

Abstract
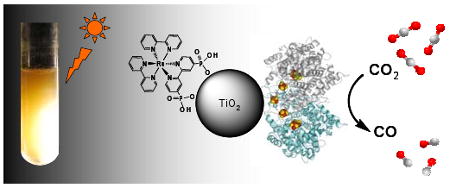
A hybrid enzyme-nanoparticle system is described for achieving clean reduction of CO2 to CO using visible light as the energy source. An aqueous dispersion of TiO2 nanoparticles modified by attachment of carbon monoxide dehydrogenase (CODH) and a Ru photosensitizer produces CO at a rate of 250 μmol CO (g TiO2)-1 h-1 when illuminated with visible light at pH 6 and 20 °C.
There is wide interest in converting the greenhouse gas CO2 into organic molecules by chemical routes1,2 and a highly desirable goal is to use solar energy to reduce CO2 to CO, efficiently and cleanly. Carbon monoxide is the feedstock for various synthetic processes, such as the d-metal catalyzed Fischer-Tropsch (production of hydrocarbons), Monsanto and Cativa (both acetic acid) processes. Carbon monoxide also has significant fuel value (ΔcH° = −283.0 kJ mol-1), and can readily be converted into methanol (e.g., by the CuO/ZnO/Al2O3-catalyzed ICI-process) for use as a liquid fuel.3 Here, we present a prototype enzyme-based system for rapid CO2 reduction to CO, driven by visible light.
The core design of a highly efficient CO2 photo-reducing system features a catalyst linked electronically to a light-capturing moiety. Since the first report of CO2 photo-reduction by a semiconductor particle suspension by Inoue et al,4 many efforts have focussed on TiO2. These systems use either high-energy irradiation (UV) for band gap excitation,5,6 or visible light following sensitization with a dye complex, for injecting electrons into the conduction band.7,8 Direct reduction of CO2 at the TiO2 surface proceeds via a thermodynamically uphill one-electron transfer to form CO2•- (E = -1.90 V vs SHE in water, corrected to pH 7).9 This high energy pathway typically leads to mixtures of products, including methane and methanol. A metallic cluster (often Cu or Pt) on the TiO2 surface can serve as the reaction center by acting as a trap for excited electrons, thereby forming a reducing site on the particle to minimize electron-hole recombination.10,11 We now report that TiO2 nanoparticles modified with a photosensitizer and the CO2-reducing enzyme CODH I from the anaerobic microbe Carboxydothermus hydrogenoformans (Ch) provide an extraordinary catalyst for CO2 photo-reduction. The enzyme bypasses the one-electron radical pathway - instead catalyzing a controlled, two-electron reduction giving CO (E = −0.46 V vs SHE at pH 6) as a clean product. The system is represented schematically in Figure 1.
Figure 1.
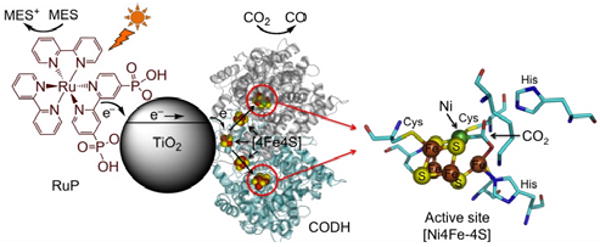
Cartoon representation of the CO2 photo-reduction system using Ch CODH I attached to RuP-modified TiO2 nanoparticles. A catalytic intermediate of the active site of the closely related enzyme CODH II with bound substrate (CO2, indicated with an arrow),16 is also shown. The oxidized photosensitizer is recovered by the sacrificial electron donor MES. The enzyme structure used in the cartoon is CODH II, created using PyMOL.
