

ERO1-β has an unexpected and selective function for oxidative protein folding in insulin-producing cells.
Abstract
Mammals have two genes encoding homologues of the endoplasmic reticulum (ER) disulfide oxidase ERO1 (ER oxidoreductin 1). ERO1-β is greatly enriched in the endocrine pancreas. We report in this study that homozygosity for a disrupting allele of Ero1lb selectively compromises oxidative folding of proinsulin and promotes glucose intolerance in mutant mice. Surprisingly, concomitant disruption of Ero1l, encoding the other ERO1 isoform, ERO1-α, does not exacerbate the ERO1-β deficiency phenotype. Although immunoglobulin-producing cells normally express both isoforms of ERO1, disulfide bond formation and immunoglobulin secretion proceed at nearly normal pace in the double mutant. Moreover, although the more reducing environment of their ER protects cultured ERO1-β knockdown Min6 cells from the toxicity of a misfolding-prone mutant Ins2Akita, the diabetic phenotype and islet destruction promoted by Ins2Akita are enhanced in ERO1-β compound mutant mice. These findings point to an unexpectedly selective function for ERO1-β in oxidative protein folding in insulin-producing cells that is required for glucose homeostasis in vivo.
Introduction
Protein disulfide isomerases (PDIs) catalyze disulfide bond formation in itinerant proteins in the ER, promoting protein folding in the secretory pathway. In the process, the PDIs are reduced by their ER clients and must be reoxidized to sustain disulfide bond formation. In yeast, PDI reoxidation is performed by the enzyme ERO1 (ER oxidoreductin 1). Thus, electrons recovered from reduced cysteines on polypeptides that are translocated into the lumen of the yeast ER are channeled to their ultimate acceptor in a relay involving the luminal PDIs and ERO1 (for reviews see Tu and Weissman, 2004; Sevier and Kaiser, 2008).
Yeast and simple metazoans such as worms and flies have a single copy of the ERO1 gene, which is essential (Frand and Kaiser, 1998; Pollard et al., 1998; Tien et al., 2008). In contrast, mammals have two genes encoding proteins homologous to yeast Ero1p, known as ERO1-α (or Ero1l; Cabibbo et al., 2000) and ERO1-β (or Ero1lb; Pagani et al., 2000). Conservation of key residues involved in enzymatic activity and its regulation and mammalian overexpression studies and trans-specific complementation experiments leave little room for doubt that the α isoform of the mammalian enzyme is able to promote disulfide bond formation in the ER (Cabibbo et al., 2000; Mezghrani et al., 2001; Appenzeller-Herzog et al., 2008; Baker et al., 2008). ERO1-α’s role in disulfide bond formation in mammals is further supported by the observation that its mRNA is found in many tissues (Cabibbo et al., 2000) and by the diverse consequences of experimental attenuation of the enzyme’s level of expression (for examples see May et al., 2005; Qiang et al., 2007; Li et al., 2009).
The β isoform is less well characterized, and its expression pattern is less well understood. An earlier study has called attention to the role of ER stress, which is a physiological condition arising from a mismatch between ER unfolded protein load and the organelle’s capacity to cope with such load, in activating ERO1-β expression via the so-called unfolded protein response (UPR; Pagani et al., 2000). More recently, a study has called attention to the high basal levels of ERO1-β protein in the pancreatic islets of Langerhans (Dias-Gunasekara et al., 2005), suggesting a measure of tissue specificity to the protein’s expression.
In simple eukaryotes such as yeast and worms, partial lowering of ERO1 activity promotes resistance to the lethal effects of high levels of ER stress (Haynes et al., 2004; Marciniak et al., 2004), and in worms, knockdown of that species’ single isoform, ero-1, in postembryonic animals significantly prolongs adult lifespan (Curran and Ruvkun, 2007). The insulin-producing cells of the islets of Langerhans are especially sensitive to the consequences of ER stress and the attendant oxidative stress (for review see Eizirik et al., 2008). Thus, the proposed induction of ERO1-β by the UPR (Pagani et al., 2000) and the report of its selective expression in the islets of Langerhans (Dias-Gunasekara et al., 2005) raised our curiosity as to the functional significance of ERO1-β expression under normal conditions and under conditions of ER stress. Therefore, we have studied mice with severe disruption of ERO1-β expression and report on their phenotype under basal conditions, under conditions of cocompromised ERO1-α expression, and under conditions of severe ER stress in the insulin-producing cells of the islets of Langerhans.
Results
Pancreatic-selective expression of ERO1-β and its disruption by the P077G11 insertion
To revisit the tissue expression of ERO1 isoforms, we raised rabbit polyclonal antisera to the α and β mouse proteins. Immunoblotting of mouse tissue extracts revealed a very strong signal of ERO1-β in the pancreas. In contrast, an ERO1-α signal was found in all tissues tested (Fig. 1 A and Fig. S1). The ERO1-β signal was concentrated in the membrane fraction of expressing cells, as previously described (not depicted; Pagani et al., 2000), and analysis of the membrane fraction from several secretory tissues confirmed the pancreatic-selective expression of the protein (Fig. 1 B). Quantitative immunoblotting of the ERO1-β content in purified membrane fraction of pancreatic tissue and calibration with the signal derived from known amounts of bacterially expressed ERO1-β indicated that the protein’s concentration in pancreatic ER fraction was in the order of 12 ng/µl (Fig. 1 C). Given a predicted molecular mass of ∼50 kD, the estimated concentration of the protein in the ER is at least 0.24 µM, which is a high concentration for an enzyme. This calculation is based on a conservative assumption that the tissue is 50% cellular and that the ER is 50% of the cell’s volume and ignores the fact that ERO1-β expression is largely limited to the islets of Langerhans (Fig. 2 C). The relatively high level expression of ERO1-β in the pancreas and cross-reactivity of the anti–ERO1-α sera with ERO1-β (Fig. S1) also account for significant ERO1-β signal in the pancreatic lysate probed with the ERO1-α antiserum (Fig. 1 A, lane 1).
Figure 1.

ERO1-β is a disulfide oxidase selectively expressed in the pancreas. (A) Immunoblot of mouse tissue detergent extracts reacted with antisera raised to ERO1-β (top), ERO1-α (middle), and Ribophorin I (bottom; an ER recovery marker). Note the selective expression of ERO1-β in the pancreas and the cross-reactivity of the anti–ERO1-α serum with ERO1-β (also see Fig. S1). The asterisk marks a nonspecific band reactive with the anti–ERO1-α serum in pancreatic lysates. (B) ERO1-β immunoblot of membrane fraction derived from mouse secretory tissues. PDI serves as a recovery marker. (C) Immunoblot of ERO1-β content of pancreatic membrane fraction (Mem Fr) derived from the indicated tissue volume and of known amounts of purified, bacterially expressed protein (ERO1-βr). The relative signal intensity (rel signal) is noted under the blot. (D) In vitro disulfide oxidase activity of bacterially expressed mouse ERO1-α (ERO1-αr), mouse ERO1-α, or mouse ERO1-β purified from transfected 293T cells, using bacterially expressed reduced human PDI or reduced E. coli thioredoxin (TrxA) as substrates and H2O2 production (read kinetically in a fluorescence-based assay) as a readout. The time-dependent increase in fluorescence of each sample is reported on over a period of 60 min (t). The inset shows a Coomassie-stained gel of the purified enzymes (see Fig. S2 for additional technical control experiments validating the assay). RFU, relative fluorescent unit. (A–D) Molecular mass is indicated in kilodaltons.
