

Bacterial peptidoglycan, the major constituent of the cell wall, is a polymer comprised of repeating units of N-acetylglucosamine (NAG) and N-acetylmuramic acid (NAM). An unusual peptide stem is appended to the NAM unit, which is the site of cross-linking among neighboring peptidoglycan strands in the mature cell wall.1 In the course of the normal bacterial growth, more than 50% of the parental peptidoglycan is recycled.2,3 The recycling is initiated by the action of lytic transglycosylases, which convert the NAM unit to the 1,6-anhydromuramyl moiety, a process that results in liberation of the novel disaccharide 1 from the polymeric cell wall (Figure 1).4 The peptide in the cell wall is processed by a number of enzymes, and it is typically comprised of three to five amino acids in the mature cell wall.5 On liberation of 1, it is internalized by the membrane protein AmpG. Once in the cytoplasm, compound 1 serves as the substrate for NagZ, which converts it to 2. Compound 2, in its various peptide forms, is believed to be an important player both in entry into the ensuing peptidoglycan recycling events and in an induction event that leads to the expression of β-lactamase, a key β-lactam antibiotic resistance enzyme.6,7
Figure 1.
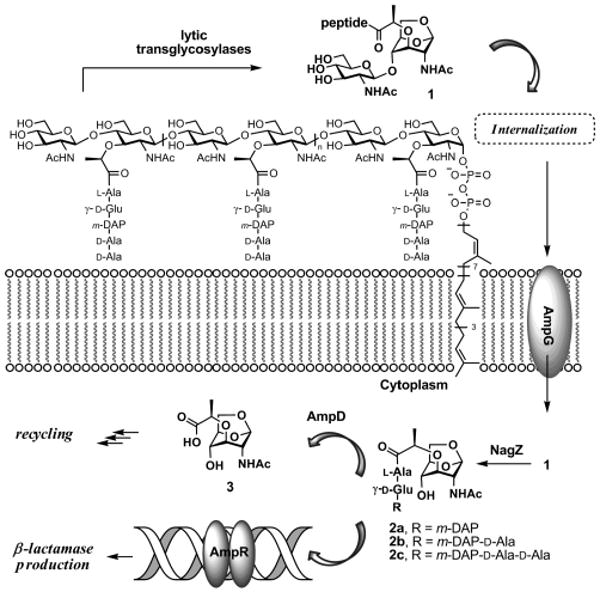
Peptidoglycan fragmentation leads to the formation of 1, which is transported across plasma membrane to initiate its recycling and the gene induction for production of β-lactamase.
The tripeptide and tetrapeptide forms of the peptide stem are prevalent in the mature peptidoglycan, because of the action of penicillin-binding proteins (PBPs) and certain bacterial cell wall endopeptidases. Hence, when 1 is internalized and processed by NagZ, the variants 2a and 2b predominate. On the other hand, when bacteria are exposed to β-lactam antibiotics, which inhibit PBPs, the result is the predominance of the form 2c.3,8 The enzyme AmpD would appear to be central to the fate of compounds 2. It has been proposed to be a peptidase that removes the peptide stem from the saccharide scaffolding of 2, initiating the recycling process.9 The enzyme is specifically implicated in processing of the tripeptide variant 2a, which makes it a player in the early stages of commitment to recycling.3,9 Binding of 2a to the dimer of the gene regulator AmpR is proposed to induce transcription of the gene for β-lactamase, unleashing resistance against β-lactam antibiotics. The notion is that exposure of the bacterium to the antibiotic would influence cell wall fragmentation and the recycling events, which down the line lead to induction of the resistance gene to counter the challenge by the antibiotic.3,10
The studies of these biochemical events have been held back by the lack of availability of authentic substrate(s) for AmpD. We describe herein the synthesis of a set of metabolites involved in cell wall recycling. Furthermore, we document that AmpD, as a gatekeeper for many of these biochemical events, is competent to turn over not merely the tripeptidyl analogue 2a but also the full-length peptide derivative 2c. Hence, it is also the key catalyst in the reversal of induction of resistance, once the challenge of the antibiotic is no longer present.
We cloned the ampD gene from Citrobacter freundii (ATCC6879) into the pET24a(+) vector under the T7 promoter by PCR amplification of the chromosomal gene. Escherichia coli BL21(DE3) cells were transformed with the vector for expression of the protein. The protein was purified to homogeneity in three chromatographic steps (see Supporting Information).
