

Abstract
The T-cell receptor (TCR)-CD3 complex serves as a central paradigm for general principles of receptor assembly, ligand recognition, and signaling in the immune system. There is no other receptor system that matches the diversity of both receptor and ligand components. The recent expansion of the immunological structural database is beginning to identify key principles of MHC and peptide recognition. The multicomponent assembly of the TCR complex illustrates general principles used by many receptors in the immune system, which rely on basic and acidic transmembrane residues to guide assembly. The intrinsic binding of the cytoplasmic domains of the CD3ε and ζ chains to the inner leaflet of the plasma membrane represents a novel mechanism for control of receptor activation: Insertion of critical CD3ε tyrosines into the hydrophobic membrane core prevents their phosphorylation before receptor engagement.
T-cell receptors are uniquely complex and display several novel characteristics, including direct binding of their two cytoplasmic domains to lipids in the inner leaflet of the membrane.
LIGAND BINDING BY THE EXTRACELLULAR DOMAINS OF T-CELL RECEPTORS: STRUCTURAL BASIS OF αβ AND γδ TCR BINDING TO A DIVERSE GROUP OF LIGANDS
The structures of many T-cell receptors (TCRs) in their ligand-bound state have now been determined, allowing some of the general rules of antigen recognition to be distilled. Though the best studied interaction is the binding of αβTCR with peptide-MHC complexes, it is highly instructive to compare the proto-typical binding to peptide-MHC with the recognition of lipid antigen-CD1 complexes and engagement of nonclassical MHC class I molecules by γδTCRs. These comparisons show that the recognition strategies for these three classes of TCR ligands are strikingly different (Fig. 1).
Figure 1.

TCR structures with different classes of ligands show distinct binding solutions. (A–C). The docking topologies are compared for the αβTCR A6 bound to a peptide-MHC class I complex (HLA-A2 with Tax P6A peptide; PDB entry 1QRN) (A), the αβTCR NKT15 bound to the complex of CD1d and the glycolipid α-galactosylceramide (PDB entry 2PO6) (B), and the γδTCR G8 bound to the nonclassical MHC class Ib molecule T22 (PDB entry 1YPZ) (C). (D–F) The placement of the TCR loops is compared for the same complexes. (D) The A6 TCR uses all six loops to contact the MHC molecule and the bound peptide. The four germline-encoded loops (α1, α2, β1, and β2) contribute to MHC binding, while the two CDR3 loops are positioned over the peptide (α3 and β3). (E) The NKT15 TCR contacts the CD1d-bound glycolipid only through germline elements encoded in the invariant TCRα chain (α1 and α3 loops). (F) The γδ TCR G8 inserts the CDR3δ chain loop into the hydrophobic groove of T22; other TCR loops do not appear to be essential for T22 binding.
Many structures of human and mouse complexes have shown a typical binding mode, which maximizes αβTCR interaction with the MHC-bound peptide (Garboczi et al. 1996; Garcia et al. 1996; Rudolph et al. 2006). This binding mode places the hypervariable loops of the TCR, the CDR3α and CDR3β loops, over the center of the bound peptide (Fig. 1A,D). Two other TCR loops, CDR1α and CDR1β, frequently also contribute to peptide recognition by binding over amino-terminal and carboxy-terminal peptide segments, respectively. Most contacts to the MHC molecule are typically made by the germline-encoded CDR1 and CDR2 loops of both TCRα and β, but the CDR3 loops can also contribute to MHC binding. Thus, six TCR loops can be involved in peptide and MHC recognition and usually provide substantial specificity for both components (Fig. 1A,D).
αβTCRs not only recognize MHC-bound peptides, but also CD1-bound lipid antigens. CD1 molecules have a similar overall fold as MHC class I molecules, but their groove is substantially wider and more hydrophobic to accommodate a diverse group of lipids, glycolipids, and lipopeptides (Barral and Brenner 2007). NK T cells recognize lipid antigens bound to CD1d, with α-galactosylceramide being the best characterized lipid antigen (Bendelac et al. 2007). In the structure of a human NK TCR, the TCR binds over the extreme end of the CD1d binding cleft (Borg et al. 2007). Furthermore, the TCR is positioned approximately parallel to the long axis of the CD1d binding groove, in contrast to the typical diagonal footprint for most αβTCRs on peptide-MHC (Fig. 1 B,E). NK T cells use a limited TCR repertoire for CD1d-lipid binding, in particular an invariant TCRα chain and a specific Jα segment (Vα24-Jα18) (Bendelac et al. 2007). In the structure only the glycosyl head group of the lipid antigen is exposed and it is contacted solely by germline-encoded loops, CDR1α (encoded by Vα24) and CDR3α (encoded by Jα18) (Fig. 1E). Only germline-encoded segments thus confer specificity for the glycolipid antigen by this NK TCR (Borg et al. 2007). This is in striking contrast to the central role of the hypervariable CDR3α and β loops in discriminating between MHC-bound peptides.
The ligands for most γδ T cells remain unknown, but in mice the closely related nonclassical MHC class Ib molecules T10 and T22 serve as ligands for a sizable population of γδ T cells in nonimmunized mice (Shin et al. 2005). The structure of the G8 γδTCR bound to T22 shows a binding mode that is entirely different from αβTCR engagement of peptide-MHC (Adams et al. 2005). The G8 TCR binds T22 sideways at a highly tilted angle (Fig. 1C), contrasting with the essentially parallel alignment of the long axes of the αβTCR and peptide-MHC. T22 does not have a bound peptide and the segment that corresponds to the amino-terminal part of the α2 helix of classical MHC class I molecules is actually unwound, exposing the hydrophobic groove. The CDR3 loop of TCRδ inserts into this hydrophobic groove and contributes a substantial fraction of the buried surface (Fig. 1F). Interestingly, transfer of this CDR3δ loop into an αβ TCR resulted in T22 binding (Adams et al. 2008). Thus, a single loop of this γδTCR appears to be sufficient for T22 binding.
The overall binding mode thus differs greatly for all three classes of ligands: αβTCRs typically bind in a diagonal orientation over most of the MHC-bound peptide, whereas the NK TCR sits over only part of the CD1d binding groove parallel to the long axis of the groove. The most extreme case is the γδTCR that binds sideways to T22 and inserts one loop into the exposed T22 groove.
