2005), Metzger et al. (2007), and Sarnat et al. (2006) are extant studies of arrhythmias using central monitor concentrations as proxies for subject exposure over large metropolitan areas, causing exposure misclassification; first study finds larger associations with vehicular emissions (BC and NO2) than with PM2.5; second study finds traffic emissions more likely cause of arrhythmias; third study finds no associations; associations in fourth study are with sulfate but not with BC; the first three studies discuss exposure misclassification as possible reason for underestimates of associations | |||
| 5. Vascular Function | Miller et al. 2009 (diesel particles reduce bioavailability of endothelium-derived NO in aortic rat rings in vitro via oxidative stress, without prior interaction with lung or vascular tissue) | Bartoli et al. 2009 (increases in mean, systolic and diastolic blood pressure found in dogs exposed to CAPs taken from near major urban roadway; BC, carbonaceous particle count associated with increases in blood pressure) | Urch et al. 2004 (significant increase in vasoconstriction in healthy human volunteers exposed to CAPs taken from near freeway associated only with EC and OC among 25 components of PM2.5 analyzed) |
| Campen et al. 2005 (fresh diesel emissions and filtered diesel exhaust cause vasoconstriction in mice ex vivo, aldehydes and alkanes most likely involved) | Urch et al. 2005 (significant increase in blood pressure in healthy human volunteers exposed to CAPs taken from near freeway, possibly associated with increase in vasoconstriction in 2004 study, related to traffic emissions) | ||
| Auchincloss et al. 2008 (in subjects aged 45–84, systolic blood pressure and pulse pressure associated with increased PM2.5 only when traffic variables (NO2 levels above median value; residence within 300 m of highway; or high density of roads near residence) were “positive,” not when traffic variables were “negative”) | |||
| Lai et al. 2005 (toll workers exposed to traffic exhaust had significantly higher levels of plasma NO, which affects vascular tone) | |||
| Peretz et al. 2007 (in healthy adult volunteers, diesel exhaust preferentially modulated genes involved in oxidative stress, inflammation, leukocyte activation and vascular homeostasis) | |||
| Peretz et al. 2008 (in adult volunteers exposed to diesel exhaust, reduction in brachial artery diameter linearly related to increasing concentration of exhaust; plasma levels of endothelin-1, a vasoconstrictor, significantly increased only at 200 μg/m3 of diesel exhaust, but not at 100 μg/m3) | |||
| 6. Inflammation | Bonvallot et al. 2001 (diesel emissions and diesel organic extracts induced increased levels of pro-inflammatory NF-κB in human bronchial epithelial cells; less intensive effects induced by stripped carbonaceous core) | McDonald et al. 2004 (increased levels of three inflammatory biomarkers (TNF-α, IL-6, and INF-γ) associated with exposure to diesel emissions, effects abolished with use of new catalyzing trap which eliminated BC completely, largely eliminated most organics, including many PAHs) | Delfino et al. 2008 (several biomarkers for inflammation [CRP, IL-6, TNF-α receptor] significantly increased with increased concentrations of BC, EC, CO, primary OC, and with increased particle number) |
| Riediker et al. 2004a (CRP elevated with increased in-vehicle PM, in study of patrol officers after 9-h shift) | |||
| Riediker 2007 (IL-6 elevated with increased in-vehicle PM, in study of patrol officers after 9-h shift) | |||
| Tornquist et al. 2007 (diesel emissions increased TNF-α, IL-6 levels in healthy human volunteers, vs. filtered air) | |||
| Zeka et al. 2006 (elevated BC levels, recorded at central monitor, associated with increased CRP levels in the obese, and in those lacking a measure of genetic protection against oxidative stress, e.g., GSTM1-null subjects. Authors discuss exposure misclassification, note that they would expect larger risks with better exposure assessment) | |||
| 7. Atherosclerosis and lipoperoxidation | See oxidative stress and inflammation sections for in vitro work relevant to atherosclerosis, caused in large part by systemic interaction of oxidative stress and inflammation | Araujo et al. 2008 (increased early atherosclerotic lesions in ApoE -/- mice breathing CAPs ambient in PAHs from near LA freeway, exposure to ultrafine PM inhibited anti- inflammatory capacity of plasma HDL) | Sharman et al. 2002 (auto mechanics, regularly exposed to higher levels of vehicular emissions than controls, had significantly higher susceptibility of plasma to oxidation) |
| Gong et al. 2007 (interaction between oxidized LDL lipids and organic diesel emission extracts affects gene expression relevant to vascular inflammation and atherosclerotic pathways in human microvascular endothelial cells; work then replicated in vivo, with similar findings – see in vivo, next column) | Gong et al. 2007 (interaction between oxidized LDL lipids and concentrated ultrafine diesel exhaust particles in Los Angeles air affects gene expression corresponding to atherosclerotic pathways in mice, viewed by authors as confirming in vitro findings in column to left) | Delfino et al. 2008 (levels of soluble P-selectin, important for platelet activation in atherosclerosis, significantly associated with increased levels of EC of outdoor origin, primary OC, in study of seniors in Los Angeles) | |
| Huang et al. 2003 (PM1.0 more likely to cause lipoperoxidation in human lung cells than larger fractions, OC and EC but not various ions associated with this effect) |
A number of the studies reviewed in this paper examined human subjects breathing ambient air. Such studies are likely to provide information most relevant to regulations designed to protect public health. Studies using genetically modified animals and highly concentrated components of ambient air, or using artificial atmospheres, are useful in generating hypotheses, but if these hypotheses are not verified in people exposed to ambient atmospheres, they may not provide an adequate basis for regulation. The database of ambient air studies does provide substantial evidence that the cardiovascular health effects associations with vehicular emissions likely reflect causality, rather than just statistical correlation.