CODH I is one of five CODH complexes expressed by Ch, with an unusual [Ni4Fe-4S] active site that catalyzes the reversible oxidation of CO to CO2.12 Turnover frequencies of up to 40 000 s-1 have been reported for CO oxidation by Ch CODHs (pH 8, 70 °C),13 and we recently harnessed some of this activity to catalyze the water-gas shift reaction by coupling CODH I to a hydrogenase on a conducting graphite micro-platelet.14 Importantly, CODH I is a highly active catalyst in both directions (oxidation and reduction), requiring little overpotential.15
All stages of preparation of our photocatalytic system were carried out at room temperature in an anaerobic glovebox. TiO2 nanoparticles (5 mg, Evonik Aeroxide P25; 21 nm diameter) were dispersed for 15 min by sonication in 2-(N-morpholino)ethanesulfonic acid (MES, 5 mL of 200 mM solution) buffer in a Pyrex pressure vessel (total volume = 9 mL). Then, Ch CODH I (20 μL of 117 μM solution) was adsorbed on the TiO2 particles by gently stirring for 20 min, whereupon the enzyme-modified TiO2 nanoparticles were sensitized by adding a solution of 0.05 mg of [RuII(bipy)2(4,4′-(PO3H2)2-bipy)]Br2 (RuP; bipy = 2,2′-bipyridine) in MES buffer (0.1 mL) to the dispersion and stirring for a further 20 min for adsorption of the dye. The vessel was then sealed tightly with a rubber septum and the headspace purged with 2% CH4 in CO2. The stirred dispersion was subsequently irradiated with visible light (tungsten-halogen lamp fitted with a 420 nm UV-light filter; light intensity 45 mW cm−2). The temperature of the suspension was controlled by immersion of the reaction vessel in a water bath. The headspace gas composition was regularly monitored by GC analysis, with the amount of produced CO quantified against the internal CH4 standard.
Figure 2 shows CO production at RuP-sensitized TiO2 nanoparticles functionalized with Ch CODH I. Panel A shows several different experiments overlaid in which the suspension (pH 6, 20 °C) was irradiated with visible light (λ > 420 nm) over a period of 4 h. Control experiments in which photosensitizer, nanoparticles, enzyme or light were excluded yielded negligible amounts of CO. Possible by-products, e.g. methane (in the absence of CH4 as internal standard), were not detected upon photo-reduction of CO2 in the fully assembled RuP-TiO2-CODH system, indicating clean CO2 to CO conversion. Panel A also shows photo-reduction of CO2 using UV band gap irradiation (UVL-28 EL Series UV lamp, 365 nm, 2 mW cm-2) with CODH I attached to TiO2 in the absence of RuP. The CO formation confirms the essential role of the TiO2 nanoparticle as an electron relay between RuP and CODH I, rather than simply acting as a common support to bring the photosensitizer and enzyme into close proximity for direct electron transfer. In all cases rates decrease with time (see below). Rates of catalysis scale with the amount of enzyme in the system (Supporting Information), strongly indicating that the enzyme activity is limiting: this is supported by the results in Panel B which show how the CO production rate varies with temperature, again consistent with enzyme-controlled activation.
Figure 2.
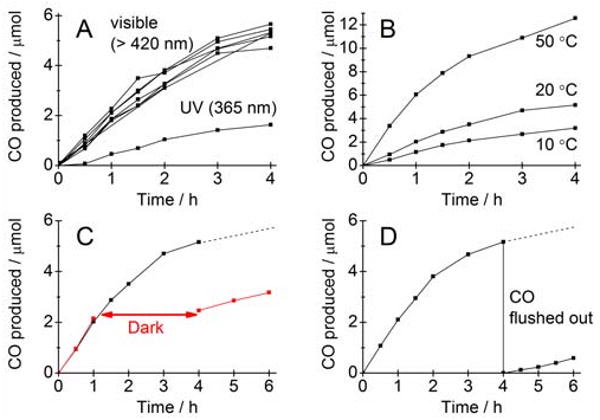
A. Photo-reduction of CO2; top: at dye-sensitized, CODH I-modified TiO2 nanoparticles using visible light (20 °C, pH 6.0, λ > 420 nm); bottom: at non-sensitized, CODH I-modified nanoparticles using band gap UV irradiation (20 °C, pH 6.0, λ = 365 nm). B. Effect of temperature on the photo-reduction rate of the dye-sensitized system (20 °C, pH 6, λ > 420 nm). C. Black trace: photo-reduction of CO2 at dye-sensitized, CODH I-modified nanoparticles with continuous visible light irradiation (20 °C, pH 6.0, λ > 420 nm); red trace: experiment in which visible light irradiation was interrupted for 3 h after 1 h of irradiation (same conditions as black trace). D. Experiment in which CO was flushed out after 4 h of visible light irradiation (20 °C, pH 6.0, λ > 420 nm) and the CO2 level was restored to the initial level (98%).
