

Abstract
Photosystem II is a reaction center protein complex located in photosynthetic membranes of plants, algae, and cyanobacteria. Using light energy, photosystem II catalyzes the oxidation of water and the reduction of plastoquinone, resulting in the release of molecular oxygen. A key component of photosystem II is cytochrome b559, a membrane-embedded heme protein with an unknown function. The cytochrome is unusual in that a heme links two separate polypeptide subunits, α and β, either as a heterodimer (αβ) or as two homodimers (α2 and β2). To determine the structural organization of cytochrome b559 in the membrane, we used site-directed mutagenesis to fuse the coding regions of the two respective genes in the cyanobacterium Synechocystis sp. PCC 6803. In this construction, the C terminus of the α subunit (9 kDa) is attached to the N terminus of the β subunit (5 kDa) to form a 14-kDa αβ fusion protein that is predicted to have two membrane-spanning α-helices with antiparallel orientations. Cells containing the αβ fusion protein grow photoautotrophically and assemble functional photosystem II complexes. Optical spectroscopy shows that the αβ fusion protein binds heme and is incorporated into photosystem II. These data support a structural model of cytochrome b559 in which one heme is coordinated to an α2 homodimer and a second heme is coordinated to a β2 homodimer. In this model, each photosystem II complex contains two cytochrome b559 hemes, with the α2 heme located near the stromal side of the membrane and the β2 heme located near the lumenal side.
Keywords: photosynthesis, photosystem II, heme protein, Synechocystis 6803, site-directed mutagenesis
In oxygenic photosynthetic organisms, the photosystem II reaction center complex uses light energy to transfer electrons from water to plastoquinone, resulting in the release of molecular oxygen into the atmosphere (1–3). The reaction center is composed of more than 25 polypeptides, but only 7 are known to be required for activity. Two of the essential polypeptides are the α and β subunits of cytochrome b559, a heme protein located near the core of the reaction center (for review, see refs. 4–6). Expression of both the α and β subunits of cytochrome b559 is required for the assembly of stable photosystem II complexes (6). Although cytochrome b559 can undergo light-induced oxidation-reduction reactions, it is not involved in the primary electron transfer reactions of photosystem II, leaving its role in the photosynthetic process uncertain. Recent proposals have focused on alternative electron transfer pathways that protect photosystem II against light-induced damage, either by wasting excess energy through an electron cycle or by deactivating potentially damaging redox states (for review, see ref. 6).
The α and β polypeptide subunits of cytochrome b559 are encoded by the psbE and psbF genes, respectively, which are cotranscribed as part of the psbEFLJ operon in the unicellular cyanobacterium Synechocystis sp. PCC 6803 (6, 7). Both polypeptides are predicted to contain one transmembrane hydrophobic domain, with a single His residue located close to the N-terminal end of each α-helix (8). Spectroscopic evidence reveals a bis-histidine ligation of the heme in the protein (9), indicating that the minimum structural unit for cytochrome b559 is a dimer of subunits cross-linked by a heme. There is controversy concerning the number of cytochrome b559 hemes in photosystem II. Some studies have measured one heme per reaction center, and others have detected two hemes (for review, see ref. 6). If there is one heme in each reaction center, then cytochrome b559 in photosystem II could only exist as an αβ heterodimer, but the presence of two hemes per reaction center offers two structural models, either a pair of αβ heterodimers or a pair of α2 and β2 homodimers.
Despite decades of research, little is known about the molecular structure of the photosystem II reaction center or of its individual subunits. Structural models of the reaction center core have been developed (10–12) based on homology with the reaction centers of the purple nonsulfur bacteria Rhodobacter sphaeroides and Rhodopseudomonas viridis, which have been resolved to the atomic level (13). However, these models are incomplete because the bacterial reaction center does not contain the manganese cluster involved in oxygen evolution, nor does it contain b-hemes analogous to cytochrome b559. To help understand the organization of photosystem II, we created a mutant in which the α and β subunits of cytochrome b559 are synthesized as a single polypeptide. This approach was used by Ma et al. (14) to determine structural features of integral membrane proteins in the bacterium Escherichia coli. Analysis of mutants of E. coli with genetic fusions between the genes encoding subunits I, II, and III, of the cytochrome bo complex established the orientation of these proteins in the membrane and revealed mutual interactions between the subunits. We used site-directed mutagenesis in the cyanobacterium Synechocystis 6803 to covalently link the α and β subunits, in a tail to head fashion, by fusing the psbE and psbF genes. This construct imposed an antiparallel orientation of the transmembrane domains of the α and β polypeptides. Mutant cells containing the fusion construct express that αβ fusion protein, assemble active photosystem II reaction centers, and grow photoautotrophically.