Studies have shown that emissions from diesel engines may be especially potent in producing adverse health outcomes (U.S. Environmental Protection Agency 2002). As of January 2007, EPA regulations require a new catalyzing trap on all new on-road diesels. These devices reduce BC levels virtually completely, while also reducing emissions of many carbonaceous species by large percentages (McDonald et al. 2004). BC is likely an important causal agent of effects with which it has been associated in many studies, both intrinsically and because many carbonaceous species co-emitted from diesels and other vehicles can be adsorbed onto the surface of BC. In addition, BC is also likely a marker for harmful carbonaceous gasses which may be co-emitted with but not adsorbed onto the particles.
The EPA regulations do not extend to retrofits, however, and older diesels are the worst emitters. Several states are taking action to reduce such diesel emissions. In California, there are now proposals to restrict diesels from prior to a particular vintage year from operating in the ports of Los Angeles and Long Beach, unless retrofitted with the new catalyzing trap. Several localities are now requiring retrofit programs on certain classes of vehicles, e.g., school buses, or are advancing the replacement date for older diesel buses. A “speciated” BC standard would be more comprehensive than a patchwork of state requirements, and would likely cause many states to require retrofits of catalyzing traps on older diesels. This would reduce not just BC, but also the associated carbonaceous emissions which adsorb onto BC and also may cause health effects per se. Further, a speciated BC standard would require states to deal with BC in areas not in violation of standards for PM2.5, but where diesel health effects would be of consequence to public health. If it made sense to require new technology on new on-road and off-road diesels—and we agree that it does—then it makes even more sense to control emissions from the older, dirtier diesels which will be in operation for perhaps another 30 years.
Disclaimer and Acknowledgements
This work reflects the views only of the authors, and does not necessarily reflect views of the Department of Energy. The authors thank JoAnn Yuill for her exceptional help, not for the first time.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
The traffic density was insignificant only when EC was also included in a multi-pollutant model; in these cases, both variables came close to significance, but traffic density appeared to be the stronger of the two.
Another vehicular emission, CO, was also so associated, but in a two pollutant model with BC was no longer significant. In studies from the 1970s and 1980s, CO was found to be significantly associated with various health endpoints, but CO levels were more than an order of magnitude higher in the 1970s than they are today.
References
- Adar SD, Kaufman JD (2007) Cardiovascular Disease and Air Pollutants: Evaluating and Improving Epidemiological Data Implicating Traffic Exposure. Inhal Toxicol 19(Supp. 1):135–149. doi:10.1080/08958370701496012 [DOI] [PubMed]
- Adar SD, Gold DR, Coull BA, Schwartz J, Stone PA, Suh H. Focused exposures to airborne traffic particles and heart rate variability in the elderly. Epidemiology. 2007;18:95–103. doi: 10.1097/01.ede.0000249409.81050.46. [DOI] [PubMed] [Google Scholar]
- Albert CM, et al. Driving and implantable cardioverter-defibrillator shocks for ventricular arrhythmias. J Am Coll Cardiol. 2007;50:2233–2240. doi: 10.1016/j.jacc.2007.06.059. [DOI] [PubMed] [Google Scholar]
- Anselme F, Loriot S, Henry J-P, Dionnet F, Napoleoni J-G, Thulliez C, Morin J-P. Inhalation of diluted diesel engine emission impacts heart rate variability and arrhythmia occurrence in a rat model of chronic ischemic heart failure. Arch Toxicol. 2007;81:299–307. doi: 10.1007/s00204-006-0147-4. [DOI] [PubMed] [Google Scholar]
- Araujo JA, Barajas B, Kleinman M, Wang X, Bennett GJ, Gong KW, Navab M, Harkema J, Sioutas C, Lusis AJ, Nel AE. Ambient particulate pollutants in the ultrafine range promote early atherosclerosis and systemic oxidative stress. Circ Res. 2008;102:589–596. doi: 10.1161/CIRCRESAHA.107.164970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auchincloss AH, Diez Roux AV, Dvonch JT, Brown PL, Barr RG, Daviglus ML, Goff DC, Jr, Kaufman JD, O’Neill MS. Associations between recent exposure to ambient fine particulate matter and blood pressure in the Multi-Ethnic Study of Atherosclerosis (MESA) Environ Health Perspect. 2008;116:486–491. doi: 10.1289/ehp.10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoli CR, Wellenius GA, Diaz EA, Lawrence J, Coull BA, Akiyama I, Lee LM, Okabe K, Verrier RL, Godleski JJ (2009) Mechanisms of inhaled fine particulate-induced arterial blood pressure changes. Environ Health Perspect 117:361–366 [DOI] [PMC free article] [PubMed]