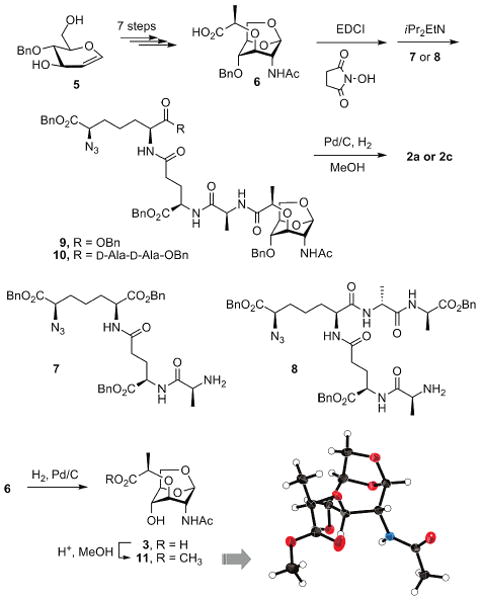
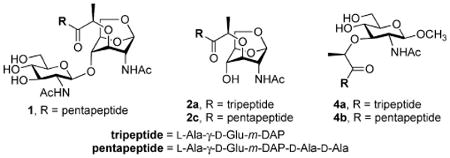
As indicated earlier, the major difficulty in studies of AmpD has been the lack of authentic substrate(s) for characterization of the enzyme.9,11 This has been an obstacle, since the putative substrates are inherently complex12,13 and they have not been available for characterization of the AmpD reaction. We have addressed this paucity of information in this study. We synthesized compounds 1 (pentapeptide), 2a, 2c, 4a, and 4b for characterization of the reaction of AmpD. We prepared 1, 2a, and 2c to explore the specificity of AmpD for its proposed 1,6-anhydromuramyl-containing substrate(s). Whereas the cytoplasmic AmpD would not come in contact with species 4a and 4b, we prepared these two compounds to explore the selectivity or specificity of AmpD for the intermediates containing the 1,6-anhy-dromuramyl moiety found in 1, 2a, and 2c.
The synthesis of compound 1 is reported elsewhere.14 Compound 6, an important intermediate in the syntheses of compounds 2a and 2c, was prepared from 4-benzyl d-glucal (5) (Scheme 1).14,15 Compound 6 was poised to receive the requisite protected peptide at its carboxylic acid, which then was put through deprotection to furnish the desired target compounds 2a and 2c. We also confirmed the structure of 1,6-anhydromuramic acid derivative by determination of the X-ray crystal structure of 11. The methyl ester of anhydromuramic acid 3 (compound 11) keeps a typical anhydropyranose structure, where all substituents are in axial positions. The details of synthetic steps leading to compounds 2a and 2c are given in the Supporting Information. The approach to synthesis of 4a and 4b, and related compounds, has been described earlier.16
Scheme 1.
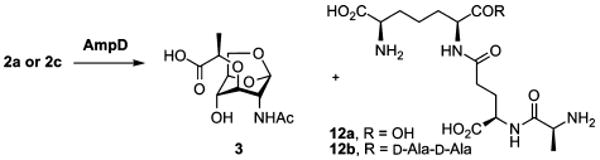
We subsequently investigated whether 2a and 2c would be processed by AmpD. The analysis by LC/MS revealed that AmpD hydrolyzed both compounds at the lactyl amide bond to generate a peptide 12a (or 12b) and the corresponding 1,6-anhydromuramyl moiety 3 (Scheme 2). Authentic synthetic samples of the peptides and of the 1,6-anhydromuramyl moiety 3 confirmed the structure assignments for the products of the AmpD reaction (see Supporting Information).
Scheme 2.
The quantitative analysis by nonlinear regression of the data for either the consumption of the substrate or the formation of the products reveals that compounds 2a and 2c were turned over by AmpD with kcat/Km values of (6.9 ± 0.4) × 104 and (2.2 ± 0.3) × 104 M−1 s−1, respectively (Table 1). In essence, the enzyme does not discriminate between the two substrates (a mere difference of 3-fold on kcat/Km). It is interesting to note that 1 (R = pentapeptide) is also turned over by AmpD, but in terms of kcat/Km, it is 300-fold worse as a substrate than 2a. But, more importantly since its Km is in the millimolar range, it is likely that AmpD would not experience saturation with this compound in vivo. This observation, compounded by the fact that the kcat value is also considerably attenuated for this substrate, indicates that it is likely that 1 is not turned over in vivo.
Table 1.