MATERIALS AND METHODS
Cell Growth and Membrane Isolation.
Synechocystis wild-type and T363 strains were grown in liquid BG11 medium (15) at 30°C under constant illumination. The T363 strain was grown in medium supplemented with 25 μg of spectinomycin per ml. For photoheterotrophic growth, 5 mM glucose was added to the BG11 medium. Cell density was determined by the optical density at 730 nm. The concentration of chlorophyll a was determined in 100% methanol, as described (16). Photosynthetic membranes were isolated according to Noren et al. (17), except that cell breakage was achieved with a bead beater using eight 30-s bursts separated by 3-min cooling periods.
Construction of the T363 Fusion Mutant.
Site-directed mutagenesis was performed according to ref. 18. As shown in Fig. 1, a 36-mer oligonucleotide primer was synthesized to span the region from the 3′ end of the psbE gene to the 5′ end of psbF gene (5′-ATTCAAGAGTTTAATCAAGCAACCCAAAATCCTAAT-3′). This primer was used to create a deletion of 42 nucleotides, which includes the stop codon of the psbE gene, a 36-nucleotide-long region between the two genes, and the start codon of the psbF gene, resulting in a translational fusion between the α and the β subunits of cytochrome b559. A spectinomycin-resistance cartridge was inserted into an NheI site, downstream of the psbEFLJ gene cluster as described (19). The resulting plasmid pSL363 was transformed into Synechocystis 6803 strain T1297, which lacks the entire psbEFLJ region (7). The resultant spectinomycin-resistance transformant was designated T363. The presence of the desired targeted deletion in these mutant cells was verified by PCR-mediated amplification.
Figure 1.
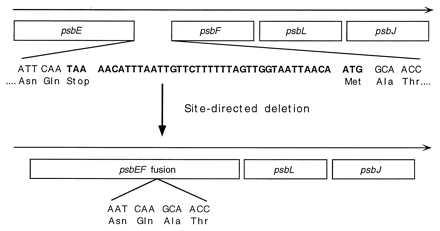
Schematic diagram illustrating how the α and β subunits of cytochrome b559 were fused by connecting the psbE (α subunit) and psbF (β subunit) genes in Synechocystis 6803. Site-directed deletion of the 42 nucleotides shown in boldface type, which includes the stop codon of the psbE and the start codon of the psbF genes, resulted in the T363 mutant strain in which the psbE and psbF genes are translationally fused together.
Production of Antibodies Against the α and β Subunits of Cytochrome b559.
The pET plasmid system (20) was used to overexpress the psbE and psbF genes of Synechocystis 6803 in E. coli cells. For this purpose, we used synthetic oligonucleotides to amplify the coding regions of each of these genes from the plasmid pKW1261 (containing the entire psbEFLJ region) by PCRs and cloned the corresponding PCR products in pET-3x plasmids as described (21). The overexpressed α and β subunits of cytochrome b559 were purified by preparative SDS/PAGE on 12% acrylamide gels and injected into rabbits. Antibodies against the α and β polypeptides were purified by using affinity chromatographic techniques as described (21).
Protein Electrophoresis and Immunodetection.
Photosynthetic membranes (loaded at 1.6 μg of chlorophyll per lane) were subjected to SDS/PAGE on 16–22% polyacrylamide gradient gels as described (22). Low molecular weight protein standards (Pharmacia) were electrophoresed on the same gel. Fractionated proteins were transferred to 0.2-μm (pore size) nitrocellulose filters (Schleicher & Schuell). Immunodetection was carried out as described (21). Alkaline phosphatase-conjugated goat anti-rabbit IgG served as secondary antibodies (Jackson Immunochemicals).
Oxygen Evolution.
The rate of oxygen evolution was measured at 30°C using a Clark-type oxygen electrode as described (23). Whole cells were suspended in BG11 medium that contained 10 mM bicarbonate. The oxygen evolution rates were light saturated and were corrected for respiration, if we assuming that the respiratory rate did not change in the light. The rate of dark respiration was typically 10–20% of the photosynthetic rate.