- Beelen R, Hoek G, van den Brandt PA, Goldbohm RA, Fischer P, Schouten LJ, Jerrett M, Hughes E, Armstrong B, Brunekreef B. Long-term effects of traffic-related air pollution on mortality in a Dutch cohort (NLCS-AIR Study) Environ Health Perspect. 2008;116:196–202. doi: 10.1289/ehp.10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilchick KC, Fetics B, Djoukeng R, Fisher SG, Fletcher RD, Singh SN, Nevo E, Berger RD. Prognostic value of heart rate variability in chronic congestive heart failure (veterans affairs’ survival trial of antiarrhythmic therapy in congestive heart failure) Am J Cardiol. 2002;90:24–28. doi: 10.1016/s0002-9149(02)02380-9. [DOI] [PubMed] [Google Scholar]
- Bonvallot V, Baeza-Squiban A, Baulig A, Brulant S, Boland S, Muzeau F, Barouki R, Marano F. Organic compounds from diesel exhaust particles elicit a proinflammatory response in human airway epithelial cells and induce cytochrome p450 1A1 Expression. Am J Respir Cell Mol Biol. 2001;25:515–521. doi: 10.1165/ajrcmb.25.4.4515. [DOI] [PubMed] [Google Scholar]
- Campen MJ, Babu NS, Helms GA, Pett S, Wernly J, Mehran R, McDonald JD. Nonparticulate components of diesel exhaust promote constriction of coronary arteries from ApoE−/− Mice. Toxicol Sci. 2005;88(1):95–102. doi: 10.1093/toxsci/kfi283. [DOI] [PubMed] [Google Scholar]
- Campen MJ, McDonald JD, Reed MD, Seagrave JC. Fresh gasoline emissions, not paved road dust, alter cardiac repolarization in ApoE−/− mice. Cardiovasc Toxicol. 2006;6:199–209. doi: 10.1385/ct:6:3:199. [DOI] [PubMed] [Google Scholar]
- Chahine T, Baccerelli A, Litonjua A, Wright RO, Suh H, Gold DR, Sparrow D, Vokonas P, Schwartz J. Particulate air pollution, oxidative stress genes, and heart rate variability in an elderly cohort. Environ Health Perspect. 2007;115:1617–1622. doi: 10.1289/ehp.10318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charniot J-C, Vignat N, Albertini J-P, Bogdanova V, Zerhouni K, Monsuew J-J, Legrand A, Artigou J-V, Bennefont-Rousselot D. Oxidative stress in patients with acute heart failure. Rejuvenation Res. 2008;11:393–398. doi: 10.1089/rej.2008.0663. [DOI] [PubMed] [Google Scholar]
- Creason J, Neas L, Walsh D, Williams R, Sheldon L, Liao D, Shy C. Particulate matter and heart rate variability among elderly retirees: the Baltimore 1998 PM study. J Environ Expos Anal Environ Epidemiol. 2001;11:116–122. doi: 10.1038/sj.jea.7500154. [DOI] [PubMed] [Google Scholar]
- de Hartog JJ, Lanki T, Timonen KL, Hoek G, Janssen NAH, Ibald-Mulli A, Peters A, Heinrich J, Tarkiainen TH, van Grieken R, van Wijnen JH, Brunekreef B, Pekkanen J. Associations between PM2.5 and heart rate variability are modified by particle composition and beta-blocker use in patients with coronary heart disease. Environ Health Perspect. 2009;117:105–111. doi: 10.1289/ehp.11062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delfino RJ, Staimer N, Tjoa T, Polidori A, Arhami M, Gillen DL, Kleinman MT, Vaziri ND, Longhurst J, Zaldivar F, Sioutas C. Circulating biomarkers of inflammation, antioxidant activity, and platelet activation are associated with primary combustion aerosols in subjects with coronary artery disease. Environ Health Perspect. 2008;116:898–906. doi: 10.1289/ehp.11189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockery DW, et al. Association of air pollution with increased incidence of ventricular tachyarrhythmias recorded by implanted cardioverter defibrillators. Environ Health Perspect. 2005;113:670–674. doi: 10.1289/ehp.7767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebelt ST, Wilson WE, Brauer M. Exposure to ambient and nonambient components of particulate matter. Epidemiology. 2005;16:396–405. doi: 10.1097/01.ede.0000158918.57071.3e. [DOI] [PubMed] [Google Scholar]
- Finkelstein M, Jerrett M, Sears MR. Traffic air pollution and mortality rate advancement periods. Am J Epidemiol. 2004;160:173–177. doi: 10.1093/aje/kwh181. [DOI] [PubMed] [Google Scholar]
- Finkelstein M, Jerrett M, Sears MR. Environmental inequality and circulatory disease mortality gradients. J Epidemiol Community Health. 2005;59:481–487. doi: 10.1136/jech.2004.026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring U, Heinrich J, Kramer U, Grote V, Hochadel M, Sugiri D, Kraft M, Rauchfuss K, Eberwein HG, Wichmann H-E. Long-term exposure to ambient air pollution and cardiopulmonary mortality in women. Epidemiology. 2006;17:545–551. doi: 10.1097/01.ede.0000224541.38258.87. [DOI] [PubMed] [Google Scholar]
- Gold DR, Litonjua AA, Zanobetti A, Coull BA, Schwartz J, MacCallum G, Verrier RL, Nearing BD, Canner MJ, Suh H, Stone PH. Air pollution and STsegment depression in elderly subjects. Environ Health Perspect. 2005;113:883–887. doi: 10.1289/ehp.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg M, Burnett R. Revised analysis of the Montreal Time-Series Study. In Revised analyses of time-series studies of air pollution and health. Boston, MA: Health Effects Institute Special Report; 2003. pp. 113–131. [Google Scholar]