Figure 2.

ERO1-β disruption by promoter trap insertion into the Ero1lb locus. (A) Ethidium bromide–stained agarose gel of genomic DNA recovered by PCR from the Ero1lb (ERO1-β) locus after digestion with PstI. Shown are samples of F2 progeny of C57BL/6; 129 F1 hybrid parents heterozygous for the ERO1-β insertion allele (i/+) from the P077G11 ES cell line and wild-type C57BL/6 and 129 DNA. Note that the mutant allele (i) tracks with the 129 version of the polymorphism, to which it is tightly linked. (B) ERO1-β immunoblot of membrane fraction of pancreas of wild-type (+/+), heterozygous (i/+), and homozygous ERO1-β mutant mice (i/i). The asterisk marks the ERO1-β–Ceo fusion protein encoded by the trapped insertion allele. Ribophorin I and immunoglobulin-binding protein (BIP) serve as ER recovery markers. (C) Fluorescent micrographs of frozen section of pancreas from wild-type (+/+) and ERO1-β homozygous mutant mice (i/i) stained with antisera to insulin (Ins), ERO1-β, and the karyophilic dye DAPI. The bottom right panel in each genotype is an overlay of the three stains. Note the colocalization of insulin and ERO1-β. Bar, 100 µm.
To date, ERO1-β’s enzymatic function had been inferred from sequence conservation with yeast Ero1p but had not been demonstrated experimentally. To critically examine this inference, ERO1-β’s ability to oxidize disulfide isomerases was measured by an in vitro fluorescent assay that detects the H2O2 produced as ERO1 passes the electron it accepts from its reduced substrates to molecular oxygen (Gross et al., 2006). The addition of Flag-tagged mouse ERO1-β or -α, purified from transfected 293T cells, to the assay resulted in a time-dependent increase in H2O2 production. Both isoforms of ERO1 had roughly similar activity in this assay, resembling that of recombinant mouse ERO1-α produced in bacteria (Fig. 1 D). Both enzymes had similar relative ability to accept electrons from a model substrate, reduced bacterial thioredoxin, and from a physiological substrate, reduced human PDI. An active site mutation ERO1-βC396A abolished all enzymatic activity in vitro (Fig. S2), as predicted (Mezghrani et al., 2001), and only background levels of H2O2 production were noted in the absence of substrate, attesting to the specificity of the assay.
The aforementioned observations confirmed the pancreatic-selective expression of ERO1-β and established definitively its ability to directly oxidize reduced PDI. To gain further insight into the physiological role of ERO1-β, we developed a mouse model with a loss of function mutation in the gene.
The P077G11 embryonic stem (ES) line contains an insertion of a replication defective promoterless retrovirus with a FlipROASCeoC-2 gene, containing a 3′ splice acceptor and the coding region for a functional signal-anchor peptide/transmembrane domain in fusion with a Neor gene and strong transcriptional terminators (De-Zolt et al., 2006). Analysis of the fusion cDNA expressed in ES cells predicted the insertion to have occurred in intron 14 and the mutant allele to direct the expression of a type I transmembrane fusion protein with an ectodomain comprised of aa 34–403 of mouse ERO1-β and a cytosolic portion with a functional Neor gene. Missing from the fusion protein are key structural elements that coordinate the FAD (flavin adenine dinucleotide) moiety required for enzymatic activity (Gross et al., 2004). Thus, the mutation is predicted to severely disrupt ERO1-β function.
Molecular analysis confirmed the insertion between exon 14 and 15 of the gene but also called attention to the existence of additional ERO1-β–homologous sequences in mouse chromosome 13 involving the 3′ portion of the gene (see Materials and methods). These homologous sequences were present in all mouse strains tested and in the sequenced genome of BL/6. The latter revealed several polymorphisms in the exonic DNA that distinguished the centromeric and telomeric versions of the duplicated sequences. Analysis of the mouse EST database and direct sequencing of cDNA from the pancreas of BL/6 mice revealed that only the centromeric ERO1-β DNA was expressed as mRNA in the pancreas (unpublished data), indicating that the telomeric sequence is likely an intron-containing pseudogene and that the insertion in P077G11 could disrupt gene function.
Chimeric males derived by injection of P077G11 ES cells into BL/6 blastocysts transmitted the mutant allele to their progeny. We exploited a polymorphism between the BL/6 and 129 genome at the 3′ end of exon 14 to mark the wild-type BL/6 allele and distinguish it from the mutant 129 allele. Thus, the presence or absence of a PstI site in genomic DNA derived by PCR from the 3′ end of intron 14 was used to distinguish F2 progeny that were wild type (+/+), heterozygous (i/+), or homozygous for the insertion (i/i; Fig. 2 A and see Materials and methods).
Immunoblotting showed that the insertion strongly compromised ERO1-β expression in the pancreas and directed expression of the predicted ERO1-β–Neor fusion protein (Fig. 2 B; the fusion protein was faintly visible at this exposure but more conspicuous in longer exposures). Immunostaining of pancreatic sections confirmed the previously described heavy islet staining of ERO1-β in the wild-type mice (the islets are recognized by insulin immunostaining; Dias-Gunasekara et al., 2005) and its absence from the homozygous mutant (i/i) mice (Fig. 2 C).
Impaired glycemic control and defective insulin biogenesis in ERO1-β mutant mice
Wild-type and mutant mice of both sexes were indistinguishable superficially, and all three genotypes were recovered at the expected frequency in the progeny of heterozygous (i/+) matings. To gauge the effect of the mutation on glycemic control, we compared the fasting blood sugar in F2 male siblings that were wild type, heterozygous, or homozygous for the mutation. By 3 mo of age, the majority of the homozygous mutant mice had a stable diabetic phenotype with mild fasting hyperglycemia (Fig. 3 A). The ratio of glucose to insulin was dramatically elevated in the homozygous mutant mice, which is consistent with a defect in insulin production/secretion (Fig. 3 B). Fasted heterozygous mutant mice had wild-type glucose levels but were relatively impaired in their ability to assimilate a glucose load in a glucose tolerance test (Fig. 3 C). This defect, too, was associated with an abnormally elevated glucose/insulin ratio (Fig. 3 D).
Figure 3.

Impaired glucose tolerance and glucose-stimulated insulin secretion in ERO1-β mutant mice. (A) Fasting blood glucose of male mice of the indicated age and indicated ERO1-β genotype. Shown are the mean and SEM in each group; also shown (in small black dots) are the glucose measurements of the individual mice in each group (*, P < 0.001). (B) Fasting blood glucose and blood glucose to serum insulin ratio of 6-mo-old male mice with the indicated ERO1-β genotype. Shown are the mean and SEM in each group (n = 5; *, P < 0.001 by two-tailed t test). (C) Blood glucose levels after intraperitoneal injection of a glucose load into 6-mo-old male otherwise-isogenic C57BL/6; 129 F1 hybrid wild-type and heterozygous ERO1-β mutant mice. Shown are the mean and SEM in each group (n = 5; *, P < 0.001 by two-way ANOVA). (D) Blood glucose to serum insulin ratio at the fasted and 15-min time point of the experiment shown in C. Shown are the mean and SEM in each group (n = 5; *, P < 0.001 by two-tailed t test) and the measurement on each individual animal in small black circles.
