

Abstract
Modern genomewide association studies are characterized by the problem of “missing heritability.” Epistasis, or genetic interaction, has been suggested as a possible explanation for the relatively small contribution of single significant associations to the fraction of variance explained. Of particular concern to investigators of genetic interactions is how to best represent and define epistasis. Previous studies have found that the use of different quantitative definitions for genetic interaction can lead to different conclusions when constructing genetic interaction networks and when addressing evolutionary questions. We suggest that instead, multiple representations of epistasis, or epistatic “subtypes,” may be valid within a given system. Selecting among these epistatic subtypes may provide additional insight into the biological and functional relationships among pairs of genes. In this study, we propose maximum-likelihood and model selection methods in a hypothesis-testing framework to choose epistatic subtypes that best represent functional relationships for pairs of genes on the basis of fitness data from both single and double mutants in haploid systems. We gauge the performance of our method with extensive simulations under various interaction scenarios. Our approach performs reasonably well in detecting the most likely epistatic subtype for pairs of genes, as well as in reducing bias when estimating the epistatic parameter (ɛ). We apply our approach to two available data sets from yeast (Saccharomyces cerevisiae) and demonstrate through overlap of our identified epistatic pairs with experimentally verified interactions and functional links that our results are likely of biological significance in understanding interaction mechanisms. We anticipate that our method will improve detection of epistatic interactions and will help to unravel the mysteries of complex biological systems.
UNDERSTANDING the nature of genetic interactions is crucial to obtaining a more complete picture of complex biological systems and their evolution. The discovery of genetic interactions has been the goal of many researchers studying a number of model systems, including but not limited to Saccharomyces cerevisiae, Caenorhabditis elegans, and Escherichia coli (You and Yin 2002; Burch et al. 2003; Burch and Chao 2004; Tong et al. 2004; Drees et al. 2005; Sanjuán et al. 2005; Segre et al. 2005; Pan et al. 2006; Zhong and Sternberg 2006; Jasnos and Korona 2007; St. Onge et al. 2007; Decourty et al. 2008). Recently, high-throughput experimental approaches, such as epistatic mini-array profiles (E-MAPs) and genetic interaction analysis technology for E. coli (GIANT-coli), have enabled the study of epistasis on a large scale (Schuldiner et al. 2005, 2006; Collins et al. 2006, 2007; Typas et al. 2008). However, it remains unclear whether the computational and statistical methods currently in use to identify these interactions are indeed the most appropriate.
The study of genetic interaction, or “epistasis,” has had a long and somewhat convoluted history. Bateson (1909) first used the term epistasis to describe the ability of a gene at one locus to “mask” the mutational influence of a gene at another locus (Cordell 2002). The term “epistacy” was later coined by Fisher (1918) to denote the statistical deviation of multilocus genotype values from an additive linear model for the value of a phenotype (Phillips 1998, 2008).
These origins are the basis for the two main current interpretations of epistasis. The first, as introduced by Bateson (1909), is the “biological,” “physiological,” or “compositional” form of epistasis, concerned with the influence of an individual's genetic background on an allele's effect on phenotype (Cheverud and Routman 1995; Phillips 1998, 2008; Cordell 2002; Moore and Williams 2005). The second interpretation, attributed to Fisher, is “statistical” epistasis, which in its linear regression framework places the phenomenon of epistasis in the context of a population (Wagner et al. 1998; Wade et al. 2001; Wilke and Adami 2001; Moore and Williams 2005; Phillips 2008). Each of these approaches is equally valid in studying genetic interactions; however, confusion still exists about how to best reconcile the methods and results of the two (Phillips 1998, 2008; Cordell 2002; Moore and Williams 2005; Liberman and Feldman 2006; Aylor and Zeng 2008).
Aside from the distinction between the statistical and the physiological definitions of epistasis, inconsistencies exist when studying solely physiological epistasis. For categorical traits, physiological epistasis is clear as a “masking” effect. When noncategorical or numerical traits are measured, epistasis is defined as the deviation of the phenotype of the multiple mutant from that expected under independence of the underlying genes.
The “expectation” of the phenotype under independence, that is, in the absence of epistasis, is not defined consistently between studies. For clarity, consider epistasis between pairs of genes and, without loss of generality, consider fitness as the phenotype. The first commonly used definition of independence, originating from additivity, defines the effect of two independent mutations to be equal to the sum of the individual mutational effects. A second, motivated by the use of fitness as a phenotype, defines the effect of the two mutations as the product of the individual effects (Elena and Lenski 1997; Desai et al. 2007; Phillips 2008). A third definition of independence has been referred to as “minimum,” where alleles at two loci are independent if the double mutant has the same fitness as the less-fit single mutant. Mani et al. (2008) claim that this has been used when identifying pairwise epistasis by searching for synthetic lethal double mutants (Tong et al. 2001, 2004; Pan et al. 2004, 2006; Davierwala et al. 2005). A fourth is the “Log” definition presented by Mani et al. (2008) and Sanjuan and Elena (2006). The less-frequently used “scaled ɛ” (Segre et al. 2005) measure of epistasis takes the multiplicative definition of independence with a scaling factor.
These different definitions of independence are partly due to distinct measurement “scales.” For some traits, a multiplicative definition of independence may be necessary to identify epistasis between two genes, whereas for other traits, additivity may be appropriate (Falconer and Mackay 1995; Wade et al. 2001; Mani et al. 2008; Phillips 2008). An interaction found under one independence definition may not necessarily be found under another, leading to different biological conclusions (Mani et al. 2008).
Mani et al. (2008) suggest that there may be an “ideal” definition of independence for all gene pairs for identifying functional relationships. However, it is plausible that different representations of independence for two genes may reflect different biological properties of the relationship (Kupper and Hogan 1978; Rothman et al. 1980). “Two categories of general interest [the additive and multiplicative definitions, respectively] are those in which etiologic factors act interchangeably in the same step in a multistep process, or alternatively act at different steps in the process” (Rothman et al. 1980, p. 468). In some cases, the discovery of epistasis may merely be an artifact of using an incorrect null model (Kupper and Hogan 1978). It may be necessary to represent “independence” differently, resulting in different statistical measures of interactions, for different pairs of genes depending on their functions.
Previous studies have suggested that different pairs of loci may have different modes of interaction and have attempted to subclassify genetic interactions into regulatory hierarchies and mutually exclusive “interaction subtypes” to elucidate underlying biological properties (Avery and Wasserman 1992; Drees et al. 2005; St. Onge et al. 2007). We suggest that epistatic relationships can be divided into several subtypes, or forms, corresponding to the aforementioned definitions of independence. As a particular gene pair may deviate from independence according to several criteria, we do not claim that these subtypes are necessarily mutually exclusive. We attempt to select the most likely epistatic subtype that is the best statistical representation of the relationship between two genes. To further subclassify interactions, epistasis among deleterious mutations can take one of two commonly used forms: positive (equivalently alleviating, antagonistic, or buffering) epistasis, where the phenotype of the double mutant is less severe than expected under independence, and negative (equivalently aggravating, synergistic, or synthetic), where the phenotype is more severe than expected (Segre et al. 2005; Collins et al. 2006; Desai et al. 2007; Mani et al. 2008).
Another objective of such distinctions is to reduce the bias of the estimator of the epistatic parameter (ɛ), which measures the extent and direction of epistasis for a given gene pair. Mani et al. (2008), assuming that the overall distribution of ɛ should be centered around 0, find that inaccurately choosing a definition of independence can result in increased bias when estimating ɛ. For example, using the minimum definition results in the most severe bias when single mutants have moderate fitness effects, and the additive definition results in the largest positive bias when at least one gene has an extreme fitness defect (Mani et al. 2008). Therefore, it is important to select an optimal estimator for ɛ for each pair of genes from among the subtypes of epistatic interactions.