- Gong KW, et al. Air-pollutant chemicals and oxidized lipids exhibit genome-wide synergistic effects on endothelial cells. Genome Biol. 2007;8:R-149. doi: 10.1186/gb-2007-8-7-r149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahame TJ (2009) Does improved exposure information for PM2.5 constituents explain differing results among epidemiological studies? Inhal Toxicol 21:381–393 [DOI] [PubMed]
- Grahame T, Hidy GM. Pinnacles and pitfalls for source apportionment of potential health effects from airborne particle exposure. Inhalation Toxicol. 2007;19(9):727–744. doi: 10.1080/08958370701399687. [DOI] [PubMed] [Google Scholar]
- Grahame T, Hidy GM. Secondary sulfate effects? Environ Health Perspect. 2007;115:A532. doi: 10.1289/ehp.10293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahame TJ, Schlesinger RB. Health effects of airborne particulate matter: do we know enough to consider regulating specific particle types or sources? Inhal Toxicol. 2007;19:457–481. doi: 10.1080/08958370701382220. [DOI] [PubMed] [Google Scholar]
- Health Effects Institute Diesel Exhaust . Critical analysis of emissions, exposure, and health effects. Special Report. Charleston, Massachusetts: Health Effects Institute; 1995. [Google Scholar]
- Health Effects Institute Research on diesel exhaust and other particles (2003) Program Summary. Health Effects Institute, Charleston, Massachusetts
- Hennekens CH, Buring JE. Epidemiology in medicine. Boston: Little, Brown and Co.; 1987. [Google Scholar]
- Hoek G, Brunekreef B, Goldbohm S, Fischer P, van den Brandt PA. Association between mortality and indicators of traffic-related air pollution in the Netherlands: a cohort study. Lancet. 2002;360:1203–1209. doi: 10.1016/S0140-6736(02)11280-3. [DOI] [PubMed] [Google Scholar]
- Hoffmann B, et al. Residence close to high traffic and prevalence of coronary heart disease. Eur Heart J. 2006;27:2696–2702. doi: 10.1093/eurheartj/ehl278. [DOI] [PubMed] [Google Scholar]
- Hoffmann B, et al. Residential exposure to traffic is associated with coronary atherosclerosis. Circulation. 2007;116:489–496. doi: 10.1161/CIRCULATIONAHA.107.693622. [DOI] [PubMed] [Google Scholar]
- Huang S-L, Hsu M-K, Chan C-C. Effects of submicrometer particle compositions on cytokine production and lipid peroxidation of human bronchial epithelial cells. Environ Health Perspect. 2003;111(4):478–482. doi: 10.1289/ehp.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibald-Mulli A, Timonen KL, Peters A, Heinrich J, Wolke G, Lanki T, Buzorius G, Kreyling WG, de Hartog J, Hoek G, ten Brink HM, Pekkanen J. Effects of particulate air pollution on blood pressure and heart rate in subjects with cardiovascular disease. Environ Health Perspect. 2004;112:369–377. doi: 10.1289/ehp.6523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iravanian S, Dudley SC., Jr Oxidative stress and the pathogenesis of atrial fibrillation. Curr Cardiol Rep. 2006;2:247–254. [Google Scholar]
- Ito K, Xue N, Thurston G. Spatial variation of PM2.5 chemical species and sourceapportioned mass concentrations in New York City. Atmos Environ. 2004;38:5269–5282. [Google Scholar]
- Janssen NAH, Schwartz J, Zanobetti Z, Suh HH. Air conditioning and source-specific particles as modifiers of the effect of PM10 on hospital admissions for heart and lung disease. Environ Health Perspect. 2002;110:43–49. doi: 10.1289/ehp.0211043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerrett M, Burnett RT, Ma R, Pope CA, Krewski D, Newbold KB, Thurston G, Shi Y, Finkelstein N, Calle EE, Thun MJ. Spatial analysis of air pollution and mortality in Los Angeles. Epidemiology. 2005;16(6):1–10. doi: 10.1097/01.ede.0000181630.15826.7d. [DOI] [PubMed] [Google Scholar]
- Kan H, et al. Prospective analysis of traffic exposure as a risk factor for incident coronary heart disease: the Atherosclerosis Risk in Communities (ARIC) Study. Environ Health Perspect. 2008;116:1463–1468. doi: 10.1289/ehp.11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Lee JH, Lim DS, Shim WJ, Ro YM, Park GH, Becker KG, Cho-Chung YS, Kim M-K. Gene expression profiling of oxidative stress on atrial fibrillation in humans. Exp Mol Med. 2003;35(5):336–349. doi: 10.1038/emm.2003.45. [DOI] [PubMed] [Google Scholar]
- Kristovich R, Knight DA, Long JF, Williams MV, Dutta PK, Waldman WJ. Macrophage-mediated endothelial inflammatory responses to airborne particulates: impact of particulate physiochemical properties. Chem Res Toxicol. 2004;17:1303–1312. doi: 10.1021/tx049893p. [DOI] [PubMed] [Google Scholar]
- Kuenzli N, Jerrett M, Mack WJ, Beckerman B, LaBree L, Gilliland F, Thomas D, Peters J, Hodis HN. Ambient air pollution and atherosclerosis in Los Angeles. Environ Health Perspect. 2005;113:201–206. doi: 10.1289/ehp.7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurl S, et al. Association of exercise-induced, silent ST-segment depression with the risk of stroke and cardiovascular diseases in men. Stroke. 2003;34:1760–1765. doi: 10.1161/01.STR.0000078564.46376.0A. [DOI] [PubMed] [Google Scholar]