The mild diabetic phenotype of the ERO1-β mutant mice was not associated with conspicuous histological changes in the islets of Langerhans (Fig. 4 A). However, insulin content in the pancreas of mutant mice was significantly decreased (Fig. 4 B and Fig. S3), and subtle disorganization of the islets, reflected in the abundance of glucagon-positive cells in their centers, was noted (Fig. 4 C). Ultrastructure of insulin-producing β cells from the homozygous mutant mice was likewise normal, except for a trend toward increased content of ER lamella. Conspicuously absent were cells with dilated ER, which are observed in other mouse models of defective protein folding in the ER (Fig. 4 D).
Figure 4.
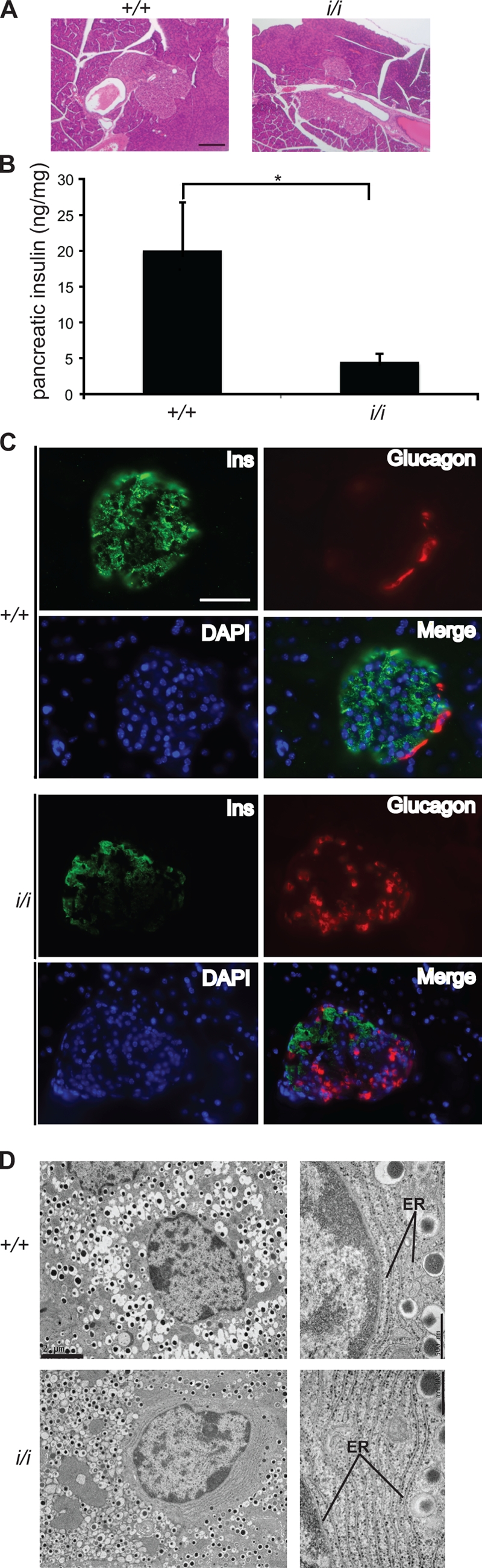
Diminished insulin stores and mildly disorganized islets in ERO1-β mutant mice. (A) Hematoxylin- and eosin-stained pancreatic sections of 3-mo-old wild-type and homozygous ERO1-β mutant mice. (B) Pancreatic insulin content of mice as in A. Shown are the mean and SEM (n = 5; *, P < 0.001 by two-tailed t test). (C) Fluorescent micrographs of frozen section of pancreas from wild-type (+/+) and ERO1-β homozygous mutant mice (i/i; as in A) stained with antisera to insulin (Ins), glucagon, and the karyophilic dye DAPI. The bottom right panel in each genotype is an overlay of the three stains. Note the presence of glucagon-positive cells in the interior of the islet of the ERO1-β mutant. (D) Electron micrographs of insulin-producing β cells from islets of Langerhans of wild-type and ERO1-β mutant mice. Note the lack of ER dilation in the higher magnification views (right) of the mutant sample. Bars: (A and C) 100 µm; (D, left) 2 µm; (D, right) 0.5 µm.
Proinsulin folds oxidatively (Anfinsen, 1973), and correct placement of three disulfide bonds is critical to the protein’s maturation in the ER (Wang et al., 1999). Pulse-chase metabolic labeling of newly synthesized protein in islets recovered from wild-type and homozygous mutant mice followed by immunoprecipitation of the labeled proinsulin and insulin from the cell lysate and the culture supernatant revealed a reproducible delay in the conversion of proinsulin to insulin (Fig. 5, A and B). This delay was associated with the notable persistence of high molecular mass (presumably oxidized) aberrant intermediates of proinsulin in the mutant cells (Fig. 5, C and D). A delay in proinsulin maturation was also observed in ERO1-β knockdown in cultured Min6 cells, an insulin-producing β cell line (Fig. S5).
Figure 5.

Delayed and inefficient oxidative maturation of proinsulin in ERO1-β mutant islets of Langerhans. (A) Autoradiograph of proinsulin (ProIns) and insulin (Ins) immunoprecipitated from lysates (top) and culture supernatant (media; bottom) of islets isolated from mice with the indicated ERO1-β genotype, metabolically labeled with [35S]methionine/cysteine for a 20-min pulse, followed by the indicated chase period. Immunopurified proteins were resolved on a reducing tris-tricine gel, and their position on the gel is indicated. (B) The ratio of mature insulin to total (proinsulin and insulin) signal of the experiment shown in A is plotted as a function of time. (C) Autoradiograph of proinsulin immunoprecipitated from lysates of islets of the indicated ERO1-β genotypes metabolically labeled as in A. The immunopurified proteins were resolved on a nonreducing tris-tricine gel. Shown are the positions of the reduced proinsulin (ProInsred; indicated by the migration of a reduced sample loaded at a distance on the same gel, lane 1), oxidized proinsulin (ProInsox), and higher molecular mass oxidized species. The bottom panel is a Coomassie blue stain of the same gel. The vertical black line indicates that intervening lanes have been spliced out. (A and C) Molecular mass is indicated in kilodaltons. (D) The ratio, as a function of time, of oxidized proinsulin to total label in the proinsulin immunoprecipitation in the experiment shown in C is shown.
Nonredundancy of the ERO1 isoforms in the endocrine pancreas and immunoglobulin-secreting cells
The aforementioned observations suggest that ERO1-β loss of function has an adverse effect on insulin biogenesis and glycemic control in mice. To determine whether ERO1-α complements the activity of the β isoform and accounts for the residual oxidative capacity of the insulin-producing cells from the ERO1-β homozygous mutant, we established compound mutant mice with mutations in both ERO1 genes by crossing the ERO1-β mutation into a strain with an insertional mutation in intron 6 of Ero1l (derived from the ES cell line XST171) that encodes a nonfunctional ERO1-α protein truncated at residue 168. Immunoblotting of pancreatic and spleen lysates from wild-type, ERO1-β mutant, ERO1-α mutant, and compound mutant mice confirmed the effect of the mutations on cognate protein expression but gave no evidence for compensatory increase in expression of one isoform when the other was lowered (Fig. 6 A). A similar observation was noted in the Min6 insulin-producing cell line (Fig. S4 A).
Figure 6.