Epistasis may be important to consider in genomic association studies, as a gene with a weak main effect may be identified only through its interaction with another gene or other genes (Frankel and Schork 1996; Culverhouse et al. 2002; Moore 2003; Cordell 2009; Moore and Williams 2009). Epistasis has also been studied extensively in the context of the evolution of sex and recombination. The mutational deterministic hypothesis proposes that the evolution of sex and recombination would be favored by negative epistatic interactions (Feldman et al. 1980; Kondrashov 1994); many other studies have also studied the importance of the form of epistasis (Elena and Lenski 1997; Otto and Feldman 1997; Burch and Chao 2004; Keightley and Otto 2006; Desai et al. 2007; MacCarthy and Bergman 2007). Indeed, according to Mani et al. (2008, p. 3466), “the choice of definition [of epistasis] alters conclusions relevant to the adaptive value of sex and recombination.”
Given fitness data from single and double mutants in haploid organisms, we implement a likelihood method to determine the subtype that is the best statistical representation of the epistatic interaction for pairs of genes. We use maximum-likelihood estimation and the Bayesian information criteria (BIC) (Schwarz 1978) with a likelihood-ratio test to select the most appropriate null or epistatic model for each putative interaction. We conduct extensive simulations to gauge the performance of our method and demonstrate that it performs reasonably well under various interaction scenarios. We apply our method to two data sets with fitness measurements obtained from yeast (Jasnos and Korona 2007; St. Onge et al. 2007), whose authors assume only multiplicative epistasis for all interactions. By examining functional links and experimentally validated interactions among epistatic pairs, we demonstrate that our results are biologically meaningful. Studying a random selection of genes, we find that minimum epistasis is more prevalent than both additive and multiplicative epistasis and that the overall distribution of ɛ is not significantly different from zero (as Jasnos and Korona 2007 suggest). For genes in a particular pathway, we advise selecting among fewer epistatic subtypes. We believe that our method of epistatic subtype classification will aid in understanding genetic interactions and their properties.
METHODS
We consider epistasis among L genes (represented by g1, g2, … , gL) given the phenotypes of each of the possible single- and double-mutant haplotypes, each with a given number of replicates. The phenotype we study is fitness scaled by wild type, obtained either through experiments or through simulation (Schuldiner et al. 2005, 2006; Segre et al. 2005; Collins et al. 2006, 2007; Jasnos and Korona 2007; St. Onge et al. 2007). The fitness of the wild-type genotype g0 is 1, and the relative fitnesses of single mutants gi (1 ≤ i ≤ L) and double mutants gij (1 ≤ i < j ≤ L) are denoted by wi and wij, respectively. Supposing that we have N replicates of fitness values for each mutant, the superscript n represents measurements in the nth replicate (1 ≤ n ≤ N). We denote by 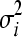 and
and  the variances in fitnesses of single mutants gi and double mutants gij, respectively. We define ɛij as the epistatic coefficient of a genetic interaction between genes gi and gj.
the variances in fitnesses of single mutants gi and double mutants gij, respectively. We define ɛij as the epistatic coefficient of a genetic interaction between genes gi and gj.
Epistatic subtype selection:
Pairwise physiological epistasis for measured numerical traits is defined as the deviation of the observed phenotype of the double mutant from the phenotype expected when the alleles at each locus act independently. As mentioned above, the additive and multiplicative models are widely used to represent independence, while the minimum model is used by investigators searching for synthetic lethal double mutants (Mani et al. 2008). Thus, we focus predominantly on these three models, as do Mani et al. (2008). The Log model is practically equivalent to the multiplicative model for deleterious mutations (Mani et al. 2008); since we generally focus on deleterious mutations, we do not consider this model. We do not include a model based on the scaled ɛ measure of epistasis (Segre et al. 2005), as it was found to be less meaningful than the multiplicative model in describing interactions (St. Onge et al. 2007).
We thus use six models: the three models of independence (additive, multiplicative, and minimum) and the three models (subtypes) of epistasis (additive, multiplicative, and minimum epistasis), which incorporate a deviation from the independence models. We have three major questions. First, when data follow one of the three models of independence, do we incorrectly infer the presence of epistasis? Second, can we detect epistasis between two genes if they interact? Third, can we accurately determine which subtype is the most appropriate for a particular pair of genes if they interact?
The three null models are as follows. If genes gi and gj act additively, i.e., there is no additive epistasis, the null model for the double-mutant fitness in the nth experiment (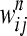 ) is a normal distribution with mean
) is a normal distribution with mean  and variance
and variance 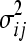 . If gi and gj act multiplicatively,
. If gi and gj act multiplicatively, 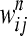 is a normal random variable with mean
is a normal random variable with mean  and variance
and variance 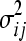 . The third model specifying the absence of epistasis assumes that
. The third model specifying the absence of epistasis assumes that 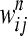 is normal with mean min(
is normal with mean min(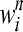 ,
,  ) and variance
) and variance  . We emphasize that the minimum model described here is a model of independence.
. We emphasize that the minimum model described here is a model of independence.
In the presence of additive epistasis ɛij, the probability distribution of the double-mutant fitness wij in the nth experiment, given  ,
, 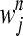 , and variance
, and variance  , is
, is
 |
(1) |
When gi and gj exhibit multiplicative epistasis ɛij,  has probability distribution
has probability distribution
 |
(2) |
This form of multiplicative epistasis is slightly different from that of other authors, who suggest wij = wiwj + ɛij (Segre et al. 2005; Jasnos and Korona 2007; St. Onge et al. 2007; Mani et al. 2008); our formulation is simply a rescaling of this formula. Our motivation for Equation 2 is that it relates more closely to the large body of research representing the fitness of an individual with k mutations as 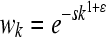 (Elena and Lenski 1997; Otto and Feldman 1997; Wilke and Adami 2001; Desai et al. 2007).
(Elena and Lenski 1997; Otto and Feldman 1997; Wilke and Adami 2001; Desai et al. 2007).
In the case of minimum epistasis,  is also distributed normally:
is also distributed normally:
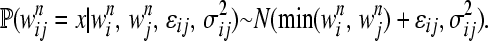 |
(3) |
An epistatic subtype refers to one of the three epistatic models shown in the equations above.
To calculate the likelihood of the observed double-mutant fitnesses, we assume that each pair of genes is independent of all other pairs; i.e., there are no interactions of higher order. The log likelihood of the data based on N replicates is
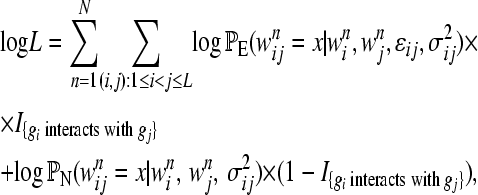 |
(4) |
where I{Φ} is an indicator function and is equal to one if Φ is true and zero otherwise, and PE and PN are the probabilities (given above) under the epistatic and null models, respectively.
Since we assume that each pair of genes is independent of all other pairs, we study each separately. We use the BIC (Schwarz 1978), a commonly used model selection criterion, to determine the most likely interaction subtype, if any, of each pair. To calculate the BIC, we first estimate the parameters ɛij and 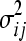 using maximum-likelihood estimation (MLE). For additive epistasis, the ML estimators (MLEs) for ɛij and
using maximum-likelihood estimation (MLE). For additive epistasis, the ML estimators (MLEs) for ɛij and  are
are
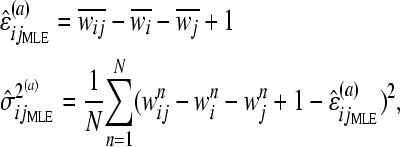 |
(5) |
where  denotes the mean fitness of genotype g· over all N replicates, and the right superscript (a) indicates that the estimator is for additive epistasis. The MLEs for multiplicative and minimum epistasis are given in supporting information, File S1.
denotes the mean fitness of genotype g· over all N replicates, and the right superscript (a) indicates that the estimator is for additive epistasis. The MLEs for multiplicative and minimum epistasis are given in supporting information, File S1.