At pH 6 and 20 °C the visible light-sensitized system produces approximately 5 μmol CO during 4 h irradiation (Figure 2A). Evaluating this on a per-gram TiO2 basis gives an average turnover rate of 250 μmol CO (g TiO2)−1 h−1 – clean, and superior to other sensitized TiO2 systems, which usually produce a range of products.17,18 On a per-mole CODH I basis, the turnover rate is approximately 530 h-1 (0.15 s−1). An experiment using natural sunlight in which the temperature varied from 17 °C to 22 °C (Oxford sky, October, Supporting Information) performed photo-reduction at a rate of approximately 0.09 s−1, i.e. 60% of the rate we observe using the artificial visible light source.
The rates and stability (loss of activity with time) are low compared to a recently reported analogous H2 production system (50 s−1) using a titaniaphilic hydrogenase under comparable conditions (pH 7, 25 °C),19 but several factors are likely to be influential, each of which are under further investigation. One factor is the smaller driving force (about 0.1 V lower) for CO2 reduction compared to H+ reduction: at pH 6, the conduction band edge of TiO2 is at approximately -0.52 V vs SHE,20 which is only just sufficient for CO2 reduction at CODH I. Another factor may be that CODH I molecules are only bound weakly to the TiO2, or lie in electro-inactive orientations: success depends upon efficient electron transfer from TiO2 into the relay of iron-sulfur clusters leading to the active site (Figure 1) which will require a more specific linking mode. Weak binding is supported by the observation that centrifugation of the particles, followed by exchange of the buffer for fresh solution and re-suspension leads to a marked decrease in activity (Supporting Information). When the suspension was kept in darkness for 3 h after irradiating for 1 h, resumption of irradiation gave a rate approximately the same as after 4 h light exposure (Figure 2C), ruling out any significant photoinstability effect (i.e. photo-degradation of RuP or CODH I). Experiments were also carried out (Figure 2D) in which, after visible light irradiation for 4 h, the system was flushed with CO2 to remove all CO and restore the CO2 level to the initial concentration. After re-starting, CO production continued at the same rate as measured after 4 h. In addition, the results were essentially unchanged when initial levels of 10, 20 and 30% CO were used (Supporting Information). Thus, neither substrate depletion nor product inhibition is responsible for the decrease in activity. The activity and stability of the system are not significantly affected by pH in the range 5.5 – 6.5 (Supporting Information) or by varying buffer concentration (50-200 mM). The lower rate of the UV system compared to the visible system is under investigation: explanations include inactivation of CODH by valence band holes and the lower power output of our UV source (2 mW cm-2 vs 45 mW cm-2 for the visible system).
In summary, we have demonstrated a heterogeneous catalyst for efficient and clean reduction of CO2 to CO driven by visible light. The sensitized hybrid enzyme-nanoparticle system (even before important refinements are made) serves as a benchmark for what must be achievable using synthetic catalysts. The results provide a strong case for CO2 activation catalysts to focus on a two-electron pathway in order to avoid the thermodynamically uphill step involved in one-electron activations.
Supplementary Material
Acknowledgments
This research was supported by UK research councils BBSRC and EPRSC (Grants BB/D52222X, BB/H003878/1 and Supergen 5 to F.A.A., EP/H00338X/1 to E.R.) and the NIH (GM39451 to S.W.R.).