Photosystem II Content.
The relative amount of photosystem II was estimated by comparing the variable chlorophyll fluorescence yield of cells as described by Chu et al. (24). Chlorophyll fluorescence was measured with a Walz PAM 100 Fluorometer (Walz, Effeltrich, Germany). The variable fluorescence yield was determined for cells incubated in the presence of dichlorobenzoquinone, ferricyanide, hydroxylamine, and DCMU.
Cytochrome Photoreduction.
The photoreduction of cytochrome b559 by continuous light was measured as described by Poulson et al. (25). Isolated photosynthetic membranes were suspended in reaction medium (20 mM Mes, pH 6.0/20 mM CaCl2/20 mM MgCl2) that was made anaerobic by bubbling with argon for 5 min in a gas-tight cuvette prior to addition of the membranes. Further depletion of oxygen was achieved adding glucose oxidase (50 units/ml), glucose (2.5 mM), and catalase (1000 units/ml). Anaerobic conditions are required for the photoaccumulation of reduced cytochrome b559 because the heme is oxidized by oxygen. Photosynthetic membranes were added to a final chlorophyll concentration of 50 μM and gently stirred for a further 5 min. Spectra were recorded prior to illumination and after illumination with white light of 10,000 μmol of photons per m2 per s. The spectra were deramped and the contribution of cytochrome b559 and cytochrome b6 to the light-induced difference spectra was determined from the best fit of a linear combination of the purified spectra of the two cytochromes by using Microsoft excel. The concentration of cytochrome b559 was determined by using a reduced minus oxidized extinction coefficient of 17.5 mM−1 cm−1 for the wavelength pairs 560 and 570 nm (26).
RESULTS
In the mutant strain T363 of Synechocystis 6803, the genes for the α and β subunits of cytochrome b559 are linked into a single ORF. Expression of this fused gene was investigated by Western blot analysis using antibodies raised against the α and β subunits. Immunostaining of membrane proteins with antibodies raised against the α subunit reveals a single band in wild-type and T363 cells (Fig. 2 Upper). The apparent molecular mass of the α subunit band for wild-type cells is 9 kDa, whereas that of the corresponding band for T363 cells is 14 kDa. Antibodies raised against the β subunit also reveals a single band in wild-type and T363 strains (Fig. 2 Lower). The apparent molecular mass of the β subunit band for wild-type cells is 5 kDa, whereas that of the corresponding band for the T363 cells is 14 kDa. These results demonstrate that the psbE-F fusion gene construct in T363 cells is expressed as a single 14-kDa polypeptide. Moreover, this fusion polypeptide is inserted into the photosynthetic membrane. The absence of any lower molecular mass bands for the T363 cells shows that the fusion polypeptide containing the α and β subunits is not modified after translation to yield separate subunits.
Figure 2.

Immunodetection of the α and β subunits of cytochrome b559 in wild type and the T363 fusion mutant of Synechocystis 6803. Isolated membranes were fractionated on a 16–22% polyacrylamide gradient gel, electrophoretically transferred to nitrocellulose filters, and probed with monospecific antibodies raised against cytochrome b559 α subunit (Upper) and β subunit (Lower). Cells were grown in light without glucose.
Fig. 3A shows that the mutant strain T363 can grow photoautotrophically, although the rate of growth (doubling time 19 h) is about half that of wild-type cells (doubling time 8.4 h). For cells grown in the absence of glucose, we found that the photosystem II content of strain T363 was less than half that of wild-type cells and that the rate of oxygen evolution was significantly lower (data not shown). The lower photosystem II content in the mutant cells is likely due to damage to the reaction center by growth light, which has been observed for other site-directed photosystem II mutants of Synechocystis 6803 (e.g., refs. 24 and 27). However, if glucose is added to the growth medium, T363 cells grow at the same rate as wild-type cells (Fig. 3B and Table 1), and the rate of oxygen evolution measured in the presence of bicarbonate is similar in the two strains (Table 1). The rate of oxygen evolution when measured under limiting light shows that the relative quantum yield of oxygen evolution is the same for T363 and WT cells grown in the presence of glucose (data not shown). For cells grown in the presence of glucose, measurements of the variable fluorescence yield in the presence of DCMU and NH2OH indicate that the amount of photosystem II in T363 cells is about 80% of that of wild-type cells (Table 1).