Disulfide bond formation despite disruption of both ERO1-α and -β. (A) Immunoblot of ERO1-β (top), ERO1-α (middle), and Ribophorin I (bottom) in pancreatic and splenic membrane fractions of mice with the indicate ERO1-α and -β genotypes. Note that the levels of one ERO1 isoform do not change in response to decline in the other isoform. The asterisk marks a nonspecific band detected by the anti–ERO1-β serum in pancreas. (B) Immunoblots of ERO1 isoforms and PDI in lysates of LPS-induced B cell blasts of the indicated genotypes. To facilitate the comparison, three different loadings of each sample were analyzed. (C) Immunoblot of IgM from lysates of wild-type and compound mutant ERO1-αi/i; ERO1-βi/i LPS blasts after 30-min exposure to DTT in vivo and the indicated period of washout. The rate of recovery of disulfide bonds in IgM is reported on by the progressive depletion of monomers (1µ) and assembly of dimers (2µ) and pentamers (5µ) on this nonreducing gel. The sample in lane 1 is from cells that had never been exposed to DTT. A reducing gel with a fraction of the same samples is presented in the bottom panel. (A–C) Molecular mass is indicated in kilodaltons. (D) Serum IgM in 3-mo-old wild-type and compound mutant ERO1-αi/i; ERO1-βi/i mice. Shown are the mean and SEM (n = 3). (E) Blood glucose concentration after a glucose injection (as in Fig. 3 C) in mice with the indicated genotypes. Shown are the mean and SEM (n = 3; *, P < 0.001 by two way ANOVA; †, no significant difference).
A similar lack of compensatory changes in isoform expression was noted in a different secretory cell type, immunoglobulin-producing lipopolysaccharide (LPS) blasts produced by exposure of spleen cells procured from individuals with wild-type and mutant ERO1 genotypes to bacterial LPS in vitro. Wild-type LPS blasts expressed both ERO1-α and -β, but LPS blasts procured from animals with mutations in one isoform did not detectably up-regulate the expression of the other isoform (Fig. 6 B). It is notable that whereas in immunoblots of reduced and denatured lysates from wild-type cells, the ERO1-β antiserum recognized two close bands of nearly equal intensity, in samples from cells lacking ERO1-α, the slower migrating form of ERO1-β predominated (Figs. 2 B and 6, A and B). The significance of this reproducible observation is presently unclear.
LPS-induced differentiation into metabolically active morphologically normal blasts (a process which takes 2–3 d) was unaffected by the deficiency in both isoforms of ERO1 (unpublished data). And oxidative folding of IgM (the major secretory product of LPS blasts) was only modestly delayed by the compound mutation; this was revealed by comparing the rate at which oxidative forms of IgM were regenerated followed by their reduction in vivo by a 30-min DTT pulse (10 mM), removal of the reducing agent, and further culture for variable period of chase before lysis in the presence of N-ethyl maleimide and nonreducing SDS-PAGE (Fig. 6 C). Serum levels of IgM were likewise similar in the wild-type and compound mutant mice (Fig. 6 D). Furthermore, in keeping with the evidence for a surprisingly minor role for ERO1 in oxidative folding in immunoglobulin-secreting cells, there was no evidence for up-regulation of UPR target genes in the compound mutant LPS blasts (Fig. S4 B).
In the endocrine pancreas, too, there was no evidence for compensation by ERO1-α for the defect imposed by ERO1-β loss of function. This was assessed by comparing glucose tolerance of 2.5-mo-old wild-type male ERO1-β mutant and compound ERO1-β; ERO1-α mutant mice. At this age, the ERO1-β mutant mice had mild fasting hyperglycemia and significant intolerance to glucose load, but these features were not enhanced by concomitant disruption of ERO1-α (Fig. 6 E).
Attenuated ERO1-β expression protects Min6 cells from the consequences of enforced expression of a misfolding-prone proinsulin but fails to protect islets of Langerhans against the same stress in vivo
Oxidative protein misfolding in the ER is deleterious to insulin-producing β cell function and viability. This pathophysiological mechanism may be especially prominent in cells expressing mutant forms of insulin that are unable to undergo proper disulfide bond formation (Wang et al., 1999; Liu et al., 2007). In yeast and worms, partial loss of function of the essential ERO1 gene enhances the organism’s ability to cope with severe ER stress (Haynes et al., 2004; Marciniak et al., 2004), which is noted in the face of significant levels of ER stress and strong activation of the UPR by compromised ERO1 function.
To test whether partial compromise of ERO1 activity may likewise protect β cells from severe ER stress, we stably lowered gene function by RNAi in Min6 cells, an insulin-producing mouse insulinoma cell line. Like yeast and worms, Min6 cells with lowered ERO1-β activity had higher levels of transcriptional targets of the UPR (Fig. 7 A) and experienced delayed maturation of proinsulin compared with the parental cells (Fig. S5). ERO1-β knockdown Min6 cells were also strongly protected from the previously noted lethal consequences of expression of a GFP-tagged misfolding-prone proinsulinAkita (with a mutation, C96Y, precluding an essential disulfide bond; Wang et al., 1999; Oyadomari et al., 2002; Liu et al., 2007). This is evinced from the observation that stable colonies expressing GFP-proinsulinAkita developed very inefficiently in wild-type (parental) Min6 cells but readily formed in the ERO1-β knockdown–derivative line KD1 (Fig. 7, B and C).
Figure 7.

Lowering ERO1-β levels protects Min6 cells from the consequences of expression of a misfolding-prone proinsulin mutant but does not preserve islet function in mice. (A) RNAi knockdown of ERO1-β in Min6 cells revealed by immunoblotting (left) or immunostaining (right). Also shown is an immunoblot of the UPR targets, GRP94, immunoglobulin-binding protein (BIP), and PDI in the parental Min6WT cells and two RNAi knockdown cell lines (Min6KD1 and Min6KD2). β-Actin serves as a loading control. Molecular mass is indicated in kilodaltons. (B) Photomicrographs of crystal violet–stained parental and ERO1-β knockdown (KD1) Min6 cells after transduction with a blasticidin resistance–marked lentivirus expressing either GFP or a fusion of GFP with a mutant proinsulinAkita GFP-INSC96Y (Liu et al., 2007). Where indicated, the cells were selected for blasticidin resistance. (C) As in B; quantification of the cell mass after transduction of the parental (Min6WT) or ERO1-β knockdown (Min6KD1) cells with the GFP lentivirus (set at 100%) or the GFP-INSC96Y lentivirus and blasticidin selection for 7 d (shown are the mean and SEM; n = 3; *, P < 0.02). (D) Fasting blood glucose of a cohort of male Akita (Ins2C96Y) mutant mice with the indicated ERO1-β genotypes at the indicated age. Shown are the mean and SEM of each group as well as the measurements in each individual mouse (in small black dots; n = 4; *, P < 0.009; and **, P < 0.003 by two-tailed t test). (E) Insulin content of pancreas from 2-mo-old male mice of the indicated Ins2 and ERO1-β genotype. Shown are the mean and SEM (n = 3; *, P < 0.005 by two-tailed t test). Bars: (A) 25 µm; (B) 1 mm.
