

Abstract
Mitofusin 2 (Mfn2) has been proposed as an important mitochondrial protein in maintaining mitochondrial network and bioenergetics. Mfn2 is highly expressed in the heart, but is downregulated in response to hypertrophic stimuli. However, little is known about how Mfn2’s expression is regulated in cardiomyocytes. Here, we have investigated how Mfn2 expression in the heart responds to fasting condition and determined if Mfn2 is one of those PPARδ-selective target genes that are involved in myocardial energy metabolism. Fasting for 48 hours in mice led to a robust increase of Mfn2 expression in the heart. On the other hand, cardiomyocyte-restricted PPARδ deficiency in mice led to substantially diminished cardiac expression of Mfn2 transcript and protein compared to that of controls. Fasting induced cardiac expression of Mfn2 was blunted in cardiomyocyte-restricted PPARδ deficient hearts. Moreover, PPARδ-selective ligand treatment in cultured cardiomyocytes induced elevated Mfn2 expression. A functional PPRE consensus sequence located at −837 to −817bp upstream of the mouse Mfn2 promoter was identified and confirmed by Electrophoretic Mobility Shift Assays and Luciferase Promoter Reporter Assays. We conclude that Mfn2 is a PPARδ-selective target, which may play an important role in regulating myocardial energy homeostasis.
Introduction
Mitochondria are essential organelles of eukaryotic cells with their capability in bioenergy generation. The heart, as a continuously working pump, is the most energy demanding organ in the body. Under normal conditions, it has been estimated that a cardiomyocyte contains more than 1,000 mitochondria [1]. Mitochondria in cardiomyocytes appear in a uniform fashion and form a network along the entire length of the myofibrillar apparatus. Mitochondria in a cardiomyocyte play a critical role in cardiac cellular energy metabolism, calcium homeostasis, biogenesis, and regulation of cell death. Mitochondrial dysfunction has been identified in majority of cardiac pathologies and the implication is complex without fully understood.
An emerging group of proteins plays crucial roles in regulating the mitochondrial network and dynamics. Mitofusins (Mfn) 1 and 2 are two of these proteins located on the outer membrane of mitochondria and involved in the dynamic formation of the mitochondrial network [2, 3]. They are the homologs of Fzo, a GTPase first discovered in D. Melanogaster [4] [5]. Knockout mouse studies unraveled that both Mfns are required for proper development ([6]). However, studies on biochemical and morphological evidences indicate that these two proteins are not functionally redundant [6, 7]. Mfn2 is highly expressed in heart, skeletal muscle and brain [8] [6] [9]. Mfn2 participates in mitochondrial fusion and contributes to the maintenance and operation of the mitochondrial network [8] [6]. Mutations in Mfn2 were reported in patients with Charco-Marie-Tooth neuropathy type 2A [10] [11]. In Drosophila, Mfn2 deficiency leads to aberrant sperm, “fuzzy onion”-like mitochondria [5].
It has been shown that repression of Mfn2 reduces glucose oxidation, mitochondrial membrane potential, and cell respiration in L6E9 myotubes [8]. The importance of Mfn2 in the heart is recently evident by a study showing that upregulation of Mfn2 is involved in H2O2-induced apoptosis in cultured rat neonatal cardiomyocytes [12]. On the other hand, it was shown that diminished Mfn2 may be involved in an early activation of apoptosis in cardiomyocytes with increased mitochondrial fragmentation and decreased mitochondrial volume [13]. Another recent report demonstrated that Mfn2 expression was downregulated in hypertrophied cardiomyocytes induced by angiotensin II and phenylephrine and in hypertrophied hearts from multiple animal models [14]. Therefore, Mfn2 expression in the heart may have important implications in cardiac pathophysiology, especially, in myocardial energetic states. It has been recently demonstrated that Mfn2 can be induced directly by the cooperative actions of PPAR-γ coactivator-1 α (PGC-1α) and estrogen related receptor (ERRα) [15] [16] [17]. However, it is not clear if other important metabolic determinants such as PPARs also govern Mfn2 expression.
In the present study, we demonstrate that fasting in mice induces cardiac expression of Mfn2 and cardiac expression of Mfn2 is determined by PPARδ signaling.
Methods
1. Culture of rat neonatal cardiomyocytes
Rat neonatal cardiomyocytes were isolated by using a cardiomyocyte isolation system (Worthington, Lakewood, NJ) with minor modifications as described previously [18] [19]. Briefly, left ventricles of 1-day-old Sprague–Dawley rats were cut into small pieces and incubated with trypsin at 4°C overnight, following by a 45-minute collagenase digestion at 37°C. The resulted cells were pre-plated for 1 hour at 37 °C in DMEM/F12, 15 mM HEPES, 5% FBS, 100 U/ml penicillin (Roche) and 100 mg/ml streptomycin (Roche, Indianapolis, IN) to allow adherence of noncardiomyocytes. Cardiomyocytes in the supernatant were re-suspended and plated at a field density of 1 × 105 cells/cm2 on 25-cm2 culture flask in culture medium [MEM with 5% bovine calf serum (Invitrogen, Carlsbad, CA), 100 U/ml penicillin and 100 mg/ml streptomycin]. Cardiomyocytes were then incubated for 24 hours at 37°C with 0.5% FBS DMEM-F12 medium before 23 hour treatments of PPARδ-selective ligand GW0742 (5 μM, Cayman Chemical, Ann Arbor, Michigan) and PPARα-selective ligand Wy14643 (10 μM, Cayman Chemical, Ann Arbor, Michigan) or DMSO. For adenoviral mediated overexpression of Mfn2 in cultured cardiomyocytes, Ad-Mfn2 (100 MOI) and Ad-LacZ (100 MOI) as control were added with 0.5% FBS DMEM-F12 medium. Prior to each experiment, the cardiomyocytes were rinsed three times with 1 ml of warm (37°C) phosphate-buffered saline (PBS).