We select the model that is the best statistical representation of the interaction for each pair of genes using the BIC, defined as
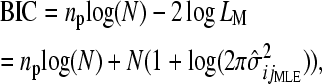 |
(6) |
where LM and np are the maximum likelihood and the number of free parameters, respectively, in the given model. For the three null models, np = 1, as only  is estimated; for the three epistatic models, np = 2, as both
is estimated; for the three epistatic models, np = 2, as both 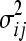 and ɛij are estimated. The model with the smallest BIC is selected as optimal, rewarding models with a higher likelihood but penalizing for additional parameters.
and ɛij are estimated. The model with the smallest BIC is selected as optimal, rewarding models with a higher likelihood but penalizing for additional parameters.
For each pair, we perform a likelihood-ratio test (LRT) of the epistatic model with the lowest BIC against its corresponding null model as an optional step to assess the significance of epistasis. The LRT statistic is given as
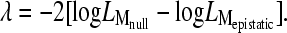 |
(7) |
Under the null distribution, λ is approximately χ2-distributed with 1 d.f. (for the one additional parameter ɛ estimated in the epistatic model).
When identifying epistasis for many gene pairs, the problem of simultaneously testing multiple hypotheses arises. We consider three commonly used multiple-testing corrections, which use the P-values obtained from the LRT above. The first is the Bonferroni correction, which sets the chosen significance level divided by the number of tests (in our case, the number of putatively epistatic gene pairs) as a P-value cutoff. This is generally considered to be the most conservative procedure to control the familywise error rate (the probability of rejecting at least one true null hypothesis among all tests). The second multiple-testing correction, proposed by Benjamini and Hochberg (1995), controls the false discovery rate (FDR, or the expected proportion of null hypotheses that are falsely rejected). Third, we control the positive false discovery rate (pFDR), which is slightly more lenient than the FDR procedure, as suggested by Storey (2002). We advocate determining significance using the FDR, which is intermediate between the other two procedures (see application to experimental data: Estimation of epistatic measures and application to experimental data). We implement our methods in R (www.r-project.org, code available upon request).
Generation of simulated data:
We carried out extensive simulations to assess the performance of our method. For simplicity, we assume that each mutation is deleterious; i.e., the fitnesses of single mutants cannot be greater than that of wild type (w0 = 1). (In Table S1, we consider relaxing this assumption to include beneficial mutations). We sample the fitnesses of single mutants gi and gj from a truncated normal distribution on [0, 1] with mean μ and standard deviation σ; i.e., wi ∼ N(μi, 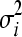 )[0,1] and wj ∼ N(μj,
)[0,1] and wj ∼ N(μj, 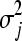 )[0,1]. Although many deletion strains found in nature have a wild-type fitness, we simulate with this support to encompass the entire range of fitness values possible for deleterious mutants. We choose the support for double-mutant fitnesses to be [0, 1.2]; since both synergistic and antagonistic epistasis may be possible, the fitness of double mutants may be >1. We simulate wij given wi and wj following the definitions presented in methods: Epistatic subtype selection. For example, if genes gi and gj act additively and independently, wij ∼ N(wi + wj – 1,
)[0,1]. Although many deletion strains found in nature have a wild-type fitness, we simulate with this support to encompass the entire range of fitness values possible for deleterious mutants. We choose the support for double-mutant fitnesses to be [0, 1.2]; since both synergistic and antagonistic epistasis may be possible, the fitness of double mutants may be >1. We simulate wij given wi and wj following the definitions presented in methods: Epistatic subtype selection. For example, if genes gi and gj act additively and independently, wij ∼ N(wi + wj – 1,  )[0,1.2]; under additive epistasis, wij ∼ N(wi + wj – 1 + ɛij,
)[0,1.2]; under additive epistasis, wij ∼ N(wi + wj – 1 + ɛij,  )[0,1.2]. Distributions under multiplicative and minimum null and epistatic models are given in File S1.
)[0,1.2]. Distributions under multiplicative and minimum null and epistatic models are given in File S1.
Analysis of experimental data:
We analyze two data sets of fitness measurements in yeast (S. cerevisiae) (Jasnos and Korona 2007; St. Onge et al. 2007). In addition to choosing the appropriate epistatic subtype on the basis of the BIC, we perform a LRT to test epistatic models against their corresponding null models and correct for multiple testing with the three methods described in Epistatic subtype selection. For all tests, we select an α significance level of 0.05.
The first data set (St. Onge et al. 2007) included 26 nonessential genes known to confer resistance to the DNA-damaging agent methyl methanesulfonate (MMS) and contained double-deletion strains for all pairs that were possible to construct (323 double-mutant strains). The authors measured the fitnesses of single and double mutants in media both with and without MMS. For both media types, while single mutants have ∼10 replicate fitness measurements, double mutants have only 2. Since our method assumes the same number of replicates for single and double mutants, we perform our method with two sampled single-mutant fitness measurements. We repeat this sampling 1000 times and select an epistatic subtype for the pairs that were chosen as epistatic in at least 900 of the 1000 replicates (see File S1 for details).
In the second study, Jasnos and Korona (2007) performed 639 random crosses among 758 randomly selected gene deletions to create a series of double mutants and assayed the growth fitnesses of each single and double mutant. As an equal number of replicates are available for both single and double mutants (n ≈ 5), we apply our method to this data set directly.
We perform several analyses to validate the use of our method. First, we examine the number of epistatic pairs sharing “specific” functional links, a term used by both St. Onge et al. (2007) and Tong et al. (2004). St. Onge et al. (2007) define these to be genes that share Gene Ontology (GO) terms (Gene Ontology Consortium 2000) that have associated with them <30 genes; Tong et al. (2004) define the cutoff for specificity to be ≤200 associated genes. We examine our data using these and several intermediate cutoffs. Second, we count epistatic pairs sharing the broader GO Slim terms (obtained from the Yeast Genome Database, www.yeastgenome.org, accessed September 11, 2009). To assess the significance of the number of specific functional links or shared GO Slim terms among inferred epistatic pairs, we implement both Fisher's exact test and a permutation procedure (File S1), which produce comparable results. Third, we examine overlap of inferred epistatic pairs with experimentally verified physical interactions in yeast [BIOGRID database (Stark et al. 2006), www.thebiogrid.org, Release 2.0.56; see File S1].
APPLICATION TO SIMULATED DATA
Estimation of epistatic measures:
We perform 10,000 simulations, following the methods described in Generation of simulated data, under each of the six models (three null models and three epistatic models) for each set of parameters shown in Table 1. For each simulation, we sample 50 fitness replicates (N = 50), obtain the BIC for each of the six models, and select the model with the lowest BIC. We compute accuracy as the proportion of simulations in which the model under which the data are simulated is selected.
TABLE 1.