To determine whether lowered levels of ERO1-β can also protect β cells against lethal levels of ER stress in mice, we bred isogenic 129 mice carrying the mutant ERO1-β insertion allele to isogenic BL/6 mice that carried the proinsulin2Akita mutation (Ins2C96Y/+) and analyzed the compound phenotype in otherwise-isogenic male F1 hybrid progeny. As reported previously (Wang et al., 1999), in the wild-type ERO1-β background, the proinsulin2Akita mutation (Ins2C96Y/+; ERO1-β+/+) resulted in a mild progressive diabetic phenotype, whereas Ins2+/+; ERO1-βi/+ mice had normal fasting blood sugars (Fig. 3 C). However, surprisingly, compounding the proinsulinAkita mutation with the ERO1-β mutation (Ins2C96Y/+; ERO1-βi/+) exacerbated glucose intolerance (Fig. 7 D). This was noted in the face of similar reduction of islet mass in pancreatic sections (not depicted) and similar lowering (compared with Ins2+/+) of pancreatic insulin content by expression of the proinsulin2Akita in Ins2C96Y/+; ERO1-β+/+ and Ins2C96Y/+; ERO1-βi/+ mice (Fig. 7 E). Thus, the benefits of a more reducing ER on cells’ ability to cope with an oxidatively misfolded mutant proinsulin were apparent in cultured insulin-producing cells but not in the endocrine pancreas of live mice.
Discussion
This paper represents the first analysis of the consequences of disruption of ERO1 isoforms in living mammals. Our findings of direct oxidation of PDI by ERO1-β in vitro and retarded oxidative folding of proinsulin in islets lacking ERO1-β are consistent with a simple model in which ERO1-β is an important disulfide oxidase in insulin-producing β cells. Insulin biosynthesis is dynamically regulated, with up to a sixfold increase in the translation rate in response to physiological excursions in glucose levels (Itoh and Okamoto, 1980). Proinsulin is the major biosynthetic product of the β cell, and its efficient oxidative folding may represent a significant quantitative challenge to the ER machinery. Thus, our findings are readily explained by a gene duplication event, giving rise to an islet-selective isoform of the ERO1 oxidase that boosts the oxidative folding capacity of insulin-producing cells through gene dosage effects.
Islets explanted from homozygous ERO1-β mutant mice (and ERO1-β–deficient Min6 cells) have a conspicuous kinetic defect (delay) in oxidative folding and proinsulin maturation. More aberrant oxidized folding intermediates of proinsulin are also observed in islets explanted from mutant mice. Furthermore, in the insulin-producing β cell line Min6 conspicuous induction of UPR targets is observed after ERO1-β knockdown. Together, these observations suggest that the enhanced load of unfolded intermediates in insulin-producing cells that lack ERO1-β challenges the protein folding environment of their ER. A similar activation of the UPR is observed in yeast and worms with partial loss of ERO1 function (Frand and Kaiser, 1998; Pollard et al., 1998; Marciniak et al., 2004).
Morphological disorganization of the islets (reflected in the presence of glucagon-producing cells in their interior) suggests ongoing degeneration. However, this feature is mild at worse, as the diabetic phenotype of the homozygous mutant, once established at ∼3 mo, does not progress. Furthermore, although the delayed maturation of proinsulin and other proteins that fold oxidatively is likely to enhance the steady-state levels of misfolded proteins in the mutant cells’ ER, the ERO1-β mutation we studied is not associated with the conspicuous ultrastructural changes that have been noted in other diseases of ER protein misfolding (Wang et al., 1999; Harding et al., 2001; Zhang et al., 2002).
The mutant phenotype may be explained by a global inefficiency in the ERO1-β mutant islet cell’s ER to pass electrons from its pool of reduced PDIs to their terminal acceptors. However, this simple model is challenged by the surprising nonredundancy of the ERO1-α and -β isoforms, although both are expressed in the pancreas (Fig. 1 A) and in Min6 cells (Fig. S4 A). Concomitant depletion of ERO1-α has no effect on glycemic control of the ERO1-β mutant. More surprising, LPS blasts (and their immunoglobulin-producing counterpart in vivo) are able to produce nearly wild-type levels of IgM despite mutation of both ERO1 isoforms. These findings point strongly to the existence of hitherto unanticipated, ERO1-independent mechanisms to generate disulfide bonds in mammalian cells. Redundant mechanisms for disulfide bond formation may also exist in more distant organisms such as in flies, as attested to by the viability of clones with homozygous deletion of that species’ single ERO1 gene (known as kiga; Tien et al., 2008) Furthermore, the differences between β cells and immunoglobulin-producing cells highlight the special role of ERO1-β in disulfide bond formation and protein folding homeostasis in the lumen of the ER of insulin-producing cells.
At present, we have few experimentally derived clues to the special features of ERO1-β that render it critical to the proper function of the β cell ER. In vitro, purified ERO1-α and -β have similar capability to accept electrons from a model substrate (reduced bacterial thioredoxin) or a physiological substrate (reduced human PDI1). However, β cells have a peculiar complement of PDIs (Fig. 7 A; Dias-Gunasekara et al., 2005) that may be better suited to work with ERO1-β than ERO1-α. Such specialization, if it exists, may have arisen because of differences in the regulation of the two ERO1 isoforms by redox. Yeast ERO1 is allosterically regulated by the oxidation of regulatory cysteines (Sevier et al., 2007). These are conserved in function in ERO1-α, but their counterpart in the ERO1-β sequence may be missing (Appenzeller-Herzog et al., 2008; Baker et al., 2008). Thus, it is tempting to speculate that the dramatic physiological excursions of proinsulin translation may have favored the evolution of a specialized nonredundant system for disulfide bond formation in β cells.
Simple eukaryotes like yeast, worms, and flies have a single ERO1 gene that maintains the oxidizing environment in their ER by transferring electrons from reduced PDI-like molecules to terminal electron acceptors. The oxidized and reduced forms of the PDIs are in equilibrium with their reduced clients and with small molecule redox buffers like reduced glutathione (Cuozzo and Kaiser, 1999). Thus, in these simple eukaryotes, ERO1 activity maintains a global redox equilibrium in the ER lumen that favors native disulfide bond formation and yet is permissive for the reshuffling and reduction of nonnative bonds.
The aforementioned model of a global redox equilibrium maintained by ERO1 is consistent with the observations that in yeast and worms, partial loss of function of the essential ERO1 gene enhances the organism’s ability to cope with severe ER stress. Thus, ero1-1 mutant yeast are better able to cope with the overexpression of a disulfide-containing, misfolding-prone mutant lysosomal peptidase, CPY*, under conditions of compromised degradation of the misfolded proteins in the ER (Haynes et al., 2004). In worms, ero-1(RNAi) enhances lifespan of adults exposed to tunicamycin, an inhibitor of N-linked glycosylation which induces severe ER stress (Marciniak et al., 2004). These benefits to the mutant accrue in spite of the ER stress that partial loss of ERO1 function promotes and have been attributed to rectification of failures of homeostasis, whereby ongoing disulfide oxidation and the attendant production of H2O2 by ERO1 challenges the cellular reserves for coping with such stress in the wild type. Additionally, the physical properties of misfolded ER proteins may be influenced by their oxidative state, with disulfide bonds stabilizing the misfolded conformation (Marciniak et al., 2004).
Consistent with these ideas, we observed that compromise of ERO1-β in cultured Min6 cells enhanced their ability to cope with enforced expression of a mutant misfolded proinsulinAkita. However, surprisingly, this benefit was not realized in mutant mice: both mild compromise to ERO1-β by heterozygosity of the P077G11 insertion allele (Fig. 7, D and E) and severe compromise by homozygosity (not depicted) accentuated the diabetic phenotype of the Ins2Akita mutation.