2. Animal studies
Breeding of cardiomyocyte-restricted PPAR-δ knockout mice: Homozygous cardiomyocyte-restricted PPARδ knockout mice (CR-PPARδ−/−) [20] were generated by crossing the transgenic Cre (α-MyHC–Cre) mice (C57/BL6) [21] with the PPARδflox/flox mice (C57/BL6) [22]. In this mouse line, PPARδ was largely eliminated in ventricular cardiomyocytes shortly after birth.
Tamoxifen-inducible cardiomyocyte-restricted PPARδ knockout line (TMPD): Homozygous TMPD mice (C57/B6) were generated by crossing a transgenic line with α-MyHC driven Mer-Cre-Mer (MCM) overexpression ([23]) and the PPARδ (PPARδflox/flox) line. Cardiomyocyte-restricted PPARδ knockout was induced in 2-month old mice by 5 days of Tamoxifen treatment (2mg/200ul in sunflower oil, intraperitoneal injection). After tamoxifen treatment ending for 5 to 10 days, PPARδ was largely eliminated in cardiomyocytes of mouse hearts from this line. PPARα knockout line (C57/B6) was obtained from commercial sourse (Jackson Lab, Bar Harbor, Maine). Animals received food and water on an ad libitum basis, and lighting was maintained on a 12-hour cycle. All experimental procedures were conducted in accordance with the Guide for Care and Use of Laboratory Animals of the National Institutes of Health, and were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham.
3. Transcript analyses
Total RNA samples were extracted using a RNA extraction kit (Qiagen, Valencia, CA). Real Time-PCR analyses (Roche LightCycler PCR system, Carlsbad, CA) were carried out to determine transcript levels of target genes. Real-Time PCR results from each gene/primer pair were normalized to β-actin, and compared across conditions. The following primers were used: Mfn2: up-AAGTCCGGGAAGCTGAAAGT, low-TCTCGGTTATGGAACCAACC; PGC-1α: up- CTCCTCCCACAACTCCTCCTCATA, low-GGGCCGTTTAGTCTTCCTTTCCTC; ERRα: AGCAAGCCCCGATGGA, Low-GAGAAGCCTGGGATGCTCTT; PPARδ: up-ATGGAACAGCCACAGGAGGA, low-ATCACAGCCCATCTGCAGCT and β-actin: up-AGATTACTGCTCTGGCTCCTA, low-CAAAGAAAGGGTGTAAAACG.
4. Protein analysis
Cytosolic protein samples were extracted using a nuclear protein extraction kit (Thermo Scientific, Rockford, IL). Protein samples (40μg) from each experiment were subjected to SDS–PAGE and electrotransferred to nitrocellulose membranes (GE Healthcare, Piscataway, NJ). Immunoblotting was performed using the enhanced chemiluminescence detection system (Thermo Scientific, Rockford, IL). A polyclonal anti-Mfn2 antibody (Sigma-Aldrich, St. Louis, MO) and a polyclonal anti-ATP synthase β-subunit (Abcam, Cambridge, MA) were used for the Western blot analyses. A Kodak Image Station 4000R (Kodak, Rochester, NY) was used for quantitative measurement of protein signals. Sample loadings were normalized by immunoblotting with an anti-actin monoclonal antibody (Sigma-Aldrich, St. Louis, MO).
5. Nuclear protein extraction and electrophoretic mobility shift assay (EMSA)
Nuclear protein was extracted from left ventricles of α-MyHC-Cre and CR-PPARδ−/− mice using a nuclear protein extraction kit (Thermo Scientific, Rockford, IL). Protein concentrations were determined with a Bio-Rad protein concentration assay kit (Bio-Rad Laboratories). Nuclear extracts were stored at −80°C until used. EMSA was performed using LightShift® Chemiluminescent EMSA Kit (Thermo Scientific, Rockford, IL) according to the manufacturer’s protocol. Double-stranded (ds) PPRE oligonucleotide (5′-TTT CAT TTA GCC CAG TTT AT-3′), which corresponds to nucleotide −837 to −817 of the mouse Mfn2 gene promoter, was synthesized and its 3′-end was labeled with Biotin (Invitrogen). Nuclear extracts (10μg protein) were incubated with 1 μg Poly (dI•dC), and the binding buffer for 20 minutes at room temperature. Bound DNA complexes were separated on a polyacrylamide gel electrophoresis and transferred to a nylon membrane (Thermo Scientific, Rockford, IL). The nylon membranes were cross-linked and chemiluminescent detection was performed.