Fractions of simulations that recover the true model (null or epistatic) among the six models (three null models and three epistatic models) with different values of the epistasis coefficient (ɛij), fitness standard deviation (σ = σi = σj = σij), and mean fitnesses of single mutants (μi and μj)
| σ | μi | μj | 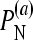 |
 |
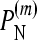 |
ɛij |  |
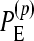 |
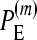 |
|---|---|---|---|---|---|---|---|---|---|
| 0.05 | 0.8 | 0.8 | −0.3 | 0.996 | 0.906 | 0.929 | |||
| 0.05 | 0.8 | 0.8 | −0.2 | 0.992 | 0.902 | 0.958 | |||
| 0.05 | 0.8 | 0.8 | 0.986 | 0.983 | 0.994 | −0.1 | 0.965 | 0.873 | 0.981 |
| 0.05 | 0.8 | 0.8 | 0.1 | 0.739 | 0.720 | 0.997 | |||
| 0.05 | 0.8 | 0.8 | 0.2 | 0.591 | 0.605 | 0.996 | |||
| 0.05 | 0.8 | 0.8 | 0.3 | 0.746 | 0.610 | 0.999 | |||
| 0.05 | 0.9 | 0.6 | −0.3 | 0.999 | 0.958 | 0.978 | |||
| 0.05 | 0.9 | 0.6 | −0.2 | 0.995 | 0.939 | 0.965 | |||
| 0.05 | 0.9 | 0.6 | 0.988 | 0.987 | 0.992 | −0.1 | 0.986 | 0.901 | 0.972 |
| 0.05 | 0.9 | 0.6 | 0.1 | 0.864 | 0.832 | 0.994 | |||
| 0.05 | 0.9 | 0.6 | 0.2 | 0.830 | 0.825 | 0.999 | |||
| 0.05 | 0.9 | 0.6 | 0.3 | 0.888 | 0.821 | 0.999 | |||
| 0.1 | 0.8 | 0.8 | −0.3 | 0.978 | 0.870 | 0.915 | |||
| 0.1 | 0.8 | 0.8 | −0.2 | 0.961 | 0.866 | 0.884 | |||
| 0.1 | 0.8 | 0.8 | 0.955 | 0.952 | 0.993 | −0.1 | 0.953 | 0.758 | 0.896 |
| 0.1 | 0.8 | 0.8 | 0.1 | 0.754 | 0.711 | 0.995 | |||
| 0.1 | 0.8 | 0.8 | 0.2 | 0.689 | 0.618 | 0.997 | |||
| 0.1 | 0.8 | 0.8 | 0.3 | 0.908 | 0.517 | 1.00 | |||
| 0.1 | 0.9 | 0.6 | −0.3 | 0.780 | 0.942 | 0.946 | |||
| 0.1 | 0.9 | 0.6 | −0.2 | 0.947 | 0.910 | 0.945 | |||
| 0.1 | 0.9 | 0.6 | 0.973 | 0.971 | 0.991 | −0.1 | 0.963 | 0.744 | 0.915 |
| 0.1 | 0.9 | 0.6 | 0.1 | 0.833 | 0.731 | 0.987 | |||
| 0.1 | 0.9 | 0.6 | 0.2 | 0.804 | 0.758 | 0.994 | |||
| 0.1 | 0.9 | 0.6 | 0.3 | 0.882 | 0.781 | 0.997 |
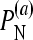 is the fraction of simulations (simulated under the additive null model) with the additive null model attaining the minimum value of the BIC among the six models.
is the fraction of simulations (simulated under the additive null model) with the additive null model attaining the minimum value of the BIC among the six models.  and
and 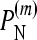 are defined in a manner similar to
are defined in a manner similar to 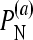 for the multiplicative and minimum models, respectively.
for the multiplicative and minimum models, respectively. 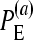 represents the power of our method, i.e., the percentage of simulations (simulated under the additive epistatic model with epistasis coefficient ɛij) with the additive epistatic model attaining the minimum value of the BIC among the six models.
represents the power of our method, i.e., the percentage of simulations (simulated under the additive epistatic model with epistasis coefficient ɛij) with the additive epistatic model attaining the minimum value of the BIC among the six models.  and
and 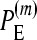 are defined in a manner similar to
are defined in a manner similar to  for the multiplicative and minimum models, respectively. Fifty replicate fitness values are simulated. See Table S6 for a similar table with 10 replicate values.
for the multiplicative and minimum models, respectively. Fifty replicate fitness values are simulated. See Table S6 for a similar table with 10 replicate values.
Table 1 demonstrates that our method can select the correct null or epistatic model for various parameter combinations. When simulating under one of the three null models, our method identifies the correct model with almost 100% accuracy. Overall, the minimum null model is identified with higher accuracy than either the additive or the multiplicative null models. Identification of the minimum epistatic subtype is consistently the most accurate (near 100%), while the accuracy of selecting the additive and multiplicative epistatic subtypes varies with the epistatic coefficient ɛij.
If two single mutants have the same mean fitness, our method is less able to detect multiplicative epistasis than either additive or minimum epistasis (Table 1). Introducing a fairly large difference between single-mutant fitness values slightly increases the accuracy of selecting multiplicative epistasis when data are simulated under this definition. For all epistatic models, the probability of selecting the appropriate model generally increases with the absolute value of ɛij: the larger the absolute value of ɛij, the larger is the difference between the expected double-mutant fitness under the correct model and the other models. We also have little ability to consistently distinguish between the additive and multiplicative models. See also Figure S1 and Figure S2.
Testing hypotheses with the LRT and FDR procedures (Benjamini and Hochberg 1995) gives nearly identical accuracy to that described above (results not shown). With data simulated under the null model, as expected, the FDR slightly decreases the false positive rate; for data simulated under an epistastic model, power decreases by at most 1%. Although the false positive rate decreases when using the Bonferroni correction, we obtain slightly lower power than with the FDR; the pFDR (Storey 2002) does not greatly improve power over the FDR (results not shown). The FDR procedure thus seems preferable.
Bias and variance of the epistatic parameter (ɛ):
Mani et al. (2008) show that using different definitions of independence results in different estimators of the epistatic parameter (ɛ), some with larger biases and variances than others. To evaluate the bias and variance of 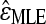 estimated with our method, we perform simulations and compute its mean squared error (MSE) under each epistatic model. Because the FDR procedure does not greatly affect the accuracy of our inference (see above), we do not implement it in these simulations. For the nth simulation,
estimated with our method, we perform simulations and compute its mean squared error (MSE) under each epistatic model. Because the FDR procedure does not greatly affect the accuracy of our inference (see above), we do not implement it in these simulations. For the nth simulation, 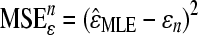 , where ɛn is the true value used in the nth simulation and
, where ɛn is the true value used in the nth simulation and 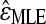 is estimated under the specific epistatic model. If the simulated |ɛ| is large, the MSE of
is estimated under the specific epistatic model. If the simulated |ɛ| is large, the MSE of  is the smallest and is nearly zero when the true epistatic model is selected (Figure S3). When the simulated |ɛ| decreases, we find that while the MSE is always small when the correct model is selected, at times the MSE is even lower under an incorrect model (Figure S3). In general, our method seems to reduce the bias and variance of ɛ.
is the smallest and is nearly zero when the true epistatic model is selected (Figure S3). When the simulated |ɛ| decreases, we find that while the MSE is always small when the correct model is selected, at times the MSE is even lower under an incorrect model (Figure S3). In general, our method seems to reduce the bias and variance of ɛ.
APPLICATION TO EXPERIMENTAL DATA
Epistasis has been studied extensively in yeast (S. cerevisiae), a model organism for which generating a large number of mutants and examining growth phenotypes are experimentally tractable. Of interest are studies measuring single- and double-mutant fitnesses on a continuous scale. With our proposed method, we reanalyze two data sets by detecting epistasis and its subtype for each pair of mutants (Jasnos and Korona 2007; St. Onge et al. 2007).
St. Onge et al. (2007) data set:
St. Onge et al. (2007) examined 26 nonessential genes known to confer resistance to MMS, constructed double-deletion strains for 323 double-mutant strains (all but two of the total possible pairs), and assumed the multiplicative form of epistasis for all interactions (see Methods: Analysis of experimental data). Following these authors, we focus on single- and double-mutant fitnesses measured in the presence of MMS. (For results in the absence of MMS, see File S1 and File S1_2.)
Using the resampling method described in Analysis of experimental data and File S1, 222 gene pairs pass the cutoff of having epistasis inferred in at least 900 of 1000 replicates. This does not include 5 synthetic lethal gene pairs. Hypothesis testing and a multiple-testing procedure (for 222 simultaneous hypotheses) are necessary to determine the final epistatic pairs.