Structural Insights Into the Mechanism of MHC Restriction
There has been much debate on the fascinating problem of why αβTCRs are “MHC restricted,” and the structural database is now large enough to extract key principles. The overall binding mode of most crystallized αβTCRs with peptide-MHC is similar, even though there is substantial variation in the binding angle relative to the long axis of the MHC molecule (Rudolph et al. 2006; Garcia et al. 2009). Importantly, the general location of the TCR chains on the peptide-MHC ligand is similar in all studied structures: The TCRβ chain is positioned over the α1 helix of the MHC molecule and the TCRα chain over the other MHC helix (α2 in MHC class I, which corresponds to β1 in MHC class II). TCRs have a similar overall fold to antibody Fab segments, but if there were no inherent rules to TCR binding to peptide-MHC, the opposite placement (TCRα rather than TCRβ over the MHC α1 helix) should have been seen by now.
Genes of the MHC are extremely polymorphic. In man, there are >800 MHC class I alleles and >600 alleles of MHC class II (Robinson et al. 2009). If TCRs are prebiased to interact with MHC proteins, essential genetic elements have to be shared between all MHC proteins. It has been appreciated for some time that the overall structure of MHC proteins and their bound peptide is quite similar (Stern and Wiley 1994). Likely because of selective pressure by pathogens, the vast majority of allelic variation between MHC proteins is located within the peptide binding groove (Bjorkman et al. 1987). Conventional alignment of the α1 domain of MHC class I and II molecules failed to show substantial homology in solvent-exposed residues that could be contacted by the TCR. However, if the alignment is shifted by a single turn of an α-helix substantial homology is evident among solvent exposed residues available for interaction with TCRs (Huseby et al. 2005; Huseby et al. 2008). For example, when the surface exposed side chains of H2-Kb and I-Ab are compared, there is substantial conservation between these MHC class I and class II proteins (Table 1). Interestingly, these conserved amino acids are located within TCR binding footprints (Fig. 2). Thus, conserved/chemically similar amino acids on the helices of all MHC proteins may be the elements that the TCR V-domains are genetically encoded to bind.
Table 1.
Alignment of solvent accessible murine MHC class I and class II residues
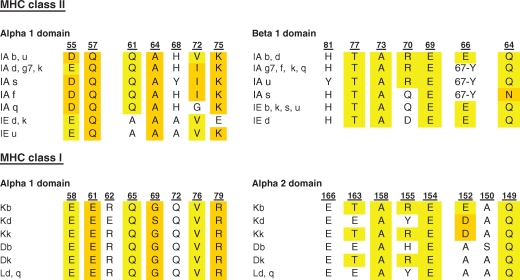 |
Helical residues of murine MHC class I and class II proteins were aligned based on a previously reported approach that takes a shift in the MHC class I and class II α1 helices into account (Huseby et al. 2005). Identical residues are highlighted in yellow, structurally similar residues in orange. In some MHC class II molecules, there is a deletion corresponding to one amino acid and residues 66/67 located in the same position of the β1 domain are indicated.
Figure 2.

Conserved helical residues of MHC class I and class II proteins are often contacted by CDR1 and CDR2 residues of TCRs. Conserved MHC helical residues are highlighted in green for structures of H-2Kb (A) and I-Ab (B). The BM3.3 TCR (H-2Kb; PDB entry 1NAM) and the B3K506 TCR (I-Ab; PDB entry 3C5Z) contact these conserved helical MHC residues. TCR CDR1β and CDR2β are colored red, CDR3α and CDR3β orange, and CDR1α and CDR2α blue. Peptide residues are colored yellow. Colored cyan are the H2-Kb α1 and IAb α1 domains, and magenta H2-Kb α2 and IAb β1 domains.
The diagonal binding orientation on the peptide-MHC surface is similar among most crystallized αβ TCRs, but because of variation in this binding angle, it has not been possible to identify MHC residues contacted by all TCRs (Baker et al. 2001; Rudolph et al. 2006). Rather, the growing structural database suggests an alternative hypothesis: Individual V-genes or groups of related V-genes have coevolved with MHC, which has resulted in preferred contacts by these V-domains with MHC (Garcia et al. 2009). This hypothesis is supported by a substantial number of crystal structures involving six different Vβ8.2 TCRs and one Vβ8.1 TCR in complex with mouse I-A molecules. These structures show close convergence of the CDR1β and CDR2β contacts with the I-A α1 chain helix (Reinherz et al. 1999; Feng et al. 2007; Dai et al. 2008; Garcia et al. 2009). This Vβ8 interaction motif has been seen in structures with different I-A alleles, Vα segments, and peptides, and mutation of these TCRβ residues reduced or abolished activation by a panel of T cells. However, at least some V-gene segments can use more than one set of interactions to bind a given MHC molecule. For example, two Vβ2-containing TCRs with different peptide specificities show distinct docking footprints on the H-2Kb α1 helix (Reiser et al. 2000; Reiser et al. 2003). The hypervariable CDR3 loops of the TCR and the MHC-bound peptide are probably involved in determining which binding interactions with the MHC helices are possible and energetically most favorable.
The identification of the conserved Vβ8 interaction motif with I-A proteins provided an opportunity to examine if these germline-encoded TCR amino acids are required for T-cell development. Particularly prominent in this interaction motif are the contributions of the CDR2β loop residues Y46, Y48, and E54. In mice expressing single, rearranged TCRβ chains, individual mutation of these CDR2β residues to alanine substantially reduced development of the entire TCR repertoire (Scott-Browne et al. 2009). The phenotype was particularly strong for mutation of the two tyrosines (Y46 and Y48) in the CDR2β loop. One of these tyrosines (Y46) is conserved in another mouse Vβ segment (Vβ6) and thymic CD4 T cells were again significantly reduced when Y46 was mutated to alanine. Interestingly, the lowered Vβ affinity appeared to be compensated by selection of TCRα chains with higher affinity for MHC. These results show that thymic selection is controlled by germline-encoded MHC contact points.
This conclusion is supported by the finding that a substantial fraction of T cells from mice with limited negative selection are highly MHC and peptide cross-reactive (Huseby et al. 2005). Interestingly, some of these TCRs are positively selected on both MHC class I and class II, which shows that some T cells can recognize conserved features of the MHC class I and II helices. Structural comparison of highly specific and highly cross‐reactive TCRs from these mice showed that the highly cross‐reactive TCR had a much smaller concentrated interaction surface that was more hydrophobic (Dai et al. 2008). The interaction with the MHC became highly focused on a limited number of TCR amino acids, which included the CDR2β residues Y46, Y48, and E54 discussed above. The germline-coded TCR contacts with MHC can thus yield T cells with a high degree of MHC and peptide cross‐reactivity, which are normally eliminated by negative selection (Huseby et al. 2008).