What may account for these differences in the outcome of ERO1-β loss of function in the two systems? The higher oxygen concentration that cultured cells were exposed to, compared with tissues, may place a premium on lowered levels of ERO1-β in the former. Differences in activation of cell death pathways (by ER stress) in immortalized cultured cells and their tissue counterparts may contribute. Insulin signaling may have an important autocrine role (Leibiger et al., 1998; Leibiger et al., 2001) that would be realized in the tissue but not in cells (growing in serum). This would attach a cost to the defect in insulin biogenesis (which is triggered by ERO1-β deficiency) that is evident in tissues but not in cells. It is also possible that the constitutive lowering of ERO1-β activity by the mutant allele perturbs developmental programs, offsetting, in the live animal, the benefit of a less oxidizing ER that is realized in stressed cultured cells. Transient inhibition of ERO1 with chemicals may provide a way to test some of these ideas.
Materials and methods
Recombinant proteins and antisera
Antisera were purchased and used in according to the vendor’s specification: anti-PDI (Stressgen), anti-KDEL (Stressgen), anti–β-actin (Sigma-Aldrich), anti–bovine insulin (Millipore), and anti–mouse IgM (Rockland). Rabbit antiserum to Ribophorin I was a gift from G. Kreibich (New York University, New York, NY).
Antisera to mouse ERO1-α and -β were raised in rabbit by immunization with mouse ERO1-α (23–464) and ERO1-β (39–467) expressed as a GST-Smt3 fusion proteins in the Escherichia coli Rosetta (D3) strain, followed by glutathione affinity chromatography, cleavage of the tag with Ulp1, and gel filtration on a Superdex 200 (GE Healthcare). The soluble ERO1-α retained its enzymatic activity (Fig. 1 D), whereas ERO1-β rapidly lost enzymatic activity but remained useful as an immunogen.
Immunopurification of Flag–ERO1-α, –ERO1-β, and –ERO1-βC396A and enzyme assays
Expression plasmids encoding ER-localized, N-terminally Flag-tagged mouse ERO1-α (23–464) and ERO1-β and ERO1-βC396A (39–467) were constructed in the pFlag-CMV1 vector (Sigma-Aldrich) and transfected into 293T cells. Cell lysate was prepared in Triton buffer (0.3% Triton X-100, 150 mM NaCl, 20 mM Hepes, pH 7.4, 10 mM CaCl2, and protease inhibitors). The Flag-tagged proteins were immunopurified with Flag M1 (Sigma-Aldrich) affinity gel overnight, eluted in 10 mM EDTA, 20 mM Tris, pH 7.5, and 0.002% Tween 20, and used in the enzyme assays shown in Fig. 1 D and Fig. S2. In brief, the indicated concentrations of ERO1, reduced E. coli thioredoxin A (TrxA) or human reduced PDI expressed in and purified from E. coli transduced with expression plasmids (gifts from D. Fass [Weizmann Institute of Science, Rehovot, Israel] and C. Thorpe [University of Delaware, Newark, DE], respectively), were combined in a 20-µl reaction in a 384-well format. After the addition of 0.1 U/ml horseradish peroxidase (Worthington Biochemical Corp.) and 10 mM Amplex Ultra red (Invitrogen), the H2O2 produced was detected as the time-dependent fluorescent signal on a microplate reader (F500; Tecan; excitation wavelength, 535 ± 20 relative fluorescent units; and emission wavelength, 590 ± 20 relative fluorescent units).
Animal breeding and genotyping
All experiments performed on mice were approved by New York University’s Institutional Animal Care and Use Committee. The P077G11 mouse ES clone with a presumptive promoter trap retroviral insertion into intron 14 of the Ero1lb (ERO1-β) locus was purchased from the German Gene Trap Consortium. After blastocyst injection, the mutant allele was transmitted to 129svev mice and maintained in that inbred background. The presence of the mutant allele was detected by genomic PCR using primers that recognize the FlipROASCeoC-2 retroviral insert (hCD2.3S, 5′-GGAGACAAGAGCCCACAGAGTAGCTAC-3′; and Neo.10AS, 5′-AGTCCCTTCCCGCTTCAGTGACAACGT-3′), giving rise to a 301-bp product.
The additional homologous sequence telomeric of the ERO1-β locus presented a challenge to distinguishing heterozygous from homozygous mutant mice. Therefore, 129svev carriers of the insertion were crossed to wild-type C57BL/6 mice and the F1 carriers identified, as shown in Fig. 2 A. To genotype F3 progeny, we exploited a sequence polymorphism in intron 14: the presence of a PstI site in the 129 but not C57BL/6 genome on a PCR fragment amplified with the primers ERO1-β.21S (5′-TGGGTGTGTCCACCGAGGCAGTGGA-3′) and ERO1-β.18AS (5′-GATCTTCAAGGCAGTTCCTAAACCCTGAG-3′) that is tightly linked to the insertion. Thus, inheritance of two 129 copies of chromosome 12 in F2 hybrids is an indication of homozygosity for the mutation, whereas inheritance of two C57BL/6 versions of the chromosome marks the animal as wild type. Meiotic recombination between the insertion and marker was excluded by measuring ERO1-β protein in mouse tissue after analysis.
In brief, germ line transmission of a promoter trap insertion in ES clone XST171 (purchased from BayGenomics) was confirmed to have occurred in intron 6 and noted to eliminate >95% of protein expression in stressed cells. The mutation was maintained by backcrossing to C57BL/6. The mutation and the wild-type alleles were distinguished by genomic PCR using the primers mERO1-α.11S (5′-CTCAAAGGTGTACAGCACGGCCAACTCATATTTTC-3′), mERO1-α.10AS (5′-AGGGTTAAGGAGTAAGTCCACATACTCAGCATCG-3′), and AMP.4AS (5′-ACCAGCGTTTCTGGGTGAGCAAAAACAGGAAGG-3′). The wild-type allele gives a mERO1-α.11S versus mERO1-α.10AS product of 220 bp, whereas the mutant allele gives an AMP.4AS versus mERO1-α.10AS product of ∼800 bp.
Akita mice bearing the Ins2C96Y mutation were purchased from The Jackson Laboratory and genotyped as previously described (Wang et al., 1999). They were crossed to 129svev heterozygous ERO1-βi/+ mice, and otherwise-isogenic C57BL/6; 129svev F1 hybrid carriers of the Akita mutation, discordant for the ERO1-β insertion, were evaluated.
Purification of microsomes from mouse tissues
Tissues were mechanically disrupted by a Teflon homogenizer in 20 mM Hepes, pH 7.4, 5 mM MgCl2, 0.3 M sucrose, and protease inhibitors. The postmitochondrial supernatant was isolated by gentle clarification at 8,000 g for 5 min, and the microsomes were recovered by pelleting at 100,000 g for 30 min. The amount of ERO1-β in the microsomes was measured by immunoblot with an infrared imaging system (Odyssey; LI-COR).
Blood glucose and insulin measurement
Blood glucose levels were measured with a portable glucose-measuring device (OneTouch Ultra; LifeScan, Inc.). Insulin levels were measured by ELISA (EZRMI-13K; Millipore), using mouse insulin as standard. Intraperitoneal glucose tolerance tests were performed on animals that had been fasted for 16 h and were injected intraperitoneally with glucose, 2 mg/g body mass.