6. Luciferase transfection assays
Mouse Mfn2 promoter fragments with or without the predicted PPRE element were subcloned into a Luciferase reporter vector (pGL3-Basic). An embryonic rat heart cell line (H9C2 cells) was seeded into 24-well plates. Vectors containing mouse Mfn2 promoter fragments with or without the putative PPRE consensus sequence were respectively transfected when the cells grew to a confluence of 50–80% after overnight culture. The above experimental groups were also co-transfected with pGL3-Basic vectors containing PPARγ co-activated α (PGC-1α) and pRL-CMV (for normalization). In addition, the above cells were infected 5 hours later with adenoviral vectors containing mouse wild type PPARδ (Ad-PPARδ, 100 MOI). As we shown previously, the above infection lead to a 2 fold overexpression of PPARδ protein [18] [19]. Finally, the above cultured H9C2 cells were treated with GW0742 (5 μM) or DMSO 23 hours before Luciferase assay. The relative Luciferase activity were tested and compared in different vectors to see whether the PPRE elements are still functionally active.
7. Statistics analyses
All data were analyzed by one factor or mixed, two-factor analysis of Variance (ANOVA) followed by post-hoc analysis with the Student–Newman–Keuls test and/or Bonferroni test to detect differences between experimental groups using GraphPad Prism software (GraphPad Software Inc., La Jolla, CA).
Results
1. Fasting induces a marked increase of Mfn2 expression in the heart
Given the importance of Mfn2 in maintaining mitochondrial architect and bioenergetics and it is highly expressed in the heart, it becomes intriguing if cardiac expression of Mfn2 is response to energy starvation conditions such as fasting. Mfn2 transcript levels in the heart were assessed by Real Time PCR measurement on samples from mice subjected to a 48 hour fasting. The results revealed that cardiac Mfn2 transcript expression was greatly increased in fasting mice compared with that of fed mice (Figure 1). In consistent with previous report [24], cardiac expression of PGC-1α was also substantially elevated in fasting mice compared with that of fed mice (Figure 1B). These results indicates that Mfn2 may be involved in signaling pathways that are coactivated by PGC-1α and potentially plays an important role in myocardial energy metabolism in starvation conditions.
Figure 1.
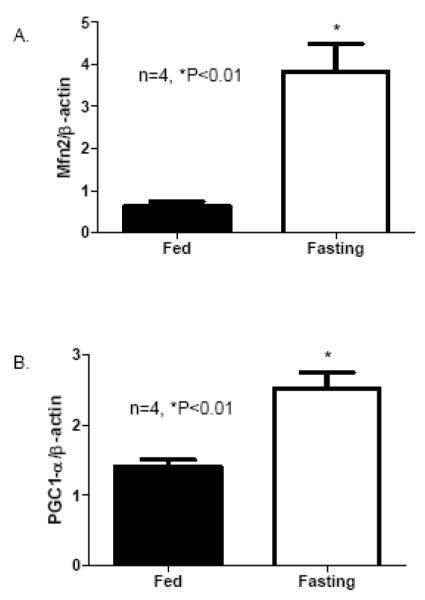
Cardiac Mfn2 and PGC-1α transcripts in response to 48 hour fasting in mice: Hearts were collected from mice with and without fasting for 48 hour. A) Real Time PCR measurement Mfn2 transcripts normalized to β-actin transcript was conducted on samples extracted from the left ventricles of mice after 48 hour fasting. Data are expressed as mean±SEM, n=4, *P<0.01. B) Real Time PCR measurement PGC-1α transcripts normalized to β-actin transcript was conducted on samples extracted from the left ventricles of mice after 48 hour fasting. Data are expressed as mean±SEM, n=4, *P<0.01.
2. Diminished Mfn2 expression in PPARδ deficient hearts
Our previous study demonstrated that myocardial energetic defect and myocardial FAO depression were the main features of mice with cardiomyocyte-restricted PPARδ deficiency (CR-PPARδ−/−) [20] [25]. To investigate cardiac Mfn2 expression in CR-PPARδ−/− hearts, expression levels of Mfn2 genes were evaluated by Real Time PCR measurement on RNA samples from CR-PPARδ−/− and control mice before overt phenotypic changes. Mfn2 transcript levels in CR-PPARδ−/− hearts were decreased by 50% relative to those of control hearts (Figure 2A.). Western blots also revealed that Mfn2 protein levels in CR-PPARδ−/− hearts were decreased by about 30% compared with those of control hearts (Figure 2B). Western blot analyses also revealed that the abundance of another mitochondrial protein, ATP synthase β subunit, were not downregulated as mfn2 did in CR-PPARδ−/− hearts (Figure 2C). These results indicate that downregulation of Mfn2 in CR-PPARδ−/− hearts is not part of a general downregulation of mitochondrial proteins.