An alternative hypothesis to explain the diagonal binding mode of the TCR with peptide-MHC proposes that this geometry is driven by the requirement to accommodate the coreceptors CD4/CD8 or by a requirement for formation of TCR dimers/oligomers before initiation of signaling (Ding et al. 1998). No structural data are yet available to test the validity of these ideas. It is important to keep in mind that these hypotheses are not mutually exclusive. For example, the requirement for formation of a higher-order structure could have defined the structural boundaries for coevolution of TCR with MHC.
A common theme emerging from all of these structural studies is that particular germline elements are used in different ways for αβ or γδTCR recognition of peptide-MHC, lipid-CD1 complexes, or nonclassical MHC class I molecules. In the case of the G8 γδTCR, a single TCR loop appears to be sufficient for binding to the nonclassical MHC class Ib molecule T22 (Adams et al. 2005). In contrast, αβTCRs typically use four germline-encoded loops for binding to the MHC helices, which enables the hypervariable CDR3 loops to probe the contents of the peptide binding groove (Rudolph et al. 2006). The recognition mode is yet again different for αβ TCRs binding to CD1d-lipid complexes (Borg et al. 2007). In this specialized case, the germline encoded Vα and Jα segments of the invariant TCRα chain are used to bind to a CD1 embedded glycolipid antigen.
ASSEMBLY OF T-CELL RECEPTOR-CD3 COMPLEXES
How is an encounter with appropriate peptide-MHC or lipid-CD1 complexes actually “read” into a T cell? The mature TCR proteins carry no signaling motifs, but rather transmit signals via tyrosine-based ITAM motifs in the cytoplasmic portions of the TCR-associated CD3γε, CD3δε, and ζζ modules (Germain and Stefanova 1999; Samelson 2002). The precise mechanisms linking MHC binding outside the cell to early biochemical events inside the cell remain an area of vigorous investigation and significant controversy. Proposed triggering models range from receptor clustering and coreceptor recruitment to an array of conformational change models in which TCR proteins are proposed to physically impinge on the CD3 modules to transmit signals (Kuhns et al. 2006; Choudhuri and van der Merwe 2007). Our ability to distinguish among these different models depends critically on a solid understanding of the spatial organization of the complete TCR-CD3 complex. However, no high-resolution structure of an intact complex is yet available. As such, we must rely on three types of available experimental data to inform our working models: (1) measurements of the stoichiometric relationships among subunits, (2) available atomic-resolution structures of folded domains, and (3) biochemical data identifying the molecular surfaces involved in intersubunit contacts.
TCR-CD3 Stoichiometry
The αβTCR is noncovalently associated with three dimeric signaling modules: CD3δε, CD3γε, and ζζ (Call and Wucherpfennig 2005). On ligand binding by the TCR, cytoplasmic ITAM motifs in the CD3 and ζ chains become phosphorylated by the Src-family kinase Lck (Weiss and Littman 1994), constituting the earliest detectable biochemical consequence of TCR ligation. Early genetic, biochemical, and immunofluorescence-based experiments established that the intact TCR-CD3 complex contains two copies of CD3ε (Blumberg et al. 1990b; de la Hera et al. 1991), consistent with a model in which both CD3δε and CD3γε are incorporated into each αβTCR complex. Similar approaches have failed to detect more than one TCRα or TCRβ chain in intact TCR-CD3 complexes (Punt et al. 1994; Call et al. 2002), indicating that the receptor is monovalent with respect to the ligand-binding module. This model is supported by direct biochemical measurements of the TCR-CD3 stoichiometry in ER-assembled complexes, in which the composition was found to be TCRαβ:CD3δε:CD3γε:ζζ (Call et al. 2004).
The composition of the γδTCR and pre-TCR complexes differ from αβTCR. Mice deficient in CD3γ, CD3ε, or ζ subunits show a near-complete arrest of thymocyte development at the CD4−CD8− (DN) stage, the point at which pre-TCR signaling is required for further maturation (reviewed in Dave 2009). However, in CD3δ-deficient mice, thymocyte development proceeded to the CD4+CD8+ (DP) stage, suggesting that pre-TCR function was intact, and the γδ T-cell compartment was unperturbed (Dave et al. 1997). Thus, whereas CD3γ, CD3ε, and ζ are required for all developmental stages and T-cell lineages, CD3δ may be dispensable for γδ and pre-TCR function in mice. A recent report has indeed shown that surface-expressed TCR-CD3 complexes on CD3δ-sufficient murine γδ T cells do not contain CD3δε modules, and quantitative flow cytometry measurements yielded a stoichiometry of TCRγδ:CD3γε(2):ζζ (Hayes and Love 2006). We note that this appears to be a difference between mouse and human γδTCR, which does incorporate CD3δε (Siegers et al. 2007). Both αβ and γδTCR thus require two CD3 modules, and based on our current understanding of TCR-CD3 assembly mechanisms (see the following discussion), the same is likely to be true for pre-TCR.
Mechanisms Directing TCR-CD3 Complex Assembly
Like the TCR, CD3δε and CD3γε heterodimers are formed through interactions between their extracellular Ig domains. The subunit interface is formed primarily through an extended contact along two β strands that terminate in the stalk regions connecting the Ig domains to the TM domains (Sun et al. 2001; Arnett et al. 2004; Kjer-Nielsen et al. 2004; Sun et al. 2004). In contrast, the disulfide-linked ζζ homodimer, having only a very small (nine amino acid) ectodomain, forms through contacts predominantly within the TM domain (Rutledge et al. 1992; Call et al. 2002; Call et al. 2006). A major remaining challenge has been to identify the molecular surfaces responsible for stabilizing contacts among these four dimeric modules.