Pancreata were removed and immediately frozen in liquid nitrogen. Protein extracts were prepared using the acid/ethanol method (http://www.amdcc.org/shared/showFile.aspx?doctypeid=3&docid=73). After neutralization, insulin in the extracts was measured by ELISA. Protein in tissue extracts was determined by the Bradford method.
Immunostaining
Mice were killed by CO2 asphyxiation, and tissues were fixed by cardiac perfusion with 4% paraformaldehyde in PBS. After rinsing in PBS, tissues were incubated overnight in 30% sucrose and embedded in optimal cutting media. Insulin and glucagon immunoreactivity were detected by incubating 5-µm thick frozen sections of mouse pancreas with anti-insulin (Millipore) diluted 1:4,000, rabbit antiglucagon (Invitrogen) diluted 1:500, or rabbit anti–ERO1-β diluted 1:300, followed by FITC-conjugated anti–guinea pig IgG or Texas red–conjugated anti-rabbit IgG (Jackson ImmunoResearch Laboratories). Slides were mounted in CitiFluor. Fluorescent images were visualized at room temperature on a microscope (Axioplan-100; Carl Zeiss, Inc.) using a 40× NA 1.3 oil Plan-NeoFluar objective (Carl Zeiss, Inc.) and acquired on a monochrome charge-coupled device camera (SPOT RT-SE6 1.4 MP; Diagnostic Instruments, Inc.) using SPOT Basic software (Diagnostic Instruments, Inc.), and overlays were created in Photoshop (Adobe).
Transmission electron microscopy
Pancreatic tissue was fixed by immersion in 2.5% glutaraldehyde in 0.1 M Sorensen buffer, postfixed in 1% osmium tetroxide, and en bloc stained with 3% uranyl acetate. The tissue was dehydrated in ethanol embedded in epon. Ultrathin sections were poststained with uranyl acetate and lead citrate and examined using an electron microscope (CM100; Philips) at 60 kV. Images were recorded digitally using a camera system (1.6 Megaplus; Kodak) with software from Advanced Microscopy Techniques Corp.
Isolation, culture, and metabolic labeling of mouse pancreatic islets
Islets of Langerhans from wild-type and ERO1-β mutant mice were isolated using collagenase-P (Roche) and purified using three benchtop sedimentations and three selections by handpicking. Islets were cultured for 20 h in RPMI-1640 (Invitrogen) containing 10% FCS (Hyclone).
Islets were preincubated in methionine-free RPMI-1640 medium for 1 h and then labeled in the same medium containing 60 µCi/ml [35S]methionine/cysteine (PerkinElmer; specific activity, >1,000 Ci/mmol). Cold chase was performed in complete (methionine containing) medium. Islets were lysed on ice for 10 min in 500 µl lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 1% Na deoxycholate, and protease inhibitors). Islet lysates were precleared by centrifugation at 4°C for 10 min at 15,000 g, followed by immunoprecipitation of the supernatant (2 h at 4°C) using 5 µl anti-insulin (which is also reactive with proinsulin). Immunoprecipitated proteins were resolved on a 15% tricine-urea-acrylamide gel and revealed by autoradiography with a phosphoimager (Typhoon; GE Healthcare).
LPS blasts
Splenic cells were cultured in RPMI-1640 at a density of 106 cells/ml and exposed to 50 µg/ml LPS from E. coli (Sigma-Aldrich), to activate B cell differentiation. Where indicated, cells were exposed to a 30-min pulse of 10 mM DTT, washed free of DTT, and cultured further until lysis (in the presence of 10 mM N-ethyl maleimide), nonreducing SDS-PAGE, and anti–mouse IgM immunoblot.
Min6, knockdown of ERO1-β, and lentiviral transduction
Min6 cells were cultured in DME supplemented with 25 mM glucose, 10% FCS, and 55 µM β-mercaptoethanol. ERO1-β knockdown was achieved using Mission short hairpin RNA (shRNA)–encoding lentiviruses directed to mouse Ero1lb mRNA (Sigma-Aldrich; GenBank/EMBL/DDBJ accession no. NM_026184) according to the manufacturer’s instructions. Knockdown clone KD1 was targeted with shRNA TRCN0000100896, and KD2 was targeted with shRNA TRCN0000100899.
Blasticidin resistance–marked lentiviruses encoding GFP (pLenti 6.3V5-TOPO; Sigma-Aldrich) or GFP-tagged human proinsulinAkita (equivalent to C96Y in mouse INS2; Liu et al., 2007) were constructed from a cDNA gift of P. Arvan (University of Michigan, Ann Arbor, MI). After transduction and selection with blasticidin at 1 µg/ml for 7 d, the cells (in triplicate wells) were fixed and stained with crystal violet. Relative cell mass was quantified by solubilizing the dye in 0.2% Triton X-100 and measuring the absorbance at 590 nm.
Statistical analysis
All results are expressed as means ± SEM. Two-tailed t tests were performed to determine p-values for paired samples, and two-way analysis of variance (ANOVA) with repeat measurements was performed to analyze continuous trends.
Online supplemental material
Fig. S1 shows experiments on the reactivity of the ERO1-α and -β sera. Fig. S2 shows experiments the on specificity of the assay for measuring the in vitro disulfide oxidase activity of ERO1. Fig. S3 shows experiments on the insulin content in the pancreas of otherwise-isogenic wild-type and heterozygous mutant ERO1-β mice. Fig. S4 is a further characterization of the ERO1-β knockdown Min6 cells, and Fig. S5 is a characterization of insulin maturation in these cells. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200911086/DC1.
Acknowledgments
We thank Debbie Fass for her role in developing the ERO1 enzymatic assay and for the E. coli TrxA expression plasmid, Colin Thorpe for the human PDI expression plasmid, Peter Arvan for the GFP-proinsulinAkita plasmid, and Gert Kreibich for the anti–Ribophorin I sera.
This work was supported by European Molecular Biology Organization long-term fellowship ALTF649-2008 to E. Zito and by National Institutes of Health grants DK47119, DK075311, and ES08681 to D. Ron.