- Laden F, Neas LM, Dockery DW, Schwartz J. Association of fine particulate matter from different sources with daily mortality in six U.S. cities. Environ. Health Perspect. 2000;108:941–947. doi: 10.1289/ehp.00108941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C-H, Liou S-H, Lin H-C, Shih T-S, Tsai P-J, Chen J-S, Yang T, Jaakkola JJK, Strickland PT. Exposure to traffic exhausts and oxidative DNA damage. Occup Environ Med. 2005;62:216–222. doi: 10.1136/oem.2004.015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanki T, Hartog J, Heinrich J, Hoek G, Janssen A, Peters A, Stolzel M, Tiomopnen K, Vallius M, Vanninen E, Pekkanen J. Can we identify sources of fine particles responsible for exercise-induced ischemia on days with elevated pollution? The ULTRA study. Environ Health Perspect. 2006;114:655–660. doi: 10.1289/ehp.8578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Nel A. The cellular impacts of diesel exhaust particles: beyond inflammation and death. Eur Respir J. 2006;27:667–668. doi: 10.1183/09031936.06.00025006. [DOI] [PubMed] [Google Scholar]
- Li N, Wang M, Oberley TD, Sempf JM, Nel AE. Comparison of the pro-oxidative and proinflammatory effects of organic diesel exhaust particle chemicals in bronchial epithelial cells and macrophages. J Epidemiol. 2002;169:4531–4541. doi: 10.4049/jimmunol.169.8.4531. [DOI] [PubMed] [Google Scholar]
- Li N, Kim S, Wang M, Froines J, Sioutas C, Nel A. Use of a stratified oxidative stress model to study the biological effects of ambient concentrated and diesel exhaust particulate matter. Inhal Toxicol. 2002;14:459–486. doi: 10.1080/089583701753678571. [DOI] [PubMed] [Google Scholar]
- Li N, Sioutas C, Cho A, Schmitz D, Misra C, Sempf J, Wang M, Oberley T, Froines J, Nel A. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ Health Perspect. 2003;111(4):455–460. doi: 10.1289/ehp.6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- Lipfert FW, Wyzga RE, Baty JD, Miller JP. Traffic density as a surrogate measure of environmental exposures in studies of air pollution health effects: Long-term mortality in a cohort of U.S. veterans. Atmos Environ. 2006;40:154–169. [Google Scholar]
- Lipfert FW, Baty JD, Wyzga RE, Miller JP. PM2.5 constituents and related air quality variables as predictors of survival in a cohort of U.S. military veterans. Inhal Toxicol. 2006;18:645–657. doi: 10.1080/08958370600742946. [DOI] [PubMed] [Google Scholar]
- Lippmann M, Ito K, Hwang J-S, Maciejczyk P, Chen LC. Cardiovascular effects of nickel in ambient Air. Environ Health Perspect. 2006;114:1662–1669. doi: 10.1289/ehp.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund AK, Knuckles TL, Akata CO, Shohet R, McDonald JD, Gigliotti A, Seagrave JC, Campen MJ. Gasoline exhaust emissions induce vascular remodeling pathways involved in atherosclerosis. Toxicol Sci. 2007;95(2):485–494. doi: 10.1093/toxsci/kfl145. [DOI] [PubMed] [Google Scholar]
- Luttmann-Gibson H, Suh HH, Coull BA, Dockery DW, Sarnat SE, Schwartz J, Stone PH, Gold DR. Short-term effects of air pollution on heart rate variability in senior adults in Steubenville, Ohio. J Occup Environ Med. 2006;48:780–788. doi: 10.1097/01.jom.0000229781.27181.7d. [DOI] [PubMed] [Google Scholar]
- Mauderly JL. Diesel emissions: is more health research still needed? Toxicol Sci. 2001;62:6–9. doi: 10.1093/toxsci/62.1.6. [DOI] [PubMed] [Google Scholar]
- Mauderly JL, Chow JC. Health effects of organic aerosols. Inhal Toxicol. 2008;20:257–288. doi: 10.1080/08958370701866008. [DOI] [PubMed] [Google Scholar]
- Maynard D, Coull BA, Gryparis A, Schwartz J. Mortality risk associated with short-term exposure to traffic particles and sulfates. Environ Health Perspect. 2007;115:751–755. doi: 10.1289/ehp.9537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JD, Harrod KS, Seagrave J, Seilkop SK, Mauderly JL. Effects of low sulfur fuel and a catalyzed particle trap on the composition and toxicity of diesel emissions. Environ Health Perspect. 2004;112:1307–1312. doi: 10.1289/ehp.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray J, Chopra M, Abdullah I, Smith WE, Dargie HJ. Evidence of oxidative stress in chronic heart failure in humans. Eur Heart J. 1993;14:1493–1498. doi: 10.1093/eurheartj/14.11.1493. [DOI] [PubMed] [Google Scholar]
- Medina-Ramon M, et al. Residential exposure to traffic-related air pollution and survival after heart failure. Environ Health Perspect. 2008;116:481–485. doi: 10.1289/ehp.10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger KB, Tolbert PE, Klein M, Peel JL, Flanders WD, Todd K, Mulholland JA, Ryan PB, Frumkin H. Ambient air pollution and cardiovascular emergency department visits. Epidemiology. 2004;15:46–56. doi: 10.1097/01.EDE.0000101748.28283.97. [DOI] [PubMed] [Google Scholar]
- Metzger KB, Klein M, Flanders WD, Peel JL, Mulholland JA, Langberg JJ, Tolbert PE. Ambient air pollution and cardiac arrhythmias in patients with implantable defibrillators. Epidemiology. 2007;18:585–592. doi: 10.1097/EDE.0b013e318124ff0e. [DOI] [PubMed] [Google Scholar]