Soluble ectodomain fragments of TCR and CD3 dimers show no measurable affinity for one another, and we now understand that the determinants for assembly are contained primarily within the TM and juxtamembrane regions. The TCR proteins have three basic amino acids within the TM domains: Two in TCRα and one in TCRβ. Similarly, CD3 and ζ proteins each contain a single aspartic or glutamic acid in their TM domains, creating a pair of acidic residues in each dimeric module. Cellular transfection studies established that at least some of these ionizable residues participate in assembly (Alcover et al. 1990; Blumberg et al. 1990a; Cosson et al. 1991; Manolios et al. 1991). A more comprehensive mutagenesis analysis using in vitro-assembled complexes revealed that each of the basic TM residues in the TCR specifically recruits one of the three signaling modules in contacts requiring both acidic TM residues (Call et al. 2002; Call et al. 2004). The picture that emerges from these studies is that of an ordered assembly process organized around three trimeric intramembrane interactions: CD3δε associates with TCRα, and CD3γε with TCRβ through the centrally placed lysine residues, and then the ζζ module associates with TCRα through an arginine residue in the upper third of the TM domain (Fig. 3A). Alanine substitution at any one of these nine positions prevents formation of a complete TCR-CD3 complex.
Figure 3.

Assembly of the TCR-CD3 complex. (A) The TCR-CD3 complex is composed of four dimeric modules: TCRαβ (or γδ), CD3δε, CD3γε, and ζζ, which associate through intramembrane contacts to form the intact complex. Each dimeric signaling module associates with the TCR through a pair of acidic TM residues that bind a specific basic TM residue in the TCR. The result is an octameric complex with the stoichiometry as shown. Ribbon structures were generated from PDB entries 1MI5 (LC13 αβTCR), 1XMW (CD3δε), and 1SY6 (CD3γε). (B) NMR structure of the ζζ TM homodimer in detergent micelles (PBD entry 2HAC). The aspartic acid pair (D6-D6) that mediates assembly with TCR is located at the helix dimer interface and stabilized by interhelical H-bonds between side-chain oxygens and backbone amide protons (indicated by dotted lines). Further interface contacts include methyl and aromatic packing (such as L9-L9) and two tyrosine-threonine H-bonds (between Y12 and T17) that are critical for dimer formation.
The identity and placement of the basic TM residues are conserved among αβTCR, γδTCR, and pre-TCR sequences, implying that the assembly mechanism described for αβTCR applies to all three complexes. This would account for the presence of two CD3 modules in γδTCR (and possibly pre-TCR) even when CD3δ is not available. Selectivity for specific TCR chains in the assembly process is likely determined by sequences in the extracellular portions of the CD3 proteins, because a chimeric CD3γ bearing the CD3δ TM domain can be incorporated into complete αβTCR-CD3 complexes and rescues surface TCR expression in CD3γ-deficient T cells (Wegener et al. 1995; Call et al. 2002). Other lines of evidence also point to important interactions among ectodomains. Mutations in the TCRα connecting peptide have adverse effects on assembly and signaling (Werlen et al. 2000), and a specific “CxxC” motif in the stalk regions of the CD3 proteins is required for association with TCR (Xu et al. 2006). The nine amino acid extracellular stalks of the ζζ dimer also appear to play a role in association with TCR, because mutations in this region adversely affect assembly (Xu and Wucherpfennig, unpubl.). Likewise, mutations in loop regions in both TCRα and TCRβ constant domains have been implicated in receptor complex stability as well as signaling functions (Kuhns and Davis 2007; Beddoe et al. 2009).
Conservation of Membrane-based Receptor Complex Assembly
The ζζ dimer is one of four homodimeric modules known to provide the signaling capacity to many activating receptors in the immune system (Call and Wucherpfennig 2007). The Fc receptor γ (Fcγ) subunit associates with a subset of Fc receptors, natural killer (NK) cell receptors, and activating receptors expressed on osteoclasts and platelets. DAP10 and DAP12 associate with NK cell receptors (Lanier 2009). These four signaling proteins share three common features: They are all disulfide-linked homodimers, all bear an aspartic acid pair in the dimeric TM domains, and all carry tyrosine-based cytoplasmic motifs that are phosphorylated on receptor triggering.
With very few exceptions, the receptors that associate with these signaling modules have a basic residue in the TM domain that is required for assembly. In a series of biochemical studies that included a broad representation of receptor complexes, a set of common features was identified (Feng et al. 2005; Garrity et al. 2005; Feng et al. 2006; Feng et al. 2007). First, in every case investigated, the association required both aspartic acid residues; alanine substitution of only one in the pair invariably resulted in dramatic assembly defects. Second, the determinants for intramembrane assembly are contained almost entirely within this focused contact site, because the TM domains of receptors could be substituted with poly-leucine or poly-valine sequences without disrupting assembly, as long as the basic residue remained in its native position (Feng et al. 2006). Finally, extracellular and intracellular domains were not required for the intramembrane assembly, because short peptides encompassing TM and juxtamembrane sequences were sufficient to build receptor complexes.
Structural Basis for Intramembrane Assembly
Precise structural information is required to understand why this particular intramembrane arrangement is desirable. The only atomic-resolution structure to emerge so far is the solution NMR structure of the ζζ TM dimer (Call et al. 2006). Consistent with the requirement for a pair of acidic TM residues, the sidechains of the two aspartic acids are colocalized at the helix dimer interface (Fig. 3B). The close apposition of two acidic groups is stabilized by a hydrogen-bonding network that includes both intra- and interhelical hydrogen bonds. The specific geometry of the di-aspartate site seems to be critical for binding to the receptor, because even the chemically conservative substitution of glutamic acid can result in major assembly defects (Call et al. 2002; Call et al. 2006).
Functional Ramifications of Understanding Receptor Assembly
At the level of TCR-CD3 complex formation within the ER, the intramembrane assembly mechanism provides an important quality control checkpoint. The basic and acidic residues that organize and stabilize the complex also act as signals for degradation of subunits that remain unassembled (Bonifacino et al. 1990; Call and Wucherpfennig 2005; Call et al. 2006). Assembly and destruction are therefore directly competing processes that function together to ensure that only intact, functional receptor complexes reach the T-cell surface.
The sequestration of the most critical stabilizing contacts within the membrane may provide a requisite degree of conformational freedom allowing rearrangements among extracellular and intracellular domains to transmit signals across the plasma membrane. The highly focused and polar nature of these contacts (especially in the hydrophobic bilayer interior where competing ions are lacking) may even allow for reorientations of TM helices around fixed points to mechanically communicate conformational changes to the intracellular signaling domains (Engelman 2003). These are difficult hypotheses to test, and will require sophisticated new experimental approaches for monitoring molecular motions in living cells receiving activating signals.