Figure 2.



Mfn2 expression in long-term PPAR deficient hearts: A). Real Time PCR measurement of Mfn2 transcript normalized to β-actin transcript in samples from 2-month-old CR-PPARδ−/− and α-MyHC–Cre hearts. Data are expressed as mean±SEM, n=4, *P<0.05. B) Western blot analyses of Mfn2 protein abundance in CR-PPARδ−/− and α-MyHC–Cre hearts. The relative amount of Mfn2 was normalized to actin protein. Data are expressed as mean±SEM, n=4, *P<0.01. C) Western blot analyses of ATP synthase β subunit in CR-PPARδ−/− and α-MyHC–Cre hearts. The relative amount of ATP synthase β subunit was normalized to actin protein. Data are expressed as mean±SEM, n=4, P>0.05. D) Real Time PCR measurement of Mfn2 transcript normalized to β-actin transcript in cardiac samples from 2-month-old CR-PPARα−/− and wild type mice. Data are expressed as mean±SEM, n=6, P>0.05. E) Real Time PCR measurement of PGC-1α transcript normalized to β-actin in samples from 2-month-old CR-PPARδ−/− and α-MyHC–Cre hearts. Data are expressed as mean±SEM, n=4, P>0.05. F) Real Time PCR measurement of PGC-1α transcript normalized to β-actin transcript in samples from 2-month-old CR-PPARδ−/− and α-MyHC–Cre hearts. Data are expressed as mean±SEM, n=4, P>0.05.
We further investigate whether Mfn2 expression is also downregulated in PPARα knockout heart. Real Time PCR measurement revealed that Mfn2 transcript levels in 2-month-old PPARα knockout hearts were not altered relative to those of control hearts (Figure 2D). To determine if the downregulation of Mfn2 expression in CR-PPARδ−/− hearts is due to potential downregulation of PGC-1α and ERRα with PPARδ deficiency, transcript expression of PGC-1α and ERRα was assessed. Real Time PCR measurement revealed that the expression of both PGC-1α and ERRα in CR-PPARδ−/− hearts was not changed compared to controls (Figure 2E and F). Therefore, it is evident that the downregulation of Mfn2 in CR-PPARδ−/− hearts should be the consequence of PPARδ deficiency.
In CR-PPARδ−/− mice, PPARδ is knocked out from ventricular cardiomyocytes shortly after birth. To exclude the possibility that the observed change of Mfn2 expression in CR-PPARδ−/− heart is due to secondary responses to long-term knockout of PPARδ in cardiomyocytes, cardiac expression of Mfn2 transcript was also assessed by Real Time PCR measurement on RNA samples from mice with short-term (5-10 days) cardiomyocyte-restricted PPARδ knockout. Cardiomyocyte-restricted PPARδ knockout was induced in 2-month old mice by Tamoxifen treatment. These mice (TMPD) were compared with age- and gender-matched MCM mice with the same Tamoxifen treatment, which designated as TMCM mice. At 10 days after Tamoxifen treatment, PPARδ was largely eliminated on RNA samples extracted from left ventricles of the TMPD mice compared with samples from control mice (TMCM) (Figure 3A). With this short-term PPARδ deficiency in the young adult mice, transcript levels of Mfn2 in TMPD hearts were decreased by about 50% compared with those of the TMCM mice (Figure 3B). While fasting-induced transcript expression of Mfn2 in the heart from TMCM and TMPD groups was consistent, it was blunted in TMPD mice compared with TMCM mice (Figure 3B). It is noted that the expression of PPARδ in the heart was not elevated by the same fasting condition (Figure 3C). Therefore, it is clear that PPARδ is essential for the heart to maintain normal Mfn2 transcript levels in basal and at least partially in fasting conditions.
Figure 3.

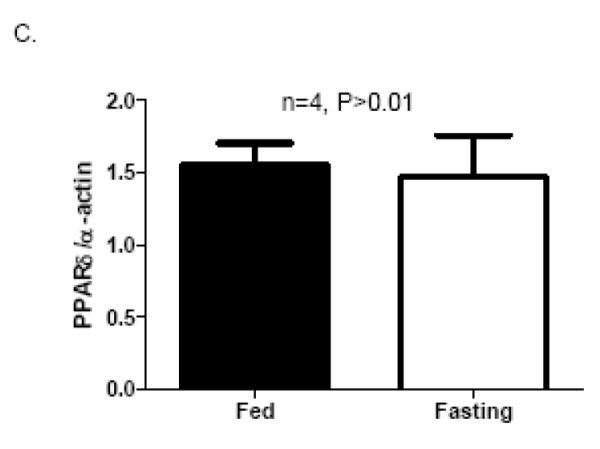
Mfn2 expression in short-term PPARδ deficient hearts: A) Real Time PCR measurement of PPARδ transcript normalized to β-actin transcript in samples from TMPD and TMCM hearts. Data are expressed as mean±SEM, n=4, *P<0.01.B) Real Time PCR measurement of Mfn2 transcript normalized to β-actin transcript in samples left ventricles from TMPD and TMCM mice in Fed and fasting conditions, respectively. Data are expressed as mean±SEM, n=5, *P<0.05 (TMCM vs TMPD), # P<0.05 (Fasting vs Fed). C) Real Time PCR measurement of PPARδ transcript normalized to β-actin transcript in left ventricles from mice in Fed and fasting condition. Data are expressed as mean±SEM, n=4, P>0.05.