Footnotes
Supporting Information Available: Effect of lower enzyme concentration (Fig. S1), CO2 photo-reduction using natural sunlight (Fig. S2), effect of centrifuging particles and exchanging buffer solution (Fig. S3), effect of pH (Fig. S4), UV-visible study of CODH I adsorption onto TiO2 nanoparticles (Fig. S5), effect of initial CO concentration (Fig. S6), approximate calculation for CODH I loading on TiO2 nanoparticles, full list of authors for ref. 12. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Sakakura T, Choi JC, Yasuda H. Chem Rev. 2007;107:2365–2387. doi: 10.1021/cr068357u. [DOI] [PubMed] [Google Scholar]
- 2.Song CS. Catal Today. 2006;115:2–32. [Google Scholar]
- 3.Waugh KC. Catal Today. 1992;15:51–75. [Google Scholar]
- 4.Inoue T, Fujishima A, Konishi S, Honda K. Nature. 1979;277:637–638. [Google Scholar]
- 5.Anpo M, Yamashita H, Ichihashi Y, Fujii Y, Honda M. J Phys Chem B. 1997;101:2632–2636. [Google Scholar]
- 6.Wu JCS, Lin HM, Lai CL. Appl Catal, A. 2005;296:194–200. [Google Scholar]
- 7.Nguyen TV, Wu JCS, Chiou CH. Catal Commun. 2008;9:2073–2076. [Google Scholar]
- 8.Ozcan O, Yukruk F, Akkaya EU, Uner D. Top Catal. 2007;44:523–528. [Google Scholar]
- 9.Koppenol WH, Rush JD. J Phys Chem. 1987;91:4429–4430. [Google Scholar]
- 10.Tseng IH, Chang WC, Wu JCS. Appl Catal, B. 2002;37:37–48. [Google Scholar]
- 11.Yamashita H, Fujii Y, Ichihashi Y, Zhang SG, Ikeue K, Park DR, Koyano K, Tatsumi T, Anpo M. Catal Today. 1998;45:221–227. [Google Scholar]
- 12.Wu M, et al. PloS Genet. 2005;1:563–574. [Google Scholar]
- 13.Svetlitchnyi V, Peschel C, Acker G, Meyer O. J Bacteriol. 2001;183:5134–5144. doi: 10.1128/JB.183.17.5134-5144.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lazarus O, Woolerton TW, Parkin A, Lukey MJ, Reisner E, Seravalli J, Pierce E, Ragsdale SW, Sargent F, Armstrong FA. J Am Chem Soc. 2009;131:14154–5. doi: 10.1021/ja905797w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parkin A, Seravalli J, Vincent KA, Ragsdale SW, Armstrong FA. J Am Chem Soc. 2007;129:10328–10329. doi: 10.1021/ja073643o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeoung JH, Dobbek H. Science. 2007;318:1461–1464. doi: 10.1126/science.1148481. [DOI] [PubMed] [Google Scholar]; Jeoung JH, Dobbek H. J Am Chem Soc. 2009;131:9922–9923. doi: 10.1021/ja9046476. [DOI] [PubMed] [Google Scholar]
- 17.Zhao ZH, Fan JM, Xie MM, Wang ZZ. J Cleaner Prod. 2009;17:1025–1029. [Google Scholar]
- 18.Indrakanti VP, Kubicki JD, Schobert HH. Energy Environ Sci. 2009;2:745–758. [Google Scholar]
- 19.Reisner E, Powell DJ, Cavazza C, Fontecilla-Camps JC, Armstrong FA. J Am Chem Soc. 2009;131:18457–18466. doi: 10.1021/ja907923r. [DOI] [PubMed] [Google Scholar]
- 20.Rothenberger G, Fitzmaurice D, Grätzel M. J Phys Chem. 1992;96:5983–5986. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.