LIPID BINDING BY THE CD3ε AND ζ CYTOPLASMIC DOMAINS
Each ITAM has two tyrosines and two aliphatic residues (YxxL/Ix6-12YxxL/I) and phosphorylation of both tyrosines by Lck or Fyn is required for binding of the tandem SH2 domains of ZAP-70 (Reth 1989; Weiss and Littman 1994; Hatada et al. 1995). Most textbooks show the cytoplasmic domains of the CD3 subunits as flexible chains in the cytosol, a rendering that suggests that they would be continuously accessible to tyrosine kinases. Is this model correct?
Lipid Binding of ITAMS
The inner leaflet of the plasma membrane has a negative charge because phosphatidylserine (PS) is almost exclusively localized to the inner leaflet in live cells. Phosphatidylserine is the most abundant anionic lipid in cellular membrane (representing ∼20% of inner leaflet lipids), and its asymmetric distribution is maintained by an ATP-dependent lipid flippase referred to as aminophospholipid translocase (Devaux 1991; Fridriksson et al. 1999; Devaux et al. 2008). This lipid asymmetry is lost when cells become apoptotic, enabling detection of such cells by labeling with the PS binding protein annexin V. Other anionic lipids present at significantly lower densities contribute to the negative charge, including phosphatidylglycerol, phosphatidic acid, and a variety of phosphoinositides in different phosphorylation states (Fridriksson et al. 1999).
The cytoplasmic domains of both CD3ε and ζ chains have a net positive charge, which raised the question whether they could bind to the inner leaflet of the plasma membrane through electrostatic interactions. Using in vitro assays with synthetic lipid vesicles, the ζ chain cytoplasmic domain was shown to bind to vesicles with a net negative charge, but not to vesicles lacking such a charge (Aivazian and Stern 2000). Binding resulted in a substantial increase in α-helical content detected by circular dichroism measurements, and these authors proposed that the three ITAMs fold into a α-helical structure upon lipid binding. Binding to synthetic lipid vesicles was later also shown for the CD3ε cytoplasmic domain as well as the cytoplasmic domain of Fcγ, which serves as a signaling module for activating Fc receptors (Sigalov et al. 2006; Xu et al. 2008; Deford-Watts et al. 2009). CD3ε has a higher net positive charge than ζ and bound acidic lipid vesicles with higher affinity. Mutation of two clusters of basic residues in the amino-terminal part of the CD3ε cytoplasmic domain abrogated lipid binding, confirming the importance of electrostatic interactions (Xu et al. 2008). Little or no binding was detected for the cytoplasmic domains of the CD3γ and CD3δ chains in these in vitro assays, which lack a net positive charge, although it remains possible that they bind cooperatively with ζ and CD3ε to cellular membranes.
It was critical to establish that such lipid binding actually occurs in cells because all binding studies had been performed in vitro with synthetic lipid vesicle preparations. A cellular fluorescence resonance energy transfer (FRET) assay was developed to directly address this question (Xu et al. 2008). In a fully extended conformation, the 57 amino acid cytoplasmic domain of CD3ε is at a substantial distance from the plasma membrane (∼200 Å). This distance is outside of the range for FRET because energy transfer from donor to acceptor fluorophore is highly distance dependent, with the upper limit being ∼100 Å (Kenworthy 2001). Efficient FRET between a fluorescent protein attached to the carboxy-terminus of CD3ε and a fluorescent dye in the plasma membrane is therefore only expected in the lipid-bound state of CD3ε, but not when the cytoplasmic domain has dissociated from the membrane. The validity of this FRET-based approach was tested with constructs using flexible linkers of increasing length (3, 25, and 50 amino acids). A high FRET signal was observed with the wild-type CD3ε cytoplasmic domain, but not with a mutant that failed to show binding in the in vitro assay described above (Sigalov et al. 2006; Xu et al. 2008).
Structure of the Membrane-bound Itam
Determination of the structure of the CD3ε cytoplasmic domain required a lipid bilayer surface sufficiently large in size to enable proper binding of the cytoplasmic domain. Detergent micelles with the appropriate lipid headgroups would not be suitable because of their high curvature. The solution to this problem was to use bicelles, which represent flat discs formed using a mixture of short- and long-chain lipids. The long-chain lipids form the bilayer core of such bicelles, whereas the short chain lipids associate at the rim. Bicelles have a size similar to small proteins and thus tumble quickly enough for solution NMR studies (Prosser et al. 2006). This approach enabled determination of the structure of the CD3ε cytoplasmic domain in a lipid-bound state (Xu et al. 2008). A surprising finding was that the cytoplasmic domain was actually inserted into the bilayer, with the peptide backbone being located at the interface between the hydrophilic headgroup region and the hydrophobic acyl chain layer. This overall position of the backbone enabled insertion of all hydrophobic side chains into the hydrophobic core of the bilayer. The structure of the ITAM itself shows insertion of all four key residues, the two tyrosines, and the two aliphatic residues, into the acyl chain region of the bilayer (Fig. 4). Helical structure was confined to short segments centered on the tyrosines, and circular dichroism experiments confirmed the presence of a small amount of α-helical structure that was lost when the tyrosines and aliphatic residues of the ITAM were mutated. Interestingly, the amino-terminal part of the CD3ε cytoplasmic domain is rather flexible, indicating that the ITAM is connected to the TM domain through a membrane-bound flexible linker (Xu et al. 2008). Functional studies showed that lipid binding by CD3ε or ζ prevented phosphorylation by Lck (Aivazian and Stern 2000; Xu et al. 2008). This means that dissociation of the ITAMs from the membrane is required as one of the initial events in TCR activation.
Figure 4.

Structure of the CD3ε ITAM in the lipid-bound state. The two tyrosines (Y38 and Y49) and the two aliphatic residues (I41 and L52) of the ITAM protrude into the hydrophobic acyl chain region of the lipid bilayer. The peptide backbone of the ITAM resides primarily at the interface between the hydrated lipid headgroup region and the hydrophobic bilayer interior. The hydrophobic layer of the bilayer is shaded light blue and the hydrated lipid headgroup region in dark blue. The POPG structure graphic to the right shows the location of the hydrophilic headgroup and the hydrophobic acyl chains of the lipid (PDB entry 2K4F).
Possible Mechanisms for ITAM Release from the Membrane
The mechanisms resulting in dissociation of the CD3ε and ζ cytoplasmic domains from the membrane on TCR triggering remain unknown. It is unlikely that subtle conformational change in the extracellular domains of the TCR-CD3 complex during peptide-MHC binding are sufficient due to the flexible nature of the N-terminal part of the cytoplasmic domain (Aivazian and Stern 2000; Xu et al. 2008). Other mechanisms must therefore account for the change in tyrosine accessibility during TCR triggering.