Kinetic Parameters for Turnover of Substrates by AmpD at pH 7.0
 | |||
|---|---|---|---|
| kcat (s−1) | Km (μM) | kcat/Km (M−1 s−1) | |
| 114 | 0.4 ± 0.1 | 1760 ± 210 | (2.3 ± 0.1) × 102 |
| 2a | 25 ± 2.0 | 360 ± 10 | (6.9 ± 0.4) × 104 |
| 2c | 11 ± 1 | 500 ± 60 | (2.2 ± 0.3) × 104 |
A more striking finding is that compounds 4a and 4b are not substrates for AmpD. Evolution of the AmpD function as a peptidase has clearly been driven by the atypical structure of the peptide, which includes features such as d-Ala, d-Glu, meso-diaminopimelate, and a peptide bond through the side chain of d-Glu. But it also has evolved to recognize the structurally distinct 1,6-anhydromuramyl moiety. This moiety is comprised of the sterically encumbered bicyclo system, with all its substituents in the axial positions, which is in sharp contrast to the muramyl ring found in the peptidoglycan (and in 4a and 4b) with its all-equatorial substituents.17
In summary, AmpD is capable of turning over 1,6-anhydromuramyl species 2a and 2c equally well. The importance of this finding is two-fold. AmpD is the gatekeeper for entry into the peptidoglycan recycling events by its turnover of 2a. Equally importantly, since it has the ability to turn over 2c—the resultant predominant species after exposure of bacteria to the β-lactam antibiotics involved in antibiotic resistance gene expression3,8—AmpD is the catalyst that reverses the recruitment of bacterial resources in induction of β-lactamase, the antibiotic resistance enzyme. It is likely that 1, the immediate product of fragmentation of peptidoglycan, does not experience effective turnover in vivo, if at all. Furthermore, AmpD has evolved to recognize both the atypical peptidoglycan peptide stem and the 1,6-anhydromuramyl moiety; hence it is a peptidase with a unique function in bacterial physiology.
Supplementary Material
Footnotes
Supporting Information Available: Experimental procedures of cloning, enzyme purification, kinetics, mass spectrometry, syntheses of the new compounds, and crystallographic information file (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Suvorov M, Fisher JF, Mobashery S. Bacterial Cell Wall: Morphology and Biochemistry. In: Goldman E, Green LH, editors. Practical Handbook of Microbiology. 2nd. CRC Press; 2008. pp. 153–183. [Google Scholar]
- 2.de Pedro MA, Donachie WD, Höltje JV, Schwarz H. J Bacteriol. 2001;183:4115–4126. doi: 10.1128/JB.183.14.4115-4126.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park JT, Uehara T. Microbiol Mol Biol Rev. 2008;72:211–227. doi: 10.1128/MMBR.00027-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suvorov M, Lee M, Hesek D, Boggess B, Mobashery S. J Am Chem Soc. 2008;130:11878–11879. doi: 10.1021/ja805482b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glauner B. Anal Biochem. 1988;172:451–464. doi: 10.1016/0003-2697(88)90468-x. [DOI] [PubMed] [Google Scholar]
- 6.Jaeger T, Mayer C. Cell Mol Life Sci. 2008;65:928–939. doi: 10.1007/s00018-007-7399-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiedemann B, Pfeifle D, Wiegand I, Janas E. Drug Resist Updat. 1998;1:223–226. doi: 10.1016/s1368-7646(98)80002-2. [DOI] [PubMed] [Google Scholar]
- 8.Uehara T, Park JT. J Bacteriol. 2008;190:3914–3922. doi: 10.1128/JB.00207-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobs C, Joris B, Jamin M, Klarsov K, Vanbeeumen J, Mengin-lecreulx D, Vanheijenoort J, Park JT, Normark S, Frère JM. Mol Microbiol. 1995;15:553–559. doi: 10.1111/j.1365-2958.1995.tb02268.x. [DOI] [PubMed] [Google Scholar]
- 10.Jacobs C, Frère JM, Normark S. Cell. 1997;88:823–832. doi: 10.1016/s0092-8674(00)81928-5. [DOI] [PubMed] [Google Scholar]
- 11.Généreux C, Dehareng D, Devreese B, Van Beeumen J, Frère JM, Joris B. Biochem J. 2004;377:111–120. doi: 10.1042/BJ20030862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawasaki A, Karasudani Y, Otsuka Y, Hasegawa M, Inohara N, Fujimoto Y, Fukase K. Chem—Eur J. 2008;14:10318–10330. doi: 10.1002/chem.200801121. [DOI] [PubMed] [Google Scholar]
- 13.Kubasch N, Schmidt RR. Eur J Org Chem. 2002:2710–2726. [Google Scholar]
- 14.Hesek D, Lee M, Zhang W, Noll BC, Mobashery S. J Am Chem Soc. 2009;131:5187–5193. doi: 10.1021/ja808498m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paulsen H, Himpkamp P, Peters T. Liebigs Ann Chem. 1986:664–674. [Google Scholar]
- 16.Hesek D, Suvorov M, Morio KI, Lee M, Brown S, Vakulenko SB, Mobashery S. J Org Chem. 2004;69:778–784. doi: 10.1021/jo035397e. [DOI] [PubMed] [Google Scholar]
- 17.Meroueh SO, Bencze KZ, Hesek D, Lee M, Fisher JF, Stemmler TL, Mobashery S. Proc Natl Acad Sci USA. 2006;103:4404–4409. doi: 10.1073/pnas.0510182103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.