Figure 3.
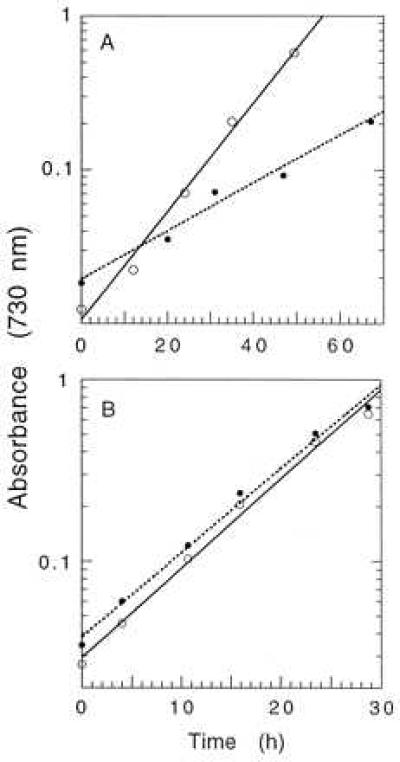
Growth curves of Synechocystis 6803 wild-type (○) and T363 (•) cells. (A) Cells grown in the absence of glucose in white light (100 μE per m2 per s). (B) Cells grown in the presence of glucose (5 mM) in white light (200 μE per m2 per s).
Table 1.
Comparison of growth rates, photosynthetic electron transport rates, and photosystem II content, of wild-type and T363 strains of Synechocystis 6803 grown in the presence of glucose
| Parameter | WT | T363 |
|---|---|---|
| Doubling times*, h | 6.2 | 6.5 |
| Electron transport rate† | 370 ± 30 | 350 ± 10 |
| Relative PSII content‡ | 1.00 | 0.79 ± 0.14 |
WT, wild type; PSII, photosystem II.
Data from Fig. 3.
Rate of oxygen evolution (μmol of O2 per mg of chlorophyll per h) measured in the presence of bicarbonate. Values are the average ± SD of three or more measurements. Cells were grown at a light intensity of 25 μE per m2 per s.
Relative photosystem II content was estimated by the ratio of the variable fluorescence divided by the maximum fluorescence [(Fmax − F0)/Fmax] (24). The average value of Fv/Fm was 0.44 for WT cells and 0.35 for cells of T363. Cells were grown at a light intensity of 25 μE per m2 per s.
The presence of photoactive cytochrome b559 in Synechocystis 6803 was investigated by optical spectroscopy. In an earlier study, using photosynthetic membranes isolated from chloroplasts, we showed that illumination by high-intensity light results in the reduction of cytochrome b559 by photosystem II (25). Fig. 4 shows that this technique is also effective in identifying heme containing cytochrome b559 in membranes isolated from Synechocystis 6803. For these experiments wild-type and mutant cells were grown in the presence of glucose to increase the concentration of photosystem II (as discussed above), thereby making it easier to detect cytochrome b559. Fig. 4A shows the light-induced spectrum of photosynthetic membranes isolated from wild-type cells and the reduced minus oxidized spectrum of purified cytochrome b559. The fact that the two spectra are virtually identical shows that the light-induced spectrum is due to the reduction of cytochrome b559. By using the Beer–Lambert law, we calculate that the amount of cytochrome b559 undergoing photoreduction in membranes isolated from wild-type cells is one heme per 420 chlorophyll molecules. It should be noted that the photoaccumulation assay, although highly reliable for identifying cytochrome b559, only detects the fraction of cytochrome b559 able to undergo photoreduction under anaerobic conditions. The actual concentration of cytochrome b559 in the membrane is higher. The photoaccumulation of reduced cytochrome b559 exhibited a half-time of about 5 s, similar to that observed for photosynthetic membranes isolated from spinach (25).
Figure 4.