It has been proposed that the continued movement of T cells across antigen presenting cells results in a mechanical force on the TCR-peptide-MHC recognition unit (Ma et al. 2008; Kim et al. 2009). Such a mechanical force could possibly result in dissociation of the ITAM from the membrane but would have to be strong or sustained enough to act through the flexible amino-terminal part of the cytoplasmic domain on the ITAM. A second possibility is that clustering of TCR-CD3 complexes in early microclusters at the immunological synapse results in competition among the cytoplasmic domains for membrane surface (Aivazian and Stern 2000). TCR microclusters can form before initiation of signaling because they are observed even in the presence of a Src kinase inhibitor that blocks Lck activity (Campi et al. 2005). A third hypothesis is that TCR clustering changes the lipid environment in the vicinity of the clustered TCRs, reducing the affinity of the cytoplasmic domains for the membrane. Lipid binding is highly sensitive to the density of negatively charged phospholipids due to the essential role of basic residues in the cytoplasmic domains for lipid binding.
CONCLUDING REMARKS
Structural data are now available not only for the extracellular domains, but also some of the transmembrane and cytoplasmic domains of the TCR-CD3 complex. These structural studies create a solid framework for understanding key functional aspects of the TCR-CD3 complex and many other activating receptors in the immune system.
ACKNOWLEDGMENTS
This work was supported by grants to K.W.W. to the National Institutes of Health (RO1 AI054520 and AI064177). E.S.H. was supported in part by Beckman Young Investigator and Searle Scholar awards.
Footnotes
Editors: Lawrence E. Samelson and Andrey Shaw
Additional Perspectives on Immunoreceptor Signaling available at www.cshperspectives.org
REFERENCES
- Adams EJ, Chien YH, Garcia KC 2005. Structure of a γδ T cell receptor in complex with the nonclassical MHC T22. Science 308:227–231 [DOI] [PubMed] [Google Scholar]
- Adams EJ, Strop P, Shin S, Chien YH, Garcia KC 2008. An autonomous CDR3δ is sufficient for recognition of the nonclassical MHC class I molecules T10 and T22 by γδ T cells. Nat Immunol 9:777–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aivazian D, Stern LJ 2000. Phosphorylation of T cell receptor zeta is regulated by a lipid dependent folding transition. Nat Struct Biol 7:1023–1026 [DOI] [PubMed] [Google Scholar]
- Alcover A, Mariuzza RA, Ermonval M, Acuto O 1990. Lysine 271 in the transmembrane domain of the T-cell antigen receptor β chain is necessary for its assembly with the CD3 complex but not for α/β dimerization. J Biol Chem 265:4131–4135 [PubMed] [Google Scholar]
- Arnett KL, Harrison SC, Wiley DC 2004. Crystal structure of a human CD3-ϵ/δ dimer in complex with a UCHT1 single-chain antibody fragment. Proc Natl Acad Sci 101:16268–16273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker BM, Turner RV, Gagnon SJ, Wiley DC, Biddison WE 2001. Identification of a crucial energetic footprint on the α1 helix of human histocompatibility leukocyte antigen (HLA)-A2 that provides functional interactions for recognition by tax peptide/HLA-A2-specific T cell receptors. J Exp Med 193:551–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral DC, Brenner MB 2007. CD1 antigen presentation: How it works. Nat Rev Immunol 7:929–941 [DOI] [PubMed] [Google Scholar]
- Beddoe T, Chen Z, Clements CS, Ely LK, Bushell SR, Vivian JP, Kjer-Nielsen L, Pang SS, Dunstone MA, Liu YC, et al. 2009. Antigen ligation triggers a conformational change within the constant domain of the αβ T cell receptor. Immunity 30:777–788 [DOI] [PubMed] [Google Scholar]
- Bendelac A, Savage PB, Teyton L 2007. The biology of NKT cells. Annu Rev Immunol 25:297–336 [DOI] [PubMed] [Google Scholar]
- Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC 1987. The foreign antigen binding site and T cell recognition regions of class I histocompatibility antigens. Nature 329:512–518 [DOI] [PubMed] [Google Scholar]
- Blumberg RS, Alarcon B, Sancho J, McDermott FV, Lopez P, Breitmeyer J, Terhorst C 1990a. Assembly and function of the T cell antigen receptor. Requirement of either the lysine or arginine residues in the transmembrane region of the α chain. J Biol Chem 265:14036–14043 [PubMed] [Google Scholar]
- Blumberg RS, Ley S, Sancho J, Lonberg N, Lacy E, McDermott F, Schad V, Greenstein JL, Terhorst C 1990b. Structure of the T-cell antigen receptor: Evidence for two CD3 ϵ subunits in the T-cell receptor-CD3 complex. Proc Natl Acad Sci 87:7220–7224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Cosson P, Klausner RD 1990. Colocalized transmembrane determinants for ER degradation and subunit assembly explain the intracellular fate of TCR chains. Cell 63:503–513 [DOI] [PubMed] [Google Scholar]
- Borg NA, Wun KS, Kjer-Nielsen L, Wilce MC, Pellicci DG, Koh R, Besra GS, Bharadwaj M, Godfrey DI, McCluskey J, et al. 2007. CD1d-lipid-antigen recognition by the semi-invariant NKT T-cell receptor. Nature 448:44–49 [DOI] [PubMed] [Google Scholar]
- Call ME, Wucherpfennig KW 2005. The T cell receptor: Critical role of the membrane environment in receptor assembly and function. Annu Rev Immunol 23:101–125 [DOI] [PubMed] [Google Scholar]
- Call ME, Wucherpfennig KW 2007. Common themes in the assembly and architecture of activating immune receptors. Nat Rev Immunol 7:841–850 [DOI] [PubMed] [Google Scholar]