To select one among the three multiple-testing procedures, we follow St. Onge et al. (2007) and examine gene pairs that share specific functional links (see Analysis of experimental data). The Bonferroni method is likely too conservative, yielding only 25 significantly epistatic pairs with only one functional link among them; alternatively, the pFDR procedure appears to be too lenient in rejecting independence for all 222 pairs. Therefore, we use the FDR procedure (although the number of functional links is not significant) and detect 193 epistatic pairs, of which 5 (2.6%) are synthetic lethals, 19 (9.8%) have additive epistasis, 33 (17.1%) have multiplicative epistasis, and 136 (70.5%) have minimum epistasis (Table 2, complete list in File S1_1). We find 29 gene pairs with positive (alleviating) epistasis and 159 gene pairs with negative (aggravating) epistasis.
TABLE 2.
Summary of gene pairs with the indicated epistatic subtypes, inferred using the FDR procedure with the BIC method that considers all three epistatic subtypes and their corresponding null models
| Epistatic subtype | Study S | Study J |
|---|---|---|
| All | 193 (100%) | 352 (100%) |
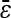 = −0.060 = −0.060 |
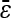 = −0.001 = −0.001 |
|
 = −0.096 = −0.096 |
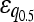 = −0.059 = −0.059 |
|
| Additive | 19 (9.8%) | 35 (9.9%) |
 = 0.115* = 0.115* |
 = 0.193*** = 0.193*** |
|
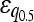 = 0.131 = 0.131 |
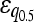 = 0.188 = 0.188 |
|
| Multiplicative | 33 (17.1%) | 63 (17.9%) |
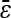 = 0.048 = 0.048 |
 = 0.017 = 0.017 |
|
 = −0.166 = −0.166 |
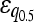 = −0.115 = −0.115 |
|
| Minimum | 136 (70.5%) | 254 (72.2%) |
 = −0.111*** = −0.111*** |
 = −0.032** = −0.032** |
|
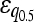 = −0.091 = −0.091 |
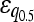 = −0.065 = −0.065 |
Numbers are the counts of each type, and percentages are given of the total number of epistatic pairs. The mean (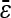 ) and median (
) and median (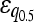 ) of the epistatic parameter (ɛ) are given for each subtype, with “*” indicating that the mean of ɛ is significantly different from 0 (*, P-value ≤0.05; **, P-value ≤0.01; ***, P-value ≤0.001). Study S refers to the St. Onge et al. (2007) data set, and study J refers to the Jasnos and Korona (2007) data set. (For study S, five of the epistatic pairs are synthetic lethals and are not shown; as a result, percentages do not sum to 100%.)
) of the epistatic parameter (ɛ) are given for each subtype, with “*” indicating that the mean of ɛ is significantly different from 0 (*, P-value ≤0.05; **, P-value ≤0.01; ***, P-value ≤0.001). Study S refers to the St. Onge et al. (2007) data set, and study J refers to the Jasnos and Korona (2007) data set. (For study S, five of the epistatic pairs are synthetic lethals and are not shown; as a result, percentages do not sum to 100%.)
To further validate the use of our method and the FDR procedure, we assess by Fisher's exact test the significance of an enrichment of both Biological Process and all GO Slim term links among epistatic pairs, neither of which are significant (Gene Ontology Consortium 2000; www.yeastgenome.org; Table 3). Our discovered epistatic interactions do overlap with experimentally verified physical interactions [BIOGRID database (Stark et al. 2006); Table 3 and Table S4]. Although some of the previously unidentified interactions that we identify could be false positives, many are likely to be new discoveries.
TABLE 3.
Comparison of validation measures for each data set for different variations of the FDR and BIC procedures, considering only a subset of epistatic subtypes with their corresponding null models: all epistatic subtypes (A, P, and M); only the additive and multiplicative subtypes (A and P); and only the additive (A), only the multiplicative (P), or only the minimum (M) subtype (see text for details)
| Subtypes considered in BIC procedure |
|||||
|---|---|---|---|---|---|
| A, P, M | A, P | A | P | M | |
| Study J | |||||
| No. found (636) | 352 | 273 | 263 | 231 | 329 |
| Functional links (25) | 19 (0.0255)* | 13 (0.2320) | 11 (0.4689) | 10 (0.4227) | 15 (0.2619) |
| GO Slim terms (Biological Process) (115) | 69 (0.1573) | 50 (0.4874) | 55 (0.0736) | 44 (0.3534) | 68 (0.04902)* |
| GO Slim terms (all) (369) | 224 (0.0009)* | 172 (0.01654)* | 160 (0.1297) | 146 (0.0273)* | 213 (0.0003)* |
| Experimentally identified (3) | 3 | 2 | 1 | 2 | 3 |
| Study S | |||||
| No. found (323) | 193 | 192 | 247 | 171 | 243 |
| Functional links (36) | 21 (0.6450) | 29 (0.0041)* | 34 (0.0031)* | 29 (0.0003)* | 24 (0.9256) |
| GO Slim terms (Biological Process) (283) | 174 (0.0657) | 174 (0.03656)* | 223 (0.0010)* | 153 (0.1825) | 213 (0.5534) |
| GO Slim terms (all) (307) | 185 (0.2866) | 182 (0.6926) | 237 (0.1472) | 162 (0.6997) | 231 (0.5908) |
| Experimentally identified (29) | 17 | 22 | 24 | 23 | 21 |
Numbers in parentheses indicate P-values by Fisher's exact test. “*” indicates significance. Study J refers to the Jasnos and Korona (2007) data set, and study S refers to the St. Onge et al. (2007) data set measured in the presence of MMS. Numbers in parentheses indicate the total number of tested pairs and the total number of each type of link found in each complete data set.
The epistatic subtypes we consider are not necessarily mutually exclusive. To more fully assess the assumptions of our method, we also consider several of the possible subsets of the epistatic subtypes (and their corresponding null models) in our procedure. As the minimum epistatic subtype was the most frequently selected in this data set, we first do not include the minimum null model or the minimum epistatic model in our procedure (i.e., we select from among four rather than six models for a pair; Table 3, column 3). In Table 3, we also examine our method's performance when it selects from only additive epistasis or independence (column 4), only multiplicative epistasis and independence (column 5), or only minimum epistasis and independence (column 6). Allowing for different subsets of subtypes does not greatly affect the number of inferred epistatic pairs with experimentally identified interactions (Table 3, Table S4). However, there are a significant number of epistatic pairs with functional links only when the minimum epistatic subtype is not included (also see Table S4 and Table S5). It is not immediately clear which epistatic subtypes are the most appropriate for these data, although including the minimum subtype may not be appropriate (Mani et al. 2008) (see discussion).
Although it may be best to consider fewer epistatic subtypes for this specific data set, we report our results including all three epistatic subtypes and their corresponding null models (Table 2). Our method identifies epistasis for 88 of the same pairs as St. Onge et al. (2007), although we identify 105 epistatic pairs not identified by the original authors (Figure S4, Table S4). St. Onge et al. (2007) find that epistatic pairs with a functional link have a positively shifted distribution of epistasis. We find no such shift in epistasis values (Table 2, Figure S5). We also demonstrate [described in application to simulated data: Bias and variance of the epistatic parameter (ɛ)] that our method seems to reduce bias of the epistatic parameter (ɛ) (Table 2). [Note that in the absence of MMS, we find that 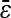 is negative and significantly different from 0 (Table S3).] When considering only a subset of the epistatic subtypes, however, we find
is negative and significantly different from 0 (Table S3).] When considering only a subset of the epistatic subtypes, however, we find 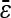 to be positive and significantly different from zero (results not shown). See File S1, Figure S6, and Figure S7 for additional discussion of the epistatic pairs we identify.
to be positive and significantly different from zero (results not shown). See File S1, Figure S6, and Figure S7 for additional discussion of the epistatic pairs we identify.