- Miller MR, Borthwick SJ, Shaw CA, McLean SG, McClure D, Mills NL, Duffin R, Donaldson K, Megson IL, Hadoke PWF, Newby DE. Direct impairment of vascular function by diesel exhaust particulate through reduced bioavailability of endothelium-derived nitric oxide induced by superoxide free radicals. Environ Health Perspect. 2009;117:611–616. doi: 10.1289/ehp.0800235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills NL, Tornquist H, Robinson SD, Gonzalez M, Darnley K, MacNee W, Boon NA, Donaldson K, Blomberg A, Sandstrom T, Newby DE. Diesel exhaust inhalation causes vascular dysfunction and impaired endogenous fibrinolysis. Circulation. 2005;112:3930–3936. doi: 10.1161/CIRCULATIONAHA.105.588962. [DOI] [PubMed] [Google Scholar]
- Mills NL, Tornquist H, Gonzalez MC, Vink E, Robinson SD, Soderberg S, Boon NA, Donaldson K, Sandstrom T, Blomberg A, Newby DE. Ischemic and thrombotic effects of dilute diesel-exhaust inhalation in men with coronary disease. N Engl J Med. 2007;357:1075–1082. doi: 10.1056/NEJMoa066314. [DOI] [PubMed] [Google Scholar]
- Nafstad P, Haheim LL, Wisloff T, Gram F, Oftedal B, Holme I, Hjermann I, Leren P. Urban air pollution and mortality in a cohort of Norwegian men. Environ Health Perspect. 2004;112:610–615. doi: 10.1289/ehp.6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman RB, Bloom HL, Shukrullah I, Darron LA, Kleinbaum D, Jones DP, Dudley SC., Jr Oxidative stress markers are associated with persistent atrial fibrillation. Clin Chem. 2007;53(9):1652–1657. doi: 10.1373/clinchem.2006.083923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SK, O’Neill MS, Vokonas PS, Sparrow D, Schwartz J. Effects of air pollution on heart rate variability: the VA normative aging study. Environ Health Perspect. 2005;113:304–309. doi: 10.1289/ehp.7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SK, O’Neill MS, Stunder BJB, Vokanas PS, Sparrow D, Koutrakis P, Schwartz J. Source location of air pollution and cardiac autonomic function: Trajectory cluster analysis for exposure assessment. J Expo Sci Environ Epidemiol. 2007;17:488–497. doi: 10.1038/sj.jes.7500552. [DOI] [PubMed] [Google Scholar]
- Pekkanen J, Peters A, Hoek G, Tiittanen P, Brunekreef B, de Hartog J, Heinrich J, Ibald-Mulli A, Kreyling WG, Lanki T, Timonen KL, Vanninen E. Particulate air pollution and risk of ST-segment depression during repeated submaximal exercise tests among subjects with coronary heart disease. Circulation. 2002;106:933–938. doi: 10.1161/01.cir.0000027561.41736.3c. [DOI] [PubMed] [Google Scholar]
- Pereira CEL, et al. Ambient particulate air pollution from vehicles promotes lipid peroxidation and inflammatory responses in rat lung. Braz J Med Biol Res. 2007;40:1353–1359. doi: 10.1590/s0100-879x2006005000164. [DOI] [PubMed] [Google Scholar]
- Peretz A, Peck EC, Bammler TK, Beyer RP, Sullivan JH, Trenga CA, Srinouanprachnah S, Farin FM, Kaufman JD. Diesel exhaust inhalation and assessment of peripheral blood mononuclear cell gene transcription effects: an exploratory study of healthy human volunteers. Inhal Toxicol. 2007;19:1107–1119. doi: 10.1080/08958370701665384. [DOI] [PubMed] [Google Scholar]
- Peretz A, Sullivan JH, Leotta DF, Tenga CA, Sands FN, Allen J, Carlsten C, Wilkinson CW, Gill EA, Kaufman JD. Diesel exhaust inhalation elicits acute vasoconstriction in vivo. Environ Health Perspect. 2008;116:937–942. doi: 10.1289/ehp.11027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Liu E, Verrier RL, Schwartz J, Gold DR, Mittleman M, Baliff J, Oh JA, Allen G, Monahan K, Dockery DW. Air pollution and incidence of cardiac arrhythmia. Epidemiology. 2000;11(1):11–17. doi: 10.1097/00001648-200001000-00005. [DOI] [PubMed] [Google Scholar]
- Peters A, von Klot S, Heier M, Trentinaglia I, Hormann A, Wichmann HE, Lowel H. Exposure to traffic and onset of myocardial infarction. N Engl J Med. 2004;351:1721–1730. doi: 10.1056/NEJMoa040203. [DOI] [PubMed] [Google Scholar]
- Pope CA, Thun MJ, Namboodiri MM, Dockery DW, Evans JS, Speizer FE, Heath CW. Particulate air pollution as a predictor of mortality in a prospective study of U.S. adults. Am. J. Crit. Care Med. 1995;151:669–674. doi: 10.1164/ajrccm/151.3_Pt_1.669. [DOI] [PubMed] [Google Scholar]
- Pope CA, Burnett RT, Thun MJ, Calle EE, Krewski D, Ito K, Thurston GD. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution. JAMA. 2002;287(9):1132–1141. doi: 10.1001/jama.287.9.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst-Hensch NM, Imboden M, Dietrich DF, Barthelemy J-C, Ackermann-Liebrich U, Berger W, Gaspoz J-M, Schwartz J. Glutathione S-Transperase polymorphisms, passive smoking, obesity, and heart rate variability in nonsmokers. Environ Health Perspect. 2008;116:1491–1499. doi: 10.1289/ehp.11402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao V, Frank N, Rush A, Dimmick F (2002) Chemical speciation of PM2.5 in urban and rural areas. Proceedings of the Air and Waste Management Association Symposium on Air Quality Measurement Methods and Technology, San Francisco
- Restrepo C, Zimmermarn R, Thurston G, Clemente J, Gorezynski J, Zhong M, Blaustein M, Chen LC. A comparison of ground-level air quality data with New York State Department of Environmental Conservation monitoring stations data in South Bronx, New York. Atmos Environ. 2004;38:5295–5304. [Google Scholar]