- Call ME, Pyrdol J, Wucherpfennig KW 2004. Stoichiometry of the T-cell receptor-CD3 complex and key intermediates assembled in the endoplasmic reticulum. Embo J 23:2348–2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Call ME, Pyrdol J, Wiedmann M, Wucherpfennig KW 2002. The organizing principle in the formation of the T cell receptor-CD3 complex. Cell 111:967–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Call ME, Schnell JR, Xu C, Lutz RA, Chou JJ, Wucherpfennig KW 2006. The structure of the zetazeta transmembrane dimer reveals features essential for its assembly with the T cell receptor. Cell 127:355–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campi G, Varma R, Dustin ML 2005. Actin and agonist MHC-peptide complex-dependent T cell receptor microclusters as scaffolds for signaling. J Exp Med 202:1031–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhuri K, van der Merwe PA 2007. Molecular mechanisms involved in T cell receptor triggering. Semin Immunol 19:255–261 [DOI] [PubMed] [Google Scholar]
- Cosson P, Lankford SP, Bonifacino JS, Klausner RD 1991. Membrane protein association by potential intramembrane charge pairs. Nature 351:414–416 [DOI] [PubMed] [Google Scholar]
- Dai S, Huseby ES, Rubtsova K, Scott-Browne J, Crawford F, Macdonald WA, Marrack P, Kappler JW 2008. Crossreactive T Cells spotlight the germline rules for αβ T cell-receptor interactions with MHC molecules. Immunity 28:324–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave VP 2009. Hierarchical role of CD3 chains in thymocyte development. Immunol Rev 232:22–33 [DOI] [PubMed] [Google Scholar]
- Dave VP, Cao Z, Browne C, Alarcon B, Fernandez-Miguel G, Lafaille J, de la Hera A, Tonegawa S, Kappes DJ 1997. CD3 δ deficiency arrests development of the α β but not the γ δ T cell lineage. Embo J 16:1360–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Hera A, Muller U, Olsson C, Isaaz S, Tunnacliffe A 1991. Structure of the T cell antigen receptor (TCR): Two CD3 ϵ subunits in a functional TCR/CD3 complex. J Exp Med 173:7–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deford-Watts LM, Tassin TC, Becker AM, Medeiros JJ, Albanesi JP, Love PE, Wulfing C, van Oers NS 2009. The cytoplasmic tail of the T cell receptor CD3 ε subunit contains a phospholipid-binding motif that regulates T cell functions. J Immunol 183:1055–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux PF 1991. Static and dynamic lipid asymmetry in cell membranes. Biochemistry 30:1163–1173 [DOI] [PubMed] [Google Scholar]
- Devaux PF, Herrmann A, Ohlwein N, Kozlov MM 2008. How lipid flippases can modulate membrane structure. Biochim Biophys Acta 1778:1591–1600 [DOI] [PubMed] [Google Scholar]
- Ding YH, Smith KJ, Garboczi DN, Utz U, Biddison WE, Wiley DC 1998. Two human T cell receptors bind in a similar diagonal mode to the HLA-A2/Tax peptide complex using different TCR amino acids. Immunity 8:403–411 [DOI] [PubMed] [Google Scholar]
- Engelman DM 2003. Electrostatic fasteners hold the T cell receptor-CD3 complex together. Mol Cell 11:5–6 [DOI] [PubMed] [Google Scholar]
- Feng D, Bond CJ, Ely LK, Maynard J, Garcia KC 2007. Structural evidence for a germline-encoded T cell receptor-major histocompatibility complex interaction ‘codon’. Nat Immunol 8:975–983 [DOI] [PubMed] [Google Scholar]
- Feng J, Call ME, Wucherpfennig KW 2006. The assembly of diverse immune receptors is focused on a polar membrane-embedded interaction site. PLoS Biol 4:e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Garrity D, Call ME, Moffett H, Wucherpfennig KW 2005. Convergence on a distinctive assembly mechanism by unrelated families of activating immune receptors. Immunity 22:427–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridriksson EK, Shipkova PA, Sheets ED, Holowka D, Baird B, McLafferty FW 1999. Quantitative analysis of phospholipids in functionally important membrane domains from RBL-2H3 mast cells using tandem high-resolution mass spectrometry. Biochemistry 38:8056–8063 [DOI] [PubMed] [Google Scholar]
- Garboczi DN, Ghosh P, Utz U, Fan QR, Biddison WE, Wiley DC 1996. Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature 384:134–141 [DOI] [PubMed] [Google Scholar]
- Garcia KC, Adams JJ, Feng D, Ely LK 2009. The molecular basis of TCR germline bias for MHC is surprisingly simple. Nat Immunol 10:143–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia KC, Degano M, Stanfield RL, Brunmark A, Jackson MR, Peterson PA, Teyton L, Wilson IA 1996. An αβ T cell receptor structure at 2.5 A and its orientation in the TCR-MHC complex. Science 274:209–219 [DOI] [PubMed] [Google Scholar]
- Garrity D, Call ME, Feng J, Wucherpfennig KW 2005. The activating NKG2D receptor assembles in the membrane with two signaling dimers into a hexameric structure. Proc Natl Acad Sci 102:7641–7646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain RN, Stefanova I 1999. The dynamics of T cell receptor signaling: complex orchestration and the key roles of tempo and cooperation. Annu Rev Immunol 17:467–522 [DOI] [PubMed] [Google Scholar]
- Hatada MH, Lu X, Laird ER, Green J, Morgenstern JP, Lou M, Marr CS, Phillips TB, Ram MK, Theriault K, et al. 1995. Molecular basis for interaction of the protein tyrosine kinase ZAP-70 with the T-cell receptor. Nature 377:32–38 [DOI] [PubMed] [Google Scholar]
- Hayes SM, Love PE 2006. Stoichiometry of the murine γδ T cell receptor. J Exp Med 203:47–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huseby ES, Kappler JW, Marrack P 2008. Thymic selection stifles TCR reactivity with the main chain structure of MHC and forces interactions with the peptide side chains. Mol Immunol 45:599–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huseby ES, White J, Crawford F, Vass T, Becker D, Pinilla C, Marrack P, Kappler JW 2005. How the T cell repertoire becomes peptide and MHC specific. Cell 122:247–260 [DOI] [PubMed] [Google Scholar]
- Kenworthy AK 2001. Imaging protein-protein interactions using fluorescence resonance energy transfer microscopy. Methods 24:289–296 [DOI] [PubMed] [Google Scholar]