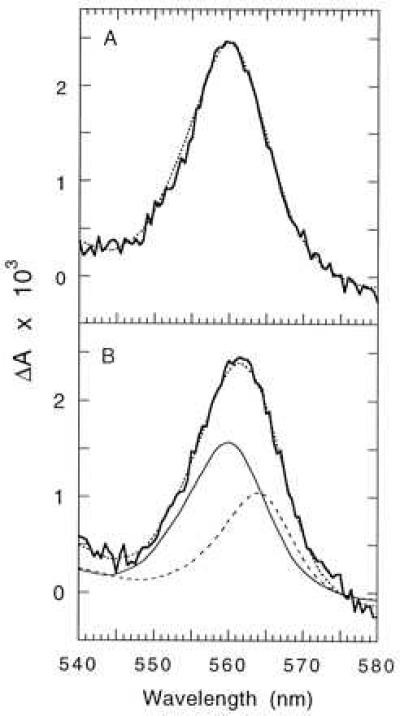
Photoaccumulation of reduced cytochrome b559 in thylakoid membranes isolated from wild type (A) and cytochrome b559 and cytochrome b6 from the T363 fusion mutant (B). Synechocystis 6803 cells were grown in the light in the presence of glucose. Thylakoid membranes were suspended under anaerobic conditions in reaction medium (20 mM Mes, pH 6.0/20 mM CaCl2/20 mM MgCl2) at a chlorophyll concentration of 50 μM and illuminated for 30 s with white light at an intensity of 10,000 μE per m2 per s. Spectra were recorded before and immediately after illumination. (A) The bold line shows the photoreduction of cytochrome b559 in wild-type cells. The dotted line shows the spectrum of purified cytochrome b559. (B) The bold line shows the photoreduction of cytochrome b559 and cytochrome b6 in cells of strain T363. The dotted line shows the best fit of the sum of purified cytochrome b559 and cytochrome b6 (fitted by using Microsoft excel). The solid line shows the contribution of cytochrome b559 (60%) and the dashed line shows the contribution of cytochrome b6 (40%) giving the best fit.
Fig. 4B shows the light-induced spectrum using membranes from strain T363 grown in the presence of glucose. Compared with wild-type cells, the absorption peak is shifted slightly to a longer wavelength because of the contribution of cytochrome b6. Deconvolution of the spectrum into contributions from cytochrome b559 and cytochrome b6 is shown in Fig. 4B. The amount of cytochrome b559 undergoing photoreduction in membranes from T363 cells is one heme per 650 chlorophyll molecules. The relative amount of cytochrome b559 in T363 cells revealed by the photoaccumulation assay is 65% of wild-type cells. Spectroscopic measurements using T363 cells grown in the absence of glucose were done to verify the presence of heme containing cytochrome b559. The experiments revealed the photoreduction of cytochrome b559, but the concentration of the cytochrome was much lower than in the photoheterotrophically grown cells (data not shown). The relative contribution of cytochrome b6 to the light-induced spectrum was much larger, creating considerable uncertainty in the deconvoluted spectra. These observations demonstrate that the cytochrome b559 fusion polypeptide in strain T363 binds heme and exhibits an absorbance peak in the reduced form at 560 nm, the same as the wild-type protein. Furthermore, the light-induced reduction of cytochrome b559 by photosystem II indicates that the fusion protein is integrated into photosystem II.
DISCUSSION
At the core of the photosystem II reaction center are two polypeptides, D1 and D2, that form a heterodimeric transmembrane complex that binds most, if not all, the cofactors required for electron transfer from water to plastoquinone. Located close to the D1/D2 reaction center core is cytochrome b599, a heme protein composed of two subunits, α and β. Beyond the fact that cytochrome b559 is needed for the stable assembly of photosystem II, little is known about its structure or functional role within the reaction center complex. It is known that each subunit of cytochrome b559 contains a single α-helix (8) and that each helix contains a single histidine residue that serves as an axial ligand to the heme (9). However, the key question of whether there is one or two hemes in each reaction center is controversial. If there is one heme, the cytochrome most likely exists as an αβ heterodimer, whereas, if there are two hemes per reaction center, the possible combinations include two αβ heterodimers or two homodimers, one α2 and one β2. Determining the number of cytochrome b559 hemes in photosystem II has proven to be a surprisingly elusive task. Reports of two hemes per reaction center are plentiful, as are reports of one heme per reaction center (for review, see ref. 6). The problem is due in part to the use of detergent in isolating photosystem II complexes, which may partially extract hemes from the reaction center (28), and in part to the uncertainty in techniques to quantify the concentration of cytochrome b559 heme and photosystem II reaction centers (6).