- Rhoden CR, Wellenius GA, Ghelfi E, Lawrence J, Gonzalez-Flecha B. PM-induced cardiac oxidative stress and dysfunction are mediated by autonomic stimulation. Biochim Biophys Acta. 2005;1725:305–313. doi: 10.1016/j.bbagen.2005.05.025. [DOI] [PubMed] [Google Scholar]
- Rich DQ, Mittleman MA, Link MS, Schwartz J, Luttmann-Gibson H, Catalano PJ, Speizer FE, Gold DR, Dockery DW. Increased risk of paroxysmal atrial fibrillation episodes associated with acute increases in ambient air pollution. Environ Health Perspect. 2006;114:120–123. doi: 10.1289/ehp.8371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Rifai N, Stampfer MJ, Hennekens CH. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation. 2000;101:1767–1772. doi: 10.1161/01.cir.101.15.1767. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Danielson E, Fonseca FAH, Genest J, Gotto AM, Kastelein JJP, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- Riediker M. Cardiovascular effects of fine particulate matter components in highway patrol officers. Inhal Toxicol. 2007;19(Supp. 1):99–105. doi: 10.1080/08958370701495238. [DOI] [PubMed] [Google Scholar]
- Riediker M, Cascio WE, Griggs TR, Herbst MC, Bromberg PA, Neas L, Williams RW, Devlin RB. Particulate matter exposure in cars is associated with cardiovascular effects in healthy young men. Am J Respir Crit Care Med. 2004;169:934–940. doi: 10.1164/rccm.200310-1463OC. [DOI] [PubMed] [Google Scholar]
- Riediker M, Devlin RB, Griggs TR, Herbst MC, Bromberg PA, Williams RW, Cascio WE. Cardiovascular effects in patrol officers are associated with fine particulate matter from brake wear and engine emissions. Particle Fibre Technol. 2004;1:2. doi: 10.1186/1743-8977-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romieu I, Garcia-Esteban R, Sunyer J, Rios C, Alcaraz-Zubeldia M, Velasco SR, Holguin R. The effect of supplementation with omega-3 polyunsaturated fatty acids on markers of oxidative stress in elderly exposed to PM2.5. Environ Health Perspect. 2008;116:1237–1242. doi: 10.1289/ehp.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenlund M, Berglind N, Pershaden G, Hallquist J, Jonson T, Bellander T. Long-term exposure to urban air pollution and myocardial infarction. Epidemiology. 2006;17:383–390. doi: 10.1097/01.ede.0000219722.25569.0f. [DOI] [PubMed] [Google Scholar]
- Ruckerl R, Greven S, Ljungman P, Aalto P, Antoniades C, Bellander T, Berglind N, Chyrsohoou C, Forasteire F, Jacquemin B, von Klot S, Koenig W, Kuchenhoff H, Lanki T, Pekkanen J, Perucci CA, Schneider A, Sunyer J, Peters A. Air pollution and inflammation (Interleukin-6, C-Reactive Protein, Fibrinogen) in myocardial infarction survivors. Environ Health Perspect. 2007;115:1072–1080. doi: 10.1289/ehp.10021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajadieh A, Nielsen OW, Rasmussen V, Hein HO, Abedini S, Hansen JF. Increased heart rate and reduced heart-rate variability are associated with subclinical inflammation in middle-aged and elderly subjects with no apparent heart disease. Eur Heart J. 2004;25:363–370. doi: 10.1016/j.ehj.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Samet JM. Traffic, Air Pollution, and Health. Inhal Toxicol. 2007;19:1021–1027. doi: 10.1080/08958370701533541. [DOI] [PubMed] [Google Scholar]
- Sarnat SE, Suh HH, Coull BA, Schwartz J, Stone PH, Gold DR. Ambient particulate air pollution and cardiac arrhythmia in a panel of older adults in Steubenville, Ohio. Occup Environ Med. 2006;63:700–706. doi: 10.1136/oem.2006.027292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarnat JA, et al. Fine particle sources and cardiorespiratory morbidity: an application of chemical mass balance and factor analytical source-apportionment methods. Environ Health Perspect. 2008;116:459–466. doi: 10.1289/ehp.10873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schunkert H, Samani NJ. Elevated C-reactive protein in atherosclerosis–chicken or egg? N Engl J Med. 2008;359:1953–1955. doi: 10.1056/NEJMe0807235. [DOI] [PubMed] [Google Scholar]
- Schwartz J. Daily deaths associated with air pollution in six U.S. cities and short-term mortality displacement in Boston. In Revised analyses of time-series studies of air pollution and health. Boston, MA: Health Effects Institute, Special Report; 2003. pp. 219–226. [Google Scholar]
- Schwartz J, Park SK, O’Neill MS, Vokonas PS, Sparrow D, Weiss S, Kelsey K. Glutathione-S-Transferase M1, obesity, statins, and autonomic effects of particles. Am M Respir Crit Care Med. 2005;172:1529–1533. doi: 10.1164/rccm.200412-1698OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz J, Litonjua A, Suh H, Verrier M, Zanobetti A, Syring M, Nearing B, Verrier R, Stone P, MacCallum G, Speizer FE, Gold DE. Traffic related pollution and heart rate variability in a panel of elderly subjects. Thorax. 2005;60:455–461. doi: 10.1136/thx.2004.024836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaton A, Soutar A, Crawford V, Elton R, McNerlan S, Cherrie J, Watt M, Agius R, Stout R. Particulate air pollution and the blood. Thorax. 1999;54:1027–1032. doi: 10.1136/thx.54.11.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharman JE, et al. Exposure to automotive pollution increases plasma susceptibility to oxidation. Arch Environ Health. 2002;57(6):536–540. doi: 10.1080/00039890209602085. [DOI] [PubMed] [Google Scholar]