- Kim ST, Takeuchi K, Sun ZY, Touma M, Castro CE, Fahmy A, Lang MJ, Wagner G, Reinherz EL 2009. The αβ T cell receptor is an anisotropic mechanosensor. J Biol Chem 284:31028–31037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjer-Nielsen L, Dunstone MA, Kostenko L, Ely LK, Beddoe T, Mifsud NA, Purcell AW, Brooks AG, McCluskey J, Rossjohn J 2004. Crystal structure of the human T cell receptor CD3 ε γ heterodimer complexed to the therapeutic mAb OKT3. Proc Natl Acad Sci 101:7675–7680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhns MS, Davis MM 2007. Disruption of extracellular interactions impairs T cell receptor-CD3 complex stability and signaling. Immunity 26:357–369 [DOI] [PubMed] [Google Scholar]
- Kuhns MS, Davis MM, Garcia KC 2006. Deconstructing the form and function of the TCR/CD3 complex. Immunity 24:133–139 [DOI] [PubMed] [Google Scholar]
- Lanier LL 2009. DAP10- and DAP12-associated receptors in innate immunity. Immunol Rev 227:150–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Janmey PA, Finkel TH 2008. The receptor deformation model of TCR triggering. FASEB J 22:1002–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolios N, Letourneur F, Bonifacino JS, Klausner RD 1991. Pairwise, cooperative and inhibitory interactions describe the assembly and probable structure of the T-cell antigen receptor. Embo J 10:1643–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser RS, Evanics F, Kitevski JL, Al-Abdul-Wahid MS 2006. Current applications of bicelles in NMR studies of membrane-associated amphiphiles and proteins. Biochemistry 45:8453–8465 [DOI] [PubMed] [Google Scholar]
- Punt JA, Roberts JL, Kearse KP, Singer A 1994. Stoichiometry of the T cell antigen receptor (TCR) complex: Each TCR/CD3 complex contains one TCR α, one TCR β, and two CD3 ε chains. J Exp Med 180:587–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinherz EL, Tan K, Tang L, Kern P, Liu J, Xiong Y, Hussey RE, Smolyar A, Hare B, Zhang R, et al. 1999. The crystal structure of a T cell receptor in complex with peptide and MHC class II. Science 286:1913–1921 [DOI] [PubMed] [Google Scholar]
- Reiser JB, Darnault C, Gregoire C, Mosser T, Mazza G, Kearney A, van der Merwe PA, Fontecilla-Camps JC, Housset D, Malissen B 2003. CDR3 loop flexibility contributes to the degeneracy of TCR recognition. Nat Immunol 4:241–247 [DOI] [PubMed] [Google Scholar]
- Reiser JB, Darnault C, Guimezanes A, Gregoire C, Mosser T, Schmitt-Verhulst AM, Fontecilla-Camps JC, Malissen B, Housset D, Mazza G 2000. Crystal structure of a T cell receptor bound to an allogeneic MHC molecule. Nat Immunol 1:291–297 [DOI] [PubMed] [Google Scholar]
- Reth M 1989. Antigen receptor tail clue. Nature 338:383–384 [PubMed] [Google Scholar]
- Robinson J, Waller MJ, Fail SC, McWilliam H, Lopez R, Parham P, Marsh SG 2009. The IMGT/HLA database. Nucleic Acids Res 37:D1013–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph MG, Stanfield RL, Wilson IA 2006. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol 24:419–466 [DOI] [PubMed] [Google Scholar]
- Rutledge T, Cosson P, Manolios N, Bonifacino JS, Klausner RD 1992. Transmembrane helical interactions: Zeta chain dimerization and functional association with the T cell antigen receptor. Embo J 11:3245–3254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samelson LE 2002. Signal transduction mediated by the T cell antigen receptor: the role of adapter proteins. Annu Rev Immunol 20:371–394 [DOI] [PubMed] [Google Scholar]
- Scott-Browne JP, White J, Kappler JW, Gapin L, Marrack P 2009. Germline-encoded amino acids in the αβ T-cell receptor control thymic selection. Nature 458:1043–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S, El-Diwany R, Schaffert S, Adams EJ, Garcia KC, Pereira P, Chien YH 2005. Antigen recognition determinants of γδ T cell receptors. Science 308:252–255 [DOI] [PubMed] [Google Scholar]
- Siegers GM, Swamy M, Fernandez-Malave E, Minguet S, Rathmann S, Guardo AC, Perez-Flores V, Regueiro JR, Alarcon B, Fisch P, et al. 2007. Different composition of the human and the mouse γδ T cell receptor explains different phenotypes of CD3γ and CD3δ immunodeficiencies. J Exp Med 204:2537–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigalov AB, Aivazian DA, Uversky VN, Stern LJ 2006. Lipid-binding activity of intrinsically unstructured cytoplasmic domains of multichain immune recognition receptor signaling subunits. Biochemistry 45:15731–15739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern LJ, Wiley DC 1994. Antigenic peptide binding by class I and class II histocompatibility proteins. Structure 2:245–251 [DOI] [PubMed] [Google Scholar]
- Sun ZY, Kim ST, Kim IC, Fahmy A, Reinherz EL, Wagner G 2004. Solution structure of the CD3εδ ectodomain and comparison with CD3εγ as a basis for modeling T cell receptor topology and signaling. Proc Natl Acad Sci 101:16867–16872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun ZJ, Kim KS, Wagner G, Reinherz EL 2001. Mechanisms contributing to T cell receptor signaling and assembly revealed by the solution structure of an ectodomain fragment of the CD3 ε γ heterodimer. Cell 105:913–923 [DOI] [PubMed] [Google Scholar]
- Wegener AM, Hou X, Dietrich J, Geisler C 1995. Distinct domains of the CD3-γ chain are involved in surface expression and function of the T cell antigen receptor. J Biol Chem 270:4675–4680 [DOI] [PubMed] [Google Scholar]
- Weiss A, Littman DR 1994. Signal transduction by lymphocyte antigen receptors. Cell 76:263–274 [DOI] [PubMed] [Google Scholar]
- Werlen G, Hausmann B, Palmer E 2000. A motif in the αβ T-cell receptor controls positive selection by modulating ERK activity. Nature 406:422–426 [DOI] [PubMed] [Google Scholar]
- Xu C, Call ME, Wucherpfennig KW 2006. A membrane-proximal tetracysteine motif contributes to assembly of CD3δε and CD3γε dimers with the T cell receptor. J Biol Chem 281:36977–36984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Gagnon E, Call ME, Schnell JR, Schwieters CD, Carman CV, Chou JJ, Wucherpfennig KW 2008. Regulation of T cell receptor activation by dynamic membrane binding of the CD3ε cytoplasmic tyrosine-based motif. Cell 135:702–713 [DOI] [PMC free article] [PubMed] [Google Scholar]