One approach to reveal the structure of cytochrome b559 in photosystem II is to determine the orientation of the α and β subunits in the membrane. There is convincing evidence based on proteolysis of exposed hydrophilic sites (29) and immunogold labeling of inside-out and right-side out vesicles (30) that the α subunit is oriented with its C terminus extending into the inner aqueous space (lumen) of the photosynthetic vesicle. This orientation would place the α subunit histidine close to the stromal surface of the photosynthetic membrane, indicating that at least one heme is located in the hydrophobic core of the membrane near the stromal side. However, the orientation of the β subunit must be known to predict the basic structure of the cytochrome. Tae and Cramer (31) investigated the accessibility of the N-terminal end of the β subunit to proteolysis by trypsin in membranes isolated from spinach and Synechocystis 6803. On the basis of these experiments, they argued that the β subunit of cytochrome b559 is likely to be oriented with its N-terminal end in the stromal phase, which would place the heme-ligand histidine near the stromal side of the membrane. This work led them to favor an αβ heterodimeric model of cytochrome b559, which could be present as one or two hemes per reaction center. It is important to note that the interpretation of the proteolysis data depends on the assumption that the isolated membranes are vesicular, right-side out, and that trypsin did not penetrate to the inner aqueous phase. However, the data show that there is some trypsin digestion of the lumen-localized manganese-stabilizing protein, albeit at a slower rate than of cytochrome b559, indicating that trypsin can infiltrate the lumen. In our view, the proteolysis data, although consistent with the heterodimeric model, leave open the possibility that the β subunit could be oriented in the opposite direction, placing the heme-ligand histidine near the lumenal phase.
To establish the orientation of the α and β subunits in the photosynthetic membrane, we created a mutant cytochrome b559 polypeptide that contains both subunits. The resulting mutant strain expressed a single 14-kDa αβ fusion protein, but neither the 9-kDa α subunit nor the 5-kDa β subunit was present (Fig. 2). The T363 mutant strain grows photoautotrophically, with a doubling time approximately half that of wild type (Fig. 3). In the presence of glucose, the growth rate of the mutant is similar to that of wild-type cells, as is the rate of oxygen evolution (Table 1). Furthermore, the amount of photosystem II reaction centers in the photosynthetic membrane was 80% of wild-type cells. These results imply that the fusion of the two cytochrome b559 subunits did not significantly affect the structural and functional integrity of photosystem II complexes. Optical spectroscopy shows that the fused cytochrome b559 polypeptide was capable of ligating heme in functional photosystem II centers. These findings and the fact that the construction of the mutant forces the transmembrane helices of the α and β subunits into an antiparallel orientation in the membrane dictate a structural model of cytochrome b559 that consists of two homodimers, α2 and β2, as shown in Fig. 5. The model is based on the demonstration that the cytochrome b559 fusion polypeptide binds heme and is present in a photosystem II reaction center that is viable and supports photoautotrophic growth. We exclude a heterodimeric model of cytochrome b559 in the mutant cells, because its assembly would require the αβ fusion subunit to insert in the membrane in two opposite orientations, one with the C-terminal end in the stromal phase and the other with its C-terminal end in the lumenal phase. This is unlikely based on our current understanding of protein insertion into membranes. The possibility that the αβ fusion protein could cross the membrane three times, which would allow for a heterodimeric model, is excluded based on hydropathy analysis, which shows that the stretch of amino acids linking the two histidine-containing α-helices is hydrophilic.
Figure 5.
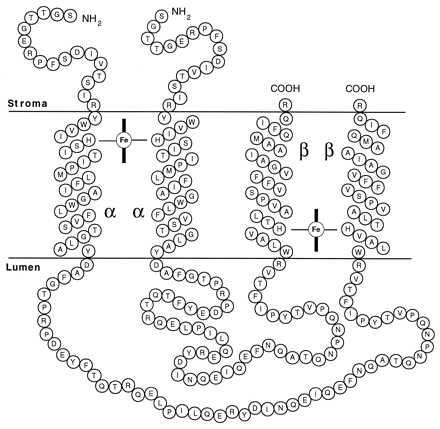
Model of two cytochrome b559 hemes in the photosynthetic membrane of strain T363 of Synechocystis 6803. See text for rationale.