Footnotes
Abbreviations used in this paper:
- ANOVA
- analysis of variance
- ES
- embryonic stem
- LPS
- lipopolysaccharide
- PDI
- protein disulfide isomerase
- shRNA
- short hairpin RNA
- TrxA
- thioredoxin A
- UPR
- unfolded protein response
References
- Anfinsen C.B. 1973. Principles that govern the folding of protein chains. Science. 181:223–230 10.1126/science.181.4096.223 [DOI] [PubMed] [Google Scholar]
- Appenzeller-Herzog C., Riemer J., Christensen B., Sørensen E.S., Ellgaard L. 2008. A novel disulphide switch mechanism in Ero1alpha balances ER oxidation in human cells. EMBO J. 27:2977–2987 10.1038/emboj.2008.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker K.M., Chakravarthi S., Langton K.P., Sheppard A.M., Lu H., Bulleid N.J. 2008. Low reduction potential of Ero1alpha regulatory disulphides ensures tight control of substrate oxidation. EMBO J. 27:2988–2997 10.1038/emboj.2008.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabibbo A., Pagani M., Fabbri M., Rocchi M., Farmery M.R., Bulleid N.J., Sitia R. 2000. ERO1-L, a human protein that favors disulfide bond formation in the endoplasmic reticulum. J. Biol. Chem. 275:4827–4833 10.1074/jbc.275.7.4827 [DOI] [PubMed] [Google Scholar]
- Cuozzo J.W., Kaiser C.A. 1999. Competition between glutathione and protein thiols for disulphide-bond formation. Nat. Cell Biol. 1:130–135 10.1038/11047 [DOI] [PubMed] [Google Scholar]
- Curran S.P., Ruvkun G. 2007. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 3:e56 10.1371/journal.pgen.0030056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De-Zolt S., Schnütgen F., Seisenberger C., Hansen J., Hollatz M., Floss T., Ruiz P., Wurst W., von Melchner H. 2006. High-throughput trapping of secretory pathway genes in mouse embryonic stem cells. Nucleic Acids Res. 34:e25 10.1093/nar/gnj026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias-Gunasekara S., Gubbens J., van Lith M., Dunne C., Williams J.A., Kataky R., Scoones D., Lapthorn A., Bulleid N.J., Benham A.M. 2005. Tissue-specific expression and dimerization of the endoplasmic reticulum oxidoreductase Ero1beta. J. Biol. Chem. 280:33066–33075 10.1074/jbc.M505023200 [DOI] [PubMed] [Google Scholar]
- Eizirik D.L., Cardozo A.K., Cnop M. 2008. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr. Rev. 29:42–61 10.1210/er.2007-0015 [DOI] [PubMed] [Google Scholar]
- Frand A.R., Kaiser C.A. 1998. The ERO1 gene of yeast is required for oxidation of protein dithiols in the endoplasmic reticulum. Mol. Cell. 1:161–170 10.1016/S1097-2765(00)80017-9 [DOI] [PubMed] [Google Scholar]
- Gross E., Kastner D.B., Kaiser C.A., Fass D. 2004. Structure of Ero1p, source of disulfide bonds for oxidative protein folding in the cell. Cell. 117:601–610 10.1016/S0092-8674(04)00418-0 [DOI] [PubMed] [Google Scholar]
- Gross E., Sevier C.S., Heldman N., Vitu E., Bentzur M., Kaiser C.A., Thorpe C., Fass D. 2006. Generating disulfides enzymatically: reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc. Natl. Acad. Sci. USA. 103:299–304 10.1073/pnas.0506448103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding H.P., Zeng H., Zhang Y., Jungries R., Chung P., Plesken H., Sabatini D.D., Ron D. 2001. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol. Cell. 7:1153–1163 10.1016/S1097-2765(01)00264-7 [DOI] [PubMed] [Google Scholar]
- Haynes C.M., Titus E.A., Cooper A.A. 2004. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell. 15:767–776 10.1016/j.molcel.2004.08.025 [DOI] [PubMed] [Google Scholar]
- Itoh N., Okamoto H. 1980. Translational control of proinsulin synthesis by glucose. Nature. 283:100–102 10.1038/283100a0 [DOI] [PubMed] [Google Scholar]
- Leibiger B., Leibiger I.B., Moede T., Kemper S., Kulkarni R.N., Kahn C.R., de Vargas L.M., Berggren P.O. 2001. Selective insulin signaling through A and B insulin receptors regulates transcription of insulin and glucokinase genes in pancreatic beta cells. Mol. Cell. 7:559–570 10.1016/S1097-2765(01)00203-9 [DOI] [PubMed] [Google Scholar]
- Leibiger I.B., Leibiger B., Moede T., Berggren P.O. 1998. Exocytosis of insulin promotes insulin gene transcription via the insulin receptor/PI-3 kinase/p70 s6 kinase and CaM kinase pathways. Mol. Cell. 1:933–938 10.1016/S1097-2765(00)80093-3 [DOI] [PubMed] [Google Scholar]
- Li G., Mongillo M., Chin K.T., Harding H., Ron D., Marks A.R., Tabas I. 2009. Role of ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 186:783–792 10.1083/jcb.200904060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M., Hodish I., Rhodes C.J., Arvan P. 2007. Proinsulin maturation, misfolding, and proteotoxicity. Proc. Natl. Acad. Sci. USA. 104:15841–15846 10.1073/pnas.0702697104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak S.J., Yun C.Y., Oyadomari S., Novoa I., Zhang Y., Jungreis R., Nagata K., Harding H.P., Ron D. 2004. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18:3066–3077 10.1101/gad.1250704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May D., Itin A., Gal O., Kalinski H., Feinstein E., Keshet E. 2005. Ero1-L alpha plays a key role in a HIF-1-mediated pathway to improve disulfide bond formation and VEGF secretion under hypoxia: implication for cancer. Oncogene. 24:1011–1020 10.1038/sj.onc.1208325 [DOI] [PubMed] [Google Scholar]
- Mezghrani A., Fassio A., Benham A., Simmen T., Braakman I., Sitia R. 2001. Manipulation of oxidative protein folding and PDI redox state in mammalian cells. EMBO J. 20:6288–6296 10.1093/emboj/20.22.6288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari S., Koizumi A., Takeda K., Gotoh T., Akira S., Araki E., Mori M. 2002. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Invest. 109:525–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani M., Fabbri M., Benedetti C., Fassio A., Pilati S., Bulleid N.J., Cabibbo A., Sitia R. 2000. Endoplasmic reticulum oxidoreductin 1-lbeta (ERO1-Lbeta), a human gene induced in the course of the unfolded protein response. J. Biol. Chem. 275:23685–23692 10.1074/jbc.M003061200 [DOI] [PubMed] [Google Scholar]
- Pollard M.G., Travers K.J., Weissman J.S. 1998. Ero1p: a novel and ubiquitous protein with an essential role in oxidative protein folding in the endoplasmic reticulum. Mol. Cell. 1:171–182 10.1016/S1097-2765(00)80018-0 [DOI] [PubMed] [Google Scholar]
- Qiang L., Wang H., Farmer S.R. 2007. Adiponectin secretion is regulated by SIRT1 and the endoplasmic reticulum oxidoreductase Ero1-L alpha. Mol. Cell. Biol. 27:4698–4707 10.1128/MCB.02279-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevier C.S., Kaiser C.A. 2008. Ero1 and redox homeostasis in the endoplasmic reticulum. Biochim. Biophys. Acta. 1783:549–556 10.1016/j.bbamcr.2007.12.011 [DOI] [PubMed] [Google Scholar]
- Sevier C.S., Qu H., Heldman N., Gross E., Fass D., Kaiser C.A. 2007. Modulation of cellular disulfide-bond formation and the ER redox environment by feedback regulation of Ero1. Cell. 129:333–344 10.1016/j.cell.2007.02.039 [DOI] [PubMed] [Google Scholar]
- Tien A.C., Rajan A., Schulze K.L., Ryoo H.D., Acar M., Steller H., Bellen H.J. 2008. Ero1L, a thiol oxidase, is required for Notch signaling through cysteine bridge formation of the Lin12-Notch repeats in Drosophila melanogaster. J. Cell Biol. 182:1113–1125 10.1083/jcb.200805001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu B.P., Weissman J.S. 2004. Oxidative protein folding in eukaryotes: mechanisms and consequences. J. Cell Biol. 164:341–346 10.1083/jcb.200311055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Takeuchi T., Tanaka S., Kubo S.K., Kayo T., Lu D., Takata K., Koizumi A., Izumi T. 1999. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J. Clin. Invest. 103:27–37 10.1172/JCI4431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P., McGrath B., Li S., Frank A., Zambito F., Reinert J., Gannon M., Ma K., McNaughton K., Cavener D.R. 2002. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol. Cell. Biol. 22:3864–3874 10.1128/MCB.22.11.3864-3874.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]