Jasnos and Korona (2007) data set:
The Jasnos and Korona (2007) data set included 758 yeast gene deletions known to cause growth defects and reports fitnesses of only a sparse subset of all possible gene pairs [≈0.2% of the possible pairwise genotypes, or 639 pairs of  ]. Because the authors do not identify epistatic pairs in a hypothesis-testing framework, we cannot explicitly compare our conclusions with theirs.
]. Because the authors do not identify epistatic pairs in a hypothesis-testing framework, we cannot explicitly compare our conclusions with theirs.
To validate our method, we examine gene pairs that have specific functional links (see methods: Analysis of experimental data). When defining a functional link using GO terms (Gene Ontology Consortium 2000) with <30 genes associated with them, only 1 of 639 tested gene pairs has a functional link. Raising the threshold of associated genes to 50 and 100, the number of tested pairs with functional links rises only to 3 and 9, respectively. Because of the large number of random genes and the sparse number of gene pairs in this data set, we follow Tong et al. (2004) and select GO terms that have associated with them ≤200 genes. Twenty-five of 639 tested pairs then have a functional link.
Only the FDR multiple-testing procedure results in a significant enrichment of functional links among epistatic pairs (Table 3); thus, it is likely the most appropriate of the three multiple-testing methods (full results in File S1). With the FDR procedure we find 352 significant epistatic pairs, of which 35 (9.9%) have additive epistasis, 63 (17.9%) have multiplicative epistasis, and 254 (72.2%) have minimum epistasis (Table 2; for a complete list, see File S1_3). These proportions of inferred subtypes suggest that the authors' original restriction to multiplicative epistasis may be inappropriate. We find 141 gene pairs with positive epistasis and 211 gene pairs with negative epistasis.
We do not find a significant number of epistatic pairs with shared GO Slim Biological Process terms (see Analysis of experimental data), but do when considering all shared GO Slim terms (Table 3). However, the significant enrichment of functional links among epistatic pairs suggests that the epistasis found with the BIC and FDR procedures is valid and of biological significance. We also find an overlap of epistatic pairs with experimentally validated physical interactions (from the BIOGRID database).
As with the St. Onge et al. (2007) data set, we also consider some of the possible subsets of the three epistatic subtypes (and their corresponding null models) in our model selection procedure (Table 3; see also Table S5). In contrast to the St. Onge et al. (2007) data set, using all three epistatic subtypes results in a significant number of epistatic pairs with functional links; this measure is not significant when using any of the other subsets of the subtypes. This suggests that our proposed method with three epistatic subtypes may indeed be the most appropriate for data sets with randomly selected genes.
We examined the distribution of the estimated values of the epistatic parameter (ɛ) for all pairs with significant epistasis. Jasnos and Korona (2007), in assuming only multiplicative epistasis, conclude that epistasis is predominantly positive. However, we find that the estimated mean of epistasis is not significantly different from zero (two-sided t-test, P-value = 0.9578; Figure 1 and Table 2). This also supports that our method reduces bias of the estimator of ɛ. We report in further detail on the epistatic pairs identified in File S1.
Figure 1.—

Distribution of the epistasis values (ɛ) for significant epistatic pairs in the Jasnos and Korona (2007) data set, determined using the FDR procedure and the BIC method including all three epistatic subtypes and their corresponding null models. Mean of ɛ is −0.0009, with a standard deviation of 0.3177; median value is −0.0587. A similar plot is shown in Figure 3 of Jasnos and Korona (2007).
DISCUSSION
We present a likelihood estimation method using the BIC, which, given fitness data from single and double mutants, determines the most appropriate epistatic subtype for pairs of genes that interact. Through extensive simulations, we demonstrate that selection of the epistatic subtype is extremely important and that our method can indeed do so accurately. We demonstrate through the analysis of two data sets that the epistasis we discover is likely biologically meaningful. Besides the detection of genetic interactions and clarification of their functions, accurate assessment of epistasis is also important for evolutionary theory. For example, the distribution of epistatic effects has important implications for the role of epistasis in the evolution of sex and recombination. We demonstrate that our method reduces bias of the epistatic parameter ɛ.
Our approach is novel for several reasons. While in reality different subtypes and representations of epistasis may be best for different pairs of loci, classical studies have usually assumed only one subtype of epistasis for all pairs. Our method selects the most likely subtype for pairs of genes, providing a more precise picture of the loci actually exhibiting epistasis and insight into their modes of genetic interaction. Second, some past studies of epistasis have not clearly distinguished between the epistatic and independence models. Jasnos and Korona (2007), for example, calculate values of ɛ without formally assessing whether the perceived epistasis is merely statistical noise. With the presented BIC and LRT procedures, we distinguish between the epistatic and null models in a hypothesis-testing framework and address the multiple-hypothesis testing problem with the FDR (Benjamini and Hochberg 1995).
Applying our method to two yeast data sets (Jasnos and Korona 2007; St. Onge et al. 2007), we detect both novel and experimentally verified epistatic interactions. We believe that an enrichment of specific functional links among epistatic pairs is likely the most direct indication of biological importance of inferred epistasis (Tong et al. 2004; St. Onge et al. 2007), as genetic interactions probably occur on a fine biological scale (see methods: Analysis of experimental data).
Including all three epistatic subtypes and their corresponding null models in our procedure results in a significant enrichment of functional links among identified epistatic pairs for the Jasnos and Korona (2007) data set, but not the St. Onge et al. (2007) data set (both with and without MMS). Performance for the St. Onge et al. (2007) data set is improved when only the additive or the multiplicative subtypes, or both, are considered (Table 3). This apparent difference between data sets may be due to the fact that the St. Onge et al. (2007) data set includes genes known to be involved in conferring resistance to MMS, whereas the Jasnos and Korona (2007) data set measured a random selection of genes and gene pairs. In examining epistasis among genes with similar functions, it thus may be more appropriate to consider fewer epistatic subtypes. This difference between the data sets may also be due to the low sample size for double mutants in the St. Onge et al. (2007) data set.
We find predominantly minimum epistasis, i.e., a deviation from the minimum null model, when considering all three subtypes in both data sets. This is likely valid for the Jasnos and Korona (2007) data set, but it remains unclear which subtypes are the most appropriate for the St. Onge et al. (2007) data set. Mani et al. (2008) suggest that the minimum definition of independence may not always be useful. Examining another data set with randomly selected genes and gene pairs will help to assess the performance of our method.
The slight overlap of epistatic pairs inferred using different subsets of the three subtypes emphasizes that the subtypes we select are not necessarily mutually exclusive (Table 3). We claim to select the statistically best-fitting epistatic subtype for pairs of genes to gain additional insight into their mode of interaction. For example, a double mutant can deviate from both additive and multiplicative independence. Selecting the additive epistatic subtype with our method suggests that additive epistasis may be the more appropriate and useful way to represent the relationship between the two genes. The different results obtained considering different subtypes again emphasizes the importance of selecting among them (Mani et al. 2008).
While Jasnos and Korona (2007) find a skew toward positive epistasis, we find estimates of epistasis to be centered around zero (Table 2). For both data sets, however, the mean of additive epistasis (ɛ) is positive and significantly different from 0, whereas the mean of minimum epistasis (ɛ) is negative and significantly different from 0 (Table 2). Applying our method to data sets with a larger number of genes may provide a more accurate view of the types and strengths of epistasis often found in nature.