3. Activation of PPARδ induces elevated Mfn2 expression in cardiomyocytes
To investigate if activation of PPARδ directly increases Mfn2 expression in cardiomyocytes, cultured neonatal cardiomyocytes were treated respectively with a PPARδ-selective ligand (GW0742, 5 μM) and with a PPARα-selective ligand (Wy14624, 10 μM), for 23 hours. Real Time PCR measurement revealed that GW0742 treatment increased Mfn2 expression by about 35% compared with vehicle treatment in neonatal cardiomyocytes (Figure 4A). On the other hand, Wy14643 treatment did not alter Mfn2 expression in cultured neonatal cardiomyocytes (Figure 4B). These results demonstrated that PPARδ activation selectively directs Mfn2 expression in cardiomyocytes and Mfn2 may be a direct PPARδ-selected gene in the heart.
Figure 4.
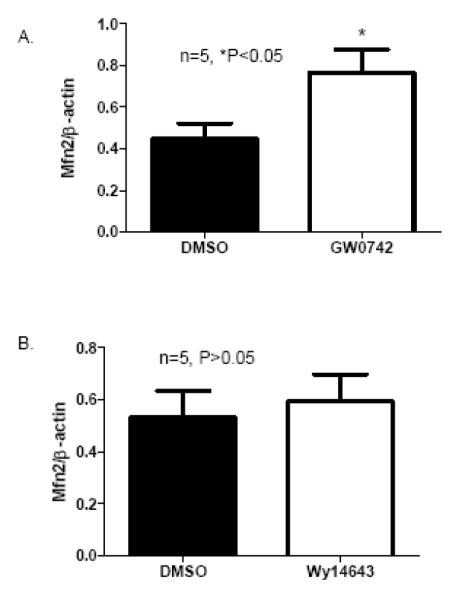
PPARδ activation induced Mfn2 expression in cultured rat neonatal cardiomyocytes: A) Real Time PCR measurement of Mfn2 transcript normalized to β-actin transcript in samples from cultured cardiomyocytes treated with a PPARδ-selective ligand, GW0742 (5 μM in DMSO) or with DMSO. Data are expressed as mean±SEM, n=5, *P<0.05. B) Real Time PCR measurement of Mfn2 transcript normalized to β-actin transcript in samples from cultured cardiomyocytes treated with a PPARα-selective ligand, WY14643 (10 μM in DMSO) or with DMSO. Data are expressed as mean±SEM, n=5, P>0.05.
4. Identify a functional PPRE in Mfn2 promoter
To further confirm that PPARδ activation directly upregulates Mfn2 expression in cardiomyocytes, we assessed the theoretical promoter regions of mouse Mfn2 gene using bioinformatics tools. A putative PPAR response element (PPRE) consensus sequence located at −837 to −817 bp upstream of the mouse Mfn2 promoter was identified. Using a LightShift chemiluminescent EMSA kit (Bio-Rad Laboratories), we confirmed that the probe derived from a fragment of this putative PPRE was strongly bound in nuclear protein extracted from control (α-MyHC-Cre) but not CR-PPARδ−/− mice in this in vitro condition (Figure 5). Similarly, no direct bound of the probe to nuclear protein extract from TMPD hearts either (data not shown). The functionality of the putative PPRE element on the Mfn2 promoter was further investigated using the Luciferase reporter assay in cultured H9C2 myocytes with PGC-1α expression and with adenoviral mediated PPARδ overexpression. Two fragments of the mouse Mfn2 promoter with (Mfn2-918) or without (Mfn2-712) the putative PPRE consensus sequence (Figure 6A), respectively, were cloned into the pGL3-Basic Luciferase reporter vector. Each of these vectors was transfected into H9C2 myocytes with treatment of GW0742 (5 μM) or DMSO. Luciferase assay revealed that treatment with GW0742, but not DMSO, induced an over 2 fold increase in Luciferase activity in those cells transfected with the Mfn2 promoter fragment containing the putative PPRE. Conversely, GW0742 treatment did not increase Luciferase activity in cells transfected with another promoter fragment without the PPRE sequence (Figure 6B). These data provide strong evidence to support that Mfn2 as a PPARδ target gene.
Figure 5.

Electrophoretic-mobility shift assays (EMSA): EMSA was performed using LightShift® Chemiluminescent EMSA Kit (Pierce) to assess the direct binding of the Biotin-labeled oligonucleotides corresponding to the putative PPRE sequence (−837 to −817) in the mouse Mfn2 promoter to endogenous PPARδ in nuclear extracts. Nuclear extracts with or without PPARδ were obtained from the α-MyHC-Cre or the CR-PPARδ−/− hearts, respectively. Experiments were performed three times with similar results.