- Steinberg D. Atherogenesis in perspective: Hypercholesterolemia and inflammation as partners in crime. Nat Med. 2002;8(11):1211–1217. doi: 10.1038/nm1102-1211. [DOI] [PubMed] [Google Scholar]
- Tapanainen JM, Thomsen PEB, Kober L, Torp-Pedersen C, Makikallio TH, Still A-M, Lindgren KS, Huikuiri HV. Fractal analysis of heart rate variability and mortality after an acute myocardial infarction. Am J Cardiol. 2002;90:347–352. doi: 10.1016/s0002-9149(02)02488-8. [DOI] [PubMed] [Google Scholar]
- Thurston GD, Spengler JD. A quantitative assessment of source contributions to inhalable particulate matter pollution in metropolitan boston. Atmos Environ. 1985;19(1):9–25. [Google Scholar]
- Thurston GD, et al. Workshop report: workshop on source apportionment of particulate matter health effects–intercomparison of results and implications. Environ. Health Perspect. 2005;113:1768–1774. doi: 10.1289/ehp.7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolbert PE, et al. Multipollutant modeling issues in a study of ambient air quality and emergency department visits in Atlanta. J Expo Sci Environ Epidemiol. 2007;17:S29–S35. doi: 10.1038/sj.jes.7500625. [DOI] [PubMed] [Google Scholar]
- Tonne C, et al. A case–control analysis of exposure to traffic and acute myocardial infarction. Environ Health Perspect. 2007;115:53–57. doi: 10.1289/ehp.9587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornquist H, Mills NL, Gonzalez M, Miller MR, Robinson SD, Megson IL, MacNee W, Donaldson K, Soderberb S, Newby DE, Sandstrom T, Blomberg A. Persistent endothelial dysfunction in humans after diesel exhaust inhalation. Am M Respir. Crit Care Med. 2007;176:395–400. doi: 10.1164/rccm.200606-872OC. [DOI] [PubMed] [Google Scholar]
- Urch B, Brook JR, Wasserstein D, Brook RD, Rajagopalan S, Corey P, Silverman F. Relative contributions of PM2.5 chemical constituents to acute arterial vasoconstriction in humans. Inhal Toxicol. 2004;16:345–352. doi: 10.1080/08958370490439489. [DOI] [PubMed] [Google Scholar]
- Urch B, Silverman F, Corey P, Brook JR, Lukic KZ, Rajagopalan S, Brook RD. Acute blood pressure responses in healthy adults during controlled air pollution exposures. Environ Health Perspect. 2005;113(8):1052–1055. doi: 10.1289/ehp.7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. Environmental Protection Agency (2002) Health assessment document for diesel engine exhaust. http://www.epa.gov/fedrgstr/EPAAIR/2002/September/Day-03/a22368.htm
- Volpato S, Guralnick JM, Ferrucci L, Balfour J, Chaves P, Fried LP, Harris TB. Cardiovascular disease, interleukin-6, and risk of mortality in older women. Circulation. 2001;103:947–953. doi: 10.1161/01.cir.103.7.947. [DOI] [PubMed] [Google Scholar]
- Wheeler A, Zanobetti A, Gold DR, Schwartz J, Stone P, Suh HH. The relationship between ambient air pollution and heart rate variability differs for individuals with heart and pulmonary disease. Environ Health Perspect. 2006;114:560–566. doi: 10.1289/ehp.8337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RH, Spengler JD, Dilwali KM, Barry BE, Samet JM. Report of workshop on traffic, health, and infrastructure planning. Arch Environ Occup Health. 2005;60:70–76. doi: 10.3200/AEOH.60.2.70-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woollard KJ, Chin-Dusting J. Therapeutic targeting of P-selectin in atherosclerosis. Inflamm Allergy Drug Targets. 2007;6:69–74. doi: 10.2174/187152807780077345. [DOI] [PubMed] [Google Scholar]
- Yan Y-H, Huang C-H, Chen W-J, Wu M-F, Cheng T-J. Effects of diesel exhaust particles on left ventricular function in isoproterenol-induced myocardial injury and healthy rats. Inhalation Toxicol. 2008;20:199–203. doi: 10.1080/08958370701861082. [DOI] [PubMed] [Google Scholar]
- Zeger SL, Thomas D, Dominici F, Samet JM, Schwartz J, Dockery D, Cohen A. Exposure measurement error in time-series studies of air pollution: concepts and consequences. Environ Health Perspect. 2000;108:419–426. doi: 10.1289/ehp.00108419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeka A, Sullivan JR, Vokonas PS, Sparrow D, Schwartz J. Inflammatory markets and particulate air pollution: characterizing the pathway to disease. Intntl J Epdiemiol. 2006;35:1347–1354. doi: 10.1093/ije/dyl132. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Hinds WG, Kim S, Sioutas C. Concentration and size distribution of ultrafine particles near a major highway. J Air Waste Manage Assoc. 2002;52:1032–1042. doi: 10.1080/10473289.2002.10470842. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Hinds WG, Kim S, Shen S, Sioutas C. Study of ultrafine particles near a major highway with heavy-duty diesel traffic. Atmos Environ. 2002;36:4323–4335. [Google Scholar]