The model shows that the reaction center of photosystem II contains two distinct cytochrome b559 hemes positioned on opposite sides of the membrane. As mentioned above, optical spectroscopy has failed to unequivocally determine the number of hemes per reaction center (6). However, recent results based on Mössbauer spectroscopy to detect heme iron in photosystem II membrane fragments indicate two b559 hemes per reaction center (32). In addition, there are several experimental observations that reveal heterogeneity in the cytochrome b559, adding further support for the presence of two distinct hemes in photosystem II. For example, Shuvalov and coworkers have shown that there are two cytochrome b559 hemes per reaction center in isolated D1/D2/cytochrome b559 photosystem II reaction centers. The cytochromes have different midpoint potentials, indicating that there are two different environments for the heme (for review, see ref. 5). It is noteworthy that cytochrome b559 exists in photosynthetic membranes with two different midpoint potentials, a high-potential form with a midpoint potential near 380 mV and a low-potential form with a midpoint potential near 60 mV (e.g., ref. 25). However, the two forms are not usually found at equal concentrations in the membrane. The accessibility of cytochrome b559 to chemical titrants also reveals two separate populations. Selak et al. (33) found that the chemical oxidation of the cytochrome by ferricyanide is strongly biphasic and that each phase exhibits the same amplitude, supporting the existence of cytochrome b559 hemes in two different environments. Biochemical support for an α2 and β2 model is provided by Moskalenko et al. (34), who examined the polypeptide organization of photosystem II reaction center preparations by chemical cross-linking, followed by gel electrophoretic analysis. Antibodies to the α subunit recognized a band corresponding to an α2 homodimer, whereas the band corresponding to the αβ heterodimer was absent or extremely weak. The amino acid composition of the β subunit makes it less likely to cross-link. The cross-linking of two α subunits shows they are close neighbors, as predicted by the homodimeric model shown in Fig. 5.
Acknowledgments
We thank Ladislav Nedbal for providing the program to deconvolute the cytochrome spectra. This work was supported by grants from the National Institutes of Health (GM45797) and the International Human Frontier Science Program to H.B.P. and by a grant from the U.S. Department of Agriculture National Research Initiative Competitive Grants Program (94-37306-0412) to J.W.
References
- 1.Pakrasi H B. Annu Rev Genet. 1995;29:755–776. doi: 10.1146/annurev.ge.29.120195.003543. [DOI] [PubMed] [Google Scholar]
- 2.Yachandra V K, Sauer K, Klein M P. Chem Rev. 1996;96:2927–2950. doi: 10.1021/cr950052k. [DOI] [PubMed] [Google Scholar]
- 3.Debus R J. Biochim Biophys Acta. 1992;1102:269–352. doi: 10.1016/0005-2728(92)90133-m. [DOI] [PubMed] [Google Scholar]
- 4.Cramer W A, Tae G-S, Furbacher P N, Böttger M. Physiologia Plantarum. 1993;88:705–711. doi: 10.1111/j.1399-3054.1993.tb01392.x. [DOI] [PubMed] [Google Scholar]
- 5.Shuvalov V A. J Bioenerg Biomembr. 1994;26:619–626. doi: 10.1007/BF00831536. [DOI] [PubMed] [Google Scholar]
- 6.Whitmarsh J, Pakrasi H B. In: Oxygenic Photosynthesis: The Light Reactions. Ort D R, Yocum C F, editors. Dordrecht, the Netherlands: Kluwer; 1996. pp. 249–264. [Google Scholar]
- 7.Pakrasi H B, Williams J G K, Arntzen C J. EMBO J. 1988;7:325–332. doi: 10.1002/j.1460-2075.1988.tb02816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Widger W R, Cramer W A, Hermodson M, Herrmann R G. FEBS Lett. 1985;191:186–190. [Google Scholar]