Figure 6.

Promoter reporter assays of PPAR regulation of Mfn2 transcriptional activities: A) A schematic diagram of the Mfn2 promoter fragments cloned into the pGL3-Basic luciferase reporter vector. Numbers in the names of plasmids represent the deletion points of the Mfn2 promoter fragments. Mfn2 −918, but not Mfn2-712, contains the putative PPRE sequence. Numbers are also given with reference to the translation start site of Luciferase (Luc). B). Luciferase assays: Cultured H9C2 cells were transfected the above reporter constructs, PGC-1α and Ad-PPARδ followed by treatment with GW0742 (5 μM) or vehicle (DMSO) alone. The results of Luciferase assays are the mean of 4 independent experiments and expressed as mean±SEM, n=4, *P<0.01.
Discussion
In this study, we investigated a role of PPARδ in transcriptional regulation of Mfn2 expression in the heart, which may be in turn involved in maintaining myocardial energy homeostasis. The current results demonstrated that cardiac Mfn2 expression was elevated in mice subjected to fasting and downregulated in mice with PPARδ deficiency. We provide further evidence to demonstrate that PPARδ directly regulates Mfn2 expression via binding to the PPRE site in its promoter. The current results further support that Mfn2 is part of PPARδ signaling in regulating myocardial energy homeostasis.
Mfn-2 is ubiquitously expressed, especially at a high level in the heart [9]. Recent evidence demonstrates that Mfn-2 may be involved in regulating cell fate of cardiomyocyte under oxidative stress [12]. However, it remains unclear how Mfn-2 expression is regulated. Its deficiency in cardiomyocytes or in hearts undergoing pathological hypertrophy has also been revealed [14]. It is not clear yet if the downregulation of Mfn2 in hearts with pathological hypertrophy contributes to the development of cardiac hypertrophy or it is one of the hypertrophic responses in response to bioenergetic deficiency. The downregulation of Mfn2 expression in the cardiomyocyte-restricted PPARδ knockout hearts before the development of cardiac hypertrophy suggests that Mfn2 deficiency may contribute to the progression of cardiac hypertrophy and heart failure in these mice [20]. It is not likely the downregulation of Mfn2 is a consequence of cardiac hypertrophy. The downregulation of Mfn2 transcript in adult hearts with induced PPARδ knockout further support this notion with minimal confounding compensatory effects. One of the main phenotypic changes in the PPARδ deficient hearts is mitochondrial abnormalities such as matrix loss and enlarged, fragmented or even dramatic depletion in the severe cases, accompanied with a large quantity of lipid droplets [20]. An essential role of Mfn2 in maintaining mitochondrial network has been well established in many other cell types [8]. While it is difficult to establish a direct cause-effect relationship between the observed downregulation of Mfn2 and mitochondrial abnormalities in the CR-PPARδ−/− heart, it is possible that Mfn2 deficiency in the CR-PPARδ−/− hearts should at least partially contribute to the fragmentation of mitochondria network in the cardiomyocyte-restricted PPARδ knockout heart as reported previously [20].
Whereas deficiency of Mfn2 may contribute to cardiac pathological development, it has been reported recently that overexpression of Mfn2 exacerbates H2O2 induced cardiomyocyte apoptosis [12]. It is conceivable that the appropriate expression of Mfn2 may be required in the heart. The finding that cardiac expression of Mfn2 is remarkably increased in response to fasting condition is intriguing. While the implication of fasting-induced cardiac Mfn2 overexpression on cardiac mitochondrial architectural integrity and energy metabolism may be complicated, it at least suggests that cardiac Mfn2 expression levels are directly correlated to the energy status of the heart. Therefore, understanding how the Mfn2 expression is regulated in the heart should provide insights into the role of Mfn2 in cardiac pathophysiology. Our results revealed that cardiac expression of PGC-1α was induced by fasting and it is consistent with previous finding [24]. It is well documented that PGC-1α is an important coactivator for a series of nuclear receptors, including PPARδ and ERRα [26, 27, 28, 29]. ERRα, another nuclear receptor has a similar effect as PPARs in regulating FAO gene expression, has been shown to directly target Mfn2 transcription expression in the presence of PGC-1α [15, 17]. It is likely that ERRα should have similar effect on controlling Mfn2 expression in the heart. Since cardiac expression of PPARδ is not induced by fasting, upregulation of cardiac Mfn2 expression may be directed by fasting-induced PGC-1α elevation, thus activating selected transcription factor signals including PPARδ and ERRα. The upregulation of Mfn2 in the heart during starvation might help optimizing mitochondrial architecture and even function for the elevating energy demand.