Inference about epistatic subtypes is affected by many factors, including the number of replicate experiments and the variance of mutant fitnesses. Accuracy clearly increases with the number of replicate measurements, but in practice <10 replications are performed (Jasnos and Korona 2007; St. Onge et al. 2007). We assess our method's accuracy when the number of replicates is decreased from 50 to 10 and to 2 (Table S2 and Table S6). As expected, accuracy decreases, but varies depending on the type of epistasis examined. Our failure to identify epistasis for several experimentally identified interacting pairs in the St. Onge et al. (2007) data set is likely due to our low power with only two double-mutant fitness measurements.
A large variance in mutant fitnesses also decreases the accuracy of our method. Our simulations use 0.05 as the standard deviation of mutant fitnesses, which is the typical value found for single and double mutants in the Jasnos and Korona (2007) data set. To assess the effect of variance of fitness on accuracy, we ran simulations with the standard deviation increased to 0.1; this slightly reduces the ability to distinguish the true epistatic model from the others (Table 1).
We also address the approximation of the distribution of the LRT statistic (λ) to the 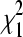 -distribution under the null hypothesis. For both data sets, very few functional links exist among epistatic pairs inferred with the Bonferroni method. This indicates that epistatic pairs with functional links have somewhat larger P-values and are significant only under less stringent multiple-testing protocols. The χ2-approximation may not always be appropriate, as the measured fitnesses sometimes deviate from normality in both data sets (Figure S8 and Figure S9). This may also explain our inability to detect some experimentally validated interactions. We urge measuring a large number of replicates to meet the normality requirement and to increase the power of identifying epistatic interactions.
-distribution under the null hypothesis. For both data sets, very few functional links exist among epistatic pairs inferred with the Bonferroni method. This indicates that epistatic pairs with functional links have somewhat larger P-values and are significant only under less stringent multiple-testing protocols. The χ2-approximation may not always be appropriate, as the measured fitnesses sometimes deviate from normality in both data sets (Figure S8 and Figure S9). This may also explain our inability to detect some experimentally validated interactions. We urge measuring a large number of replicates to meet the normality requirement and to increase the power of identifying epistatic interactions.
Our method is not limited to studying fitness as a phenotype and can be applied to any trait, although it is necessary to scale the phenotype of interest by the wild-type value. Our likelihood-based method is applicable only to studies of experimental data where multiple replicates are measured; it is not immediately applicable to studies of computationally predicted fitness values, with only one measurement (Segre et al. 2005). However, adding random noise terms to each predicted value to create multiple “replicates” could allow our method to be applied in the future. In addition, it is important to note that the epistasis identified with this method is a property of the alleles at each locus; it is conceivable that when examining other pairs of alleles at the tested loci, different results could be obtained. We also note that our method is applicable only to haploid systems. Identifying epistasis and constructing double mutants is much more difficult in diploid systems, as more complex statistical issues arise due to dominance and other effects (Otto and Feldman 1997; Wagner et al. 1998; Gregersen et al. 2006; Li et al. 2007; Flint and Mackay 2009). Since current methods to identify epistasis in haploid systems are still in their infancy, it is important to first develop and perfect these methods before addressing diploid systems.
Our method is one in a wide research field studying genetic interactions and in a long history of the study of epistasis (Hartman and Tippery 2004; Bansal et al. 2007; Roguev et al. 2008; Chipman and Singh 2009). As mentioned above, our method leaves many open questions. Not all of the genetic interactions that we find may translate into physical interactions, and many researchers have pursued the question of how genetic and protein–protein interactions are related (Lee et al. 2004; Wong et al. 2005; Schwartz et al. 2009). The translation of discoveries of epistasis into discoveries of functional interactions is a very intriguing, albeit complex, problem. We expect that our method will shed light on intrinsic properties of epistasis and may help to address these broader issues.
Acknowledgments
We thank Elhanan Borenstein for his helpful comments and suggestions, as well as two anonymous reviewers and the editor of Genetics. This research project is supported by National Institutes of Health (NIH) grant GM28016 to M.W.F., NIH grant R01 GM073059 to Hua Tang, and a Stanford Dean's postdoctoral fellowship to H.G. J.M.G. is supported by the Stanford Genome Training Program, which is funded by the NIH (academic year 2008–2009), and by the National Science Foundation Graduate Research Fellowship (2009 to present).
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.109.111120/DC1.
References
- Avery, L., and S. Wasserman, 1992. Ordering gene function: the interpretation of epistasis in regulatory hierarchies. Trends Genet. 8 312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylor, D., and Z. Zeng, 2008. From classical genetics to quantitative genetics to systems biology: modeling epistasis. PLoS Genet. 4 e1000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal, M., V. Belcastro, A. Ambesi-Impiombato and D. di Bernardo, 2007. How to infer gene networks from expression profiles. Mol. Syst. Biol. 3 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateson, W., 1909. Mendel's Principles of Heredity. Cambridge University Press, Cambridge, UK/London/New York.
- Benjamini, Y., and Y. Hochberg, 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57 289–300. [Google Scholar]
- Burch, C., and L. Chao, 2004. Epistasis and its relationship to canalization in the RNA virus phi6. Genetics 167 559–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burch, C., P. Turner and K. Hanley, 2003. Patterns of epistasis in RNA viruses: a review of the evidence from vaccine design. J. Evol. Biol. 16 1223–1235. [DOI] [PubMed] [Google Scholar]
- Cheverud, J. M., and E. J. Routman, 1995. Epistasis and its contribution to genetic variance components. Genetics 139 1455–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipman, K. C., and A. J. Singh, 2009. Predicting genetic interactions with random walks on biological networks. BMC Bioinformatics 10 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, S. R., M. Schuldiner, N. J. Krogan and J. S. Weissman, 2006. A strategy for extracting and analyzing large-scale quantitative epistatic interaction data. Genome Biol. 7 R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, S. R., K. M. Miller, N. L. Maas, A. Roguev, J. Fillingham et al., 2007. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature 446 806–810. [DOI] [PubMed] [Google Scholar]
- Cordell, H. J., 2002. Epistasis: what it means, what it doesn't mean, and statistical methods to detect it in humans. Hum. Mol. Genet. 11 2463–2468. [DOI] [PubMed] [Google Scholar]
- Cordell, H. J., 2009. Detecting gene-gene interactions that underlie human diseases. Nat. Rev. Genet. 10 392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culverhouse, R., B. Suarez, J. Lin and T. Reich, 2002. A perspective on epistasis: limits of models displaying no main effect. Am. J. Hum. Genet. 70 461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davierwala, A., J. Haynes, Z. Li, R. Brost, M. D. Robinson et al., 2005. The synthetic genetic interaction spectrum of essential genes. Nat. Genet. 37 1147–1152. [DOI] [PubMed] [Google Scholar]