- 9.Babcock G T, Widger W R, Cramer W A, Oertling W A, Metz J G. Biochemistry. 1985;24:3638–3645. doi: 10.1021/bi00335a036. [DOI] [PubMed] [Google Scholar]
- 10.Svensson B, Etchebest C, Tuffery P, Kan P v, Smith J, Styring S. Biochemistry. 1996;35:14486–14502. doi: 10.1021/bi960764k. [DOI] [PubMed] [Google Scholar]
- 11.Ruffle S V, Donnelly D, Bundell T L, Nugent J H A. Photosynth Res. 1992;34:287–300. doi: 10.1007/BF00033446. [DOI] [PubMed] [Google Scholar]
- 12.Xiong J, Subramaniam S, Govindjee Protein Sci. 1996;5:2054–2073. doi: 10.1002/pro.5560051012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deisenhofer J, Michel H. Annu Rev Cell Biol. 1991;7:1–23. doi: 10.1146/annurev.cb.07.110191.000245. [DOI] [PubMed] [Google Scholar]
- 14.Ma J, Lemieux L, Gennis R B. Biochemistry. 1993;32:7692–7697. doi: 10.1021/bi00081a013. [DOI] [PubMed] [Google Scholar]
- 15.Allen M M. J Phycol. 1968;4:1–4. doi: 10.1111/j.1529-8817.1968.tb04667.x. [DOI] [PubMed] [Google Scholar]
- 16.Lichtenthaler H K. Methods Enzymol. 1987;148:350–382. [Google Scholar]
- 17.Noren G H, Boerner R J, Barry B A. Biochemistry. 1991;30:3943–3950. doi: 10.1021/bi00230a020. [DOI] [PubMed] [Google Scholar]
- 18.Anbudurai P R, Pakrasi H B. Z Naturforsch. 1993;48c:267–274. doi: 10.1515/znc-1993-3-424. [DOI] [PubMed] [Google Scholar]
- 19.Pakrasi H B, Ciechi P D, Whitmarsh J. EMBO J. 1991;10:1619–1627. doi: 10.1002/j.1460-2075.1991.tb07684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Studier F W, Rosenberg A H, Dunn J H, Dubendorff J W. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 21.Lind L K, Shukla V K, Nyhus K J, Pakrasi H B. J Biol Chem. 1993;268:1575–1579. [PubMed] [Google Scholar]
- 22.Nyhus K J, Ikeuchi M, Inoue Y, Whitmarsh J, Pakrasi H B. J Biol Chem. 1992;267:12489–12495. [PubMed] [Google Scholar]
- 23.Nedbal L, Gibas C, Whitmarsh J. Photosynth Res. 1991;30:85–94. doi: 10.1007/BF00042006. [DOI] [PubMed] [Google Scholar]
- 24.Chu H-A, Nguyen A P, Debus R. Biochemistry. 1994;33:6137–6149. doi: 10.1021/bi00186a013. [DOI] [PubMed] [Google Scholar]
- 25.Poulson M, Samson G, Whitmarsh J. Biochemistry. 1995;34:10932–10938. doi: 10.1021/bi00034a027. [DOI] [PubMed] [Google Scholar]
- 26.Cramer W A, Theg S M, Widger W R. Photosynth Res. 1986;10:393–403. doi: 10.1007/BF00118305. [DOI] [PubMed] [Google Scholar]
- 27.Chu H-A, Nguyen A P, Debus R J. Biochemistry. 1995;34:6150–6157. doi: 10.1021/bi00186a014. [DOI] [PubMed] [Google Scholar]
- 28.van Leeuwen P J, Nieveen M C, van de Meent E J, Dekker J P, van Gorkom J. Photosynth Res. 1991;28:149–153. doi: 10.1007/BF00054128. [DOI] [PubMed] [Google Scholar]
- 29.Tae G S, Cramer W A. FEBS Lett. 1989;259:161–164. doi: 10.1016/0014-5793(89)81518-2. [DOI] [PubMed] [Google Scholar]
- 30.Vallon O, Tae G-S, Cramer W A, Simpson D, Hoyer-Hanson G, Bogorad L. Biochim Biophys Acta. 1989;975:132–141. doi: 10.1016/s0005-2728(89)80211-7. [DOI] [PubMed] [Google Scholar]
- 31.Tae G S, Cramer W A. Biochemistry. 1994;33:10060–10068. doi: 10.1021/bi00199a033. [DOI] [PubMed] [Google Scholar]
- 32.Kurreck J, Garbers A, Reifarth F, Andréasson L-E, Parak F, Renger G. FEBS Lett. 1996;381:53–57. doi: 10.1016/0014-5793(96)00061-0. [DOI] [PubMed] [Google Scholar]
- 33.Selak M E, Koch-Whitmarsh B E, Whitmarsh J. In: Advances in Photosynthesis Research. Sybesma C, editor. Vol. 1. The Hague, The Netherlands: Martinus Nijhoff/Dr. Junk; 1984. pp. 493–496. [Google Scholar]
- 34.Moskalenko A A, Barbato R, Giacometti G M. FEBS Lett. 1992;314:271–274. doi: 10.1016/0014-5793(92)81487-7. [DOI] [PubMed] [Google Scholar]