The current study supports a major regulating role of PPARδ in Mfn2 expression in the heart. We demonstrated that PPARδ binds a novel PPRE sequence within the promoter of Mfn2 and this PPRE functions as a positive response element for PPARδ-specific agonist. Subsequent analyses of the rat and human Mfn2 promoters also revealed sequences that have significant homology to the PPRE consensus sequence of the mouse one. Therefore, it is unlikely that the observed altered Mfn2 expression in response to PPARδ deficiency or activation were the consequences of indirect effects from ERRα or other unknown regulators of Mfn2 expression. Instead, it appears that PPARδ and ERRα, the two functionally similar nuclear receptors, exert their independent regulating function on Mfn2 transcription. PPARα appears not to be involved in Mfn2 expression in cardiomyocytes but the underpinning mechanism is unclear. Further study on how Mfn2 expression is regulated by PPARδ and ERRα in coordinate with their common transcription coactivator PGC-1α is warranted. This further suggests that Mfn2 may be one of the mitochondrial proteins that play important roles in maintaining myocardial energy homeostasis.
Since Mfn1 may possesses similar effects on mitochondrial fusion and function, future studies should be informative to see if Mfn1 is also regulated by PPARδ. However, previous studies have shown that the two similar proteins are not redundant [6] [7]. It has been revealed that Mfn1 and 2 have a specific functional difference in how they regulate mitochondrial fusion [30]. Mfn2 is most abundantly expressed in cardiac and skeletal muscle. Therefore, the present study only focuses on PPARδ’s role on the expression of Mfn2.
In summary, the present study demonstrates that Mfn2 expression in the heart is specifically regulated by PPARδ. Cardiomyocyte-restricted knockout of PPARδ in mice leads to Mfn2 deficiency and contributes to the development of myocardial bioenergetic defects. The discovery of the regulation effect of PPARδ on Mfn2 expression should provide mechanistic insights into gene regulation of mitochondrial dynamic and energy metabolism in the heart and new therapeutic potentials for heart disease.
ACKNOWLEDEMENTS
This work was supported by grants from NIH (1R01HL085499, 1R01HL084456 and R21 AT003734). We thank Rammasamy Subbianh for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Marin-Garcia J. Mitochondria and the heart. Springer; 2005. [Google Scholar]
- [2].Santel A, Fuller MT. Control of mitochondrial morphology by a human mitofusin. Journal of cell science. 2001 Mar;114(Pt 5):867–74. doi: 10.1242/jcs.114.5.867. [DOI] [PubMed] [Google Scholar]
- [3].Rojo M, Legros F, Chateau D, Lombes A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. Journal of cell science. 2002 Apr 15;115(Pt 8):1663–74. doi: 10.1242/jcs.115.8.1663. [DOI] [PubMed] [Google Scholar]
- [4].Rapaport D, Brunner M, Neupert W, Westermann B. Fzo1p is a mitochondrial outer membrane protein essential for the biogenesis of functional mitochondria in Saccharomyces cerevisiae. The Journal of biological chemistry. 1998 Aug 7;273(32):20150–5. doi: 10.1074/jbc.273.32.20150. [DOI] [PubMed] [Google Scholar]
- [5].Hales KG, Fuller MT. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell. 1997 Jul 11;90(1):121–9. doi: 10.1016/s0092-8674(00)80319-0. [DOI] [PubMed] [Google Scholar]
- [6].Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. The Journal of cell biology. 2003 Jan 20;160(2):189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ishihara N, Eura Y, Mihara K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. Journal of cell science. 2004 Dec 15;117(Pt 26):6535–46. doi: 10.1242/jcs.01565. [DOI] [PubMed] [Google Scholar]
- [8].Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. The Journal of biological chemistry. 2003 May 9;278(19):17190–7. doi: 10.1074/jbc.M212754200. [DOI] [PubMed] [Google Scholar]
- [9].Chen KH, Guo X, Ma D, Guo Y, Li Q, Yang D, et al. Dysregulation of HSG triggers vascular proliferative disorders. Nature cell biology. 2004 Sep;6(9):872–83. doi: 10.1038/ncb1161. [DOI] [PubMed] [Google Scholar]
- [10].Kijima K, Numakura C, Izumino H, Umetsu K, Nezu A, Shiiki T, et al. Mitochondrial GTPase mitofusin 2 mutation in Charcot-Marie-Tooth neuropathy type 2A. Hum Genet. 2005 Jan;116(12):23–7. doi: 10.1007/s00439-004-1199-2. [DOI] [PubMed] [Google Scholar]
- [11].Verhoeven K, Claeys KG, Zuchner S, Schroder JM, Weis J, Ceuterick C, et al. MFN2 mutation distribution and genotype/phenotype correlation in Charcot-Marie-Tooth type 2. Brain. 2006 Aug;129(Pt 8):2093–102. doi: 10.1093/brain/awl126. [DOI] [PubMed] [Google Scholar]
- [12].Shen T, Zheng M, Cao C, Chen C, Tang J, Zhang W, et al. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. The Journal of biological chemistry. 2007 Aug 10;282(32):23354–61. doi: 10.1074/jbc.M702657200. [DOI] [PubMed] [Google Scholar]