- Decourty, L., C. Saveanu, K. Zemam, F. Hantraye, E. Frachon et al., 2008. Linking functionally related genes by sensitive and quantitative characterization of genetic interaction profiles. Proc. Natl. Acad. Sci. USA 105 5821–5826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai, M., D. Weissman and M. Feldman, 2007. Evolution can favor antagonistic epistasis. Genetics 177 1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drees, B. L., V. Thorsson, G. W. Carter, A. W. Rives, M. Z. Raymond et al., 2005. Derivation of genetic interaction networks from quantitative phenotype data. Genome Biol. 6 R38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elena, S., and R. Lenski, 1997. Test of synergistic interactions among deleterious mutations in bacteria. Nature 390 395–398. [DOI] [PubMed] [Google Scholar]
- Falconer, D., and T. Mackay, 1995. Introduction to Quantitative Genetics. Longman, Harlow, UK. [DOI] [PMC free article] [PubMed]
- Feldman, M., F. Christiansen and L. Brooks, 1980. Evolution of recombination in a constant environment. Proc. Natl. Acad. Sci. USA 77 4838–4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, R., 1918. The correlations between relatives on the supposition of Mendelian inheritance. Trans. R. Soc. Edinb. 52 399–433. [Google Scholar]
- Flint, J., and T. F. C. Mackay, 2009. Genetic architecture of quantitative traits in mice, flies, and humans. Genome Res. 19 723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel, W., and N. Schork, 1996. Who's afraid of epistasis? Nat. Genet. 14 371–373. [DOI] [PubMed] [Google Scholar]
- Gene Ontology Consortium, 2000. Gene ontology: tool for the unification of biology. Nat. Genet. 25 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregersen, J. W., K. R. Kranc, X. Ke, P. Svendsen, L. S. Madsen, et al., 2006. Functional epistasis on a common MHC haplotype associated with multiple sclerosis. Nature 443 574–577. [DOI] [PubMed] [Google Scholar]
- Hartman, J., and N. Tippery, 2004. Systematic quantification of gene interactions by phenotypic array analysis. Genome Biol. 5 R49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasnos, L., and R. Korona, 2007. Epistatic buffering of fitness loss in yeast double deletion strains. Nat. Genet. 39 550–554. [DOI] [PubMed] [Google Scholar]
- Keightley, P. D., and S. P. Otto, 2006. Interference among deleterious mutations favours sex and recombination in finite populations. Nature 443 89–92. [DOI] [PubMed] [Google Scholar]
- Kondrashov, A., 1994. Muller's ratchet under epistatic selection. Genetics 136 1469–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupper, L., and M. Hogan, 1978. Interaction in epidemiologic studies. Am. J. Epidemiol. 108 447–453. [DOI] [PubMed] [Google Scholar]
- Lee, I., S. V. Date, A. T. Adai and E. M. Marcotte, 2004. A probabilistic functional network of yeast genes. Science 306 1555–1558. [DOI] [PubMed] [Google Scholar]
- Li, H., G. Gao, J. Li, G. P. Page and K. Zhang, 2007. Detecting epistatic interactions contributing to human gene expression using the CEPH family data. BMC Proc. 1(Suppl. 1):S67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman, U., and M. W. Feldman, 2006. Evolutionary theory for modifiers of epistasis using a general symmetric model. Proc. Natl. Acad. Sci. USA 103 19402–19406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCarthy, T., and A. Bergman, 2007. Coevolution of robustness, epistasis, and recombination favors asexual reproduction. Proc. Natl. Acad. Sci. USA 104 12801–12806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani, R., R. P. St. Onge, J. L. I. Hartman, G. Giaever and F. P. Roth, 2008. Defining genetic interaction. Proc. Natl. Acad. Sci. USA 105 3461–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, J., 2003. The ubiquitous nature of epistasis in determining susceptibility to common human diseases. Hum. Hered. 56 73–82. [DOI] [PubMed] [Google Scholar]
- Moore, J., and S. Williams, 2005. Traversing the conceptual divide between biological and statistical epistasis: systems biology and a more modern synthesis. BioEssays 27 637–646. [DOI] [PubMed] [Google Scholar]
- Moore, J., and S. Williams, 2009. Epistasis and its implications for personal genetics. Am. J. Hum. Genet. 85 309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto, S. P., and M. Feldman, 1997. Deleterious mutations, variable epistatic interactions, and the evolution of recombination. Theor. Popul. Biol. 51 134–147. [DOI] [PubMed] [Google Scholar]
- Pan, X., D. Yuan, D. Xiang, X. Wang, S. Sookhai-Mahadeo et al., 2004. A robust toolkit for functional profiling of the yeast genome. Mol. Cell 16 487–496. [DOI] [PubMed] [Google Scholar]
- Pan, X., P. Ye, D. Yuan, X. Wang, J. Bader et al., 2006. A DNA integrity network in the yeast Saccharomyces cerevisiae. Cell 124 1069–1081. [DOI] [PubMed] [Google Scholar]
- Phillips, P., 1998. The language of gene inetraction. Genetics 149 1167–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips, P., 2008. Epistasis—the essential role of gene interactions in the structure and evolution of genetic systems. Nat. Rev. Genet. 9 855–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roguev, A., S. Bandyopadhyay, M. Zofall, K. Zhang, T. Fischer et al., 2008. Conservation and rewiring of functional modules revealed by an epistasis map in fission yeast. Science 322 405–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman, K., S. Greenland and A. Walker, 1980. Concepts of interaction. Am. J. Epidemiol. 112 467–470. [DOI] [PubMed] [Google Scholar]
- Sanjuán, R., and S. Elena, 2006. Epistasis correlates to genomic complexity. Proc. Natl. Acad. Sci. USA 103 104402–104405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuán, R., J. Cuevas, A. Moya and S. Elena, 2005. Epistasis and the adaptability of an RNA virus. Genetics 170 1001–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuldiner, M., S. Collins, N. Thompson, V. Denic, A. Bhamidipati et al., 2005. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell 123 507–519. [DOI] [PubMed] [Google Scholar]
- Schuldiner, M., S. Collins, J. Weissman and N. Krogan, 2006. Quantitative genetic analysis in Saccharomyces cerevisiae using epistatic miniarray profiles (E-MAPs) and its application to chromatin functions. Methods 40 344–352. [DOI] [PubMed] [Google Scholar]
- Schwartz, A. S., J. Yu, K. R. Gardenour, R. L. Finley, Jr. and T. Ideker, 2009. Cost-effective strategies for completing the interactome. Nat. Methods 6 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, G., 1978. Estimating the dimension of a model. Ann. Stat. 6 461–464. [Google Scholar]
- Segre, D., A. DeLuna, G. Church and R. Kishony, 2005. Modular epistasis in yeast metabolism. Nat. Genet. 37 77–83. [DOI] [PubMed] [Google Scholar]
- St. Onge, R. P., R. Mani, J. Oh, M. Proctor, E. Fung et al., 2007. Systematic pathway analysis using high-resolution fitness profiling of combinatorial gene deletions. Nat. Genet. 39 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark, C., B. Breitkreutz, T. Reguly, L. Boucher, A. Breitkreutz et al., 2006. Biogrid: a general repository for interaction datasets. Nucleic Acids Res. 34 D535–D539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey, J., 2002. A direct approach to false discovery rates. J. R. Stat. Soc. B 64(part 3): 479–498. [Google Scholar]
- Tong, A., M. Evangelista, A. Parsons, H. Xu, G. Bader et al., 2001. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294 2364–2368. [DOI] [PubMed] [Google Scholar]
- Tong, A., G. Lesage, G. Bader, H. Ding, H. Xu et al., 2004. Global mapping of the yeast genetic interaction network. Science 303 808–813. [DOI] [PubMed] [Google Scholar]
- Typas, A., R. Nichols, D. Siegele, M. Shales, S. R. Collins et al., 2008. High-throughput quantitative analyses of genetic interactions in E. coli. Nat. Methods 5 781–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade, M., R. Winther, A. Agrawal and C. Goodnight, 2001. Alternative definitions of epistasis: independence and interaction. Trends Ecol. Evol. 16 498–504. [Google Scholar]
- Wagner, G. P., M. D. Laubichler and H. Bagheri-Chaichian, 1998. Genetic measurement theory of epistatic effects. Genetics 102 569–580. [PubMed] [Google Scholar]
- Wilke, C. O., and C. Adami, 2001. Interaction between directional epistasis and average mutational effects. Proc. R. Soc. Lond. Ser. B Biol. Sci. 268 1469–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, S., L. Zhang and F. P. F. Roth, 2005. Discovering functional relationships: biochemistry versus genetics. Trends Genet. 21 424–427. [DOI] [PubMed] [Google Scholar]
- You, L., and J. Yin, 2002. Dependence of epistasis on environment and mutation severity as revealed by in silico mutagenesis of phage T7. Genetics 160 1273–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, W., and P. Sternberg, 2006. Genome-wide prediction of C. elegans genetic interactions. Science 311 1481–1484. [DOI] [PubMed] [Google Scholar]