- [13].Parra V, Eisner V, Chiong M, Criollo A, Moraga F, Garcia A, et al. Changes in mitochondrial dynamics during ceramide-induced cardiomyocyte early apoptosis. Cardiovasc Res. 2008 Jan 15;77(2):387–97. doi: 10.1093/cvr/cvm029. [DOI] [PubMed] [Google Scholar]
- [14].Fang L, Moore XL, Gao XM, Dart AM, Lim YL, Du XJ. Down-regulation of mitofusin-2 expression in cardiac hypertrophy in vitro and in vivo. Life sciences. 2007 May 16;80(23):2154–60. doi: 10.1016/j.lfs.2007.04.003. [DOI] [PubMed] [Google Scholar]
- [15].Willy PJ, Murray IR, Qian J, Busch BB, Stevens WC, Jr., Martin R, et al. Regulation of PPARgamma coactivator 1alpha (PGC-1alpha) signaling by an estrogen-related receptor alpha (ERRalpha) ligand. Proceedings of the National Academy of Sciences of the United States of America. 2004 Jun 15;101(24):8912–7. doi: 10.1073/pnas.0401420101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cartoni R, Leger B, Hock MB, Praz M, Crettenand A, Pich S, et al. Mitofusins 1/2 and ERRalpha expression are increased in human skeletal muscle after physical exercise. J Physiol. 2005 Aug 15;567(Pt 1):349–58. doi: 10.1113/jphysiol.2005.092031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Soriano FX, Liesa M, Bach D, Chan DC, Palacin M, Zorzano A. Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator-activated receptor-gamma coactivator-1 alpha, estrogen-related receptor-alpha, and mitofusin 2. Diabetes. 2006 Jun;55(6):1783–91. doi: 10.2337/db05-0509. [DOI] [PubMed] [Google Scholar]
- [18].Cheng L, Ding G, Qin Q, Xiao Y, Woods D, Chen YE, et al. Peroxisome proliferator-activated receptor [delta] activates fatty acid oxidation in cultured neonatal and adult cardiomyocytes. Biochemical and Biophysical Research Communications. 2004 Jan 9;313(2):277–86. doi: 10.1016/j.bbrc.2003.11.127. 2004. [DOI] [PubMed] [Google Scholar]
- [19].Ding G, Cheng L, Qin Q, Frontin S, Yang Q. PPARdelta modulates lipopolysaccharide-induced TNFalpha inflammation signaling in cultured cardiomyocytes. Journal of molecular and cellular cardiology. 2006 Jun;40(6):821–8. doi: 10.1016/j.yjmcc.2006.03.422. [DOI] [PubMed] [Google Scholar]
- [20].Cheng L, Ding G, Qin Q, Huang Y, Lewis W, He N, et al. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med. 2004 Nov;10(11):1245–50. doi: 10.1038/nm1116. [DOI] [PubMed] [Google Scholar]
- [21].Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. The Journal of clinical investigation. 1997 Jul 1;100(1):169–79. doi: 10.1172/JCI119509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hevener AL, He W, Barak Y, Le J, Bandyopadhyay G, Olson P, et al. Muscle-specific Pparg deletion causes insulin resistance. Nat Med. 2003 Dec;9(12):1491–7. doi: 10.1038/nm956. [DOI] [PubMed] [Google Scholar]
- [23].Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, et al. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circulation research. 2001;89(1):20–5. doi: 10.1161/hh1301.092687. [DOI] [PubMed] [Google Scholar]
- [24].Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. The Journal of clinical investigation. 2000 Oct;106(7):847–56. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lee J, Hu Q, Nakamura Y, Wang X, Zhang X, Zhu X, et al. Open-chest 31P magnetic resonance spectroscopy of mouse heart at 4.7 Tesla. J Magn Reson Imaging. 2006 Dec;24(6):1269–76. doi: 10.1002/jmri.20766. [DOI] [PubMed] [Google Scholar]
- [26].Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H, et al. Peroxisome-Proliferator-Activated Receptor delta Activates Fat Metabolism to Prevent Obesity. Cell. 2003;113(2):159–70. doi: 10.1016/s0092-8674(03)00269-1. [DOI] [PubMed] [Google Scholar]
- [27].Dressel U, Allen TL, Pippal JB, Rohde PR, Lau P, Muscat GE. The peroxisome proliferator-activated receptor beta/delta agonist, GW501516, regulates the expression of genes involved in lipid catabolism and energy uncoupling in skeletal muscle cells. Mol Endocrinol. 2003 Dec;17(12):2477–93. doi: 10.1210/me.2003-0151. [DOI] [PubMed] [Google Scholar]
- [28].Hondares E, Pineda-Torra I, Iglesias R, Staels B, Villarroya F, Giralt M. PPARdelta, but not PPARalpha, activates PGC-1alpha gene transcription in muscle. Biochem Biophys Res Commun. 2007 Mar 23;354(4):1021–7. doi: 10.1016/j.bbrc.2007.01.092. [DOI] [PubMed] [Google Scholar]
- [29].Huss JM, Torra IP, Staels B, Giguere V, Kelly DP. Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Molecular and cellular biology. 2004 Oct;24(20):9079–91. doi: 10.1128/MCB.24.20.9079-9091.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cipolat S, de Brito O Martins, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proceedings of the National Academy of Sciences of the United States of America. 2004 Nov 9;101(45):15927–32. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]