

Abstract
We demonstrate the effectiveness of intrinsic Raman spectroscopy (IRS) at reducing errors caused by absorption and scattering. Physical tissue models, solutions of varying absorption and scattering coefficients with known concentrations of Raman scatterers, are studied. We show significant improvement in prediction error by implementing IRS to predict concentrations of Raman scatterers using both ordinary least squares regression (OLS) and partial least squares regression (PLS). In particular, we show that IRS provides a robust calibration model that does not increase in error when applied to samples with optical properties outside the range of calibration.
1. Introduction
In many applications of Raman spectroscopy the analytes of interest are embedded in a turbid medium, in which the absorption and elastic scattering coefficients, μa and μs, respectively, may significantly distort the measured Raman spectrum, in contrast to the intrinsic Raman spectrum obtained from a transparent medium. A prime example, of interest to us, is biological tissue analysis. The apparent contributions of the Raman spectral features, and therefore the shape of the spectrum, may be altered as a result of the wavelength dependence of μa and μs. Additionally, the overall Raman intensity may be altered as a result of turbidity-induced changes in sampling volume, defined as the overlap between the three-dimensional delivery and collection volumes, which affects the propagation of both the excitation and scattered Raman light. Time- or sample-dependent turbidity variations can further complicate the extraction of quantitative information.
In order to account for such non-analyte-specific variability in building a multivariate calibration model, samples with significant extremes of variability are incorporated into the calibration data set. However, this approach sacrifices accuracy because of increased model complexity, not to mention the added costs associated with inclusion of large sample numbers for in vivo studies. The better solution is to directly reduce the turbidity variability through prior or additional information such that a more accurate model is obtained and prediction error is reduced.
Corrections for turbidity variations in the form of spectral shape distortions have been extensively pursued in fluorescence spectroscopy [1-5]. However, in many cases the recorded spectra are normalized and information about sampling volume variations are lost, rendering the analysis semi-quantitative. For quantitative studies, variations in the sampling volume are critical because of the relationship to the absolute number of analyte molecules sampled.
For quantitative Raman spectroscopy, corrections for both shape and intensity distortions have been developed [6-9], but the existing methods are designed for media with isotropic elastic scattering, and in which only one optical property changes at a time. Thus, they are not suitable for our application to biological media, in which elastic scattering is anisotropic [10, 11] and both μa and μs can vary simultaneously and independently.
The underlying principle of all of these correction methods is that Raman scattering or fluorescence at a given wavelength is proportional to elastically scattered light (diffuse reflectance) at the same wavelength. Therefore, with the aid of a broadband light source to measure diffuse reflectance, distortions in Raman and fluorescence spectra can be removed.
In the companion paper (Part I), we present the theory for a method for correcting these distortions called intrinsic Raman spectroscopy (IRS), and conduct numerical simulations to validate the theory. As discussed in detail in Part I, diffuse reflectance spectroscopy is used to correct the observed Raman spectra. The present paper presents an experimental study of IRS, using in vitro physical tissue models of various turbidities. We demonstrate that IRS provides a marked reduction in prediction error compared with the uncorrected measurements via both explicit and implicit multivariate calibration techniques.
2. Implementation
In the companion paper (Part I: Theory and simulations), the following equation for implementing the IRS correction was derived:
| (1) |
where RamINT is the intrinsic Raman signal observed in the absence of absorption and scattering, μt is the reduced total attenuation coefficient (μt = μa + μs), RamOBS is the measured (observed) Raman signal, and f(Rd) is a parameter, the calibration function, that depends only on diffuse reflectance, Rd, sample geometry, and anisotropy, g. The functional form of f(Rd) is complex, but may be fit by a simple analytical function such as a power law or an exponential function. f(Rd) can be obtained experimentally by fixing the concentration of a known Raman scatterer and varying the optical properties of the medium, as described below. Thus, by knowing f(Rd), measuring Rd, and extracting μt, RamINT can be obtained from RamOBS.
The most general method of implementing the IRS correction is the “wavelengthdependent” approach. It is appropriate when the diffuse reflectance spectra exhibit significant spectral structure. In this case one employs Eq. (1), wavelength-by-wavelength, to obtain the intrinsic Raman spectra. However, owing to the division operation in Eq. (1), noise and minor errors applied at each wavelength may be amplified when applied over the entire spectral range, and thus introduce artifacts that lead to higher prediction errors in the subsequent multivariate calibration. Therefore, use of the λ-dependent method should be restricted to cases in which narrow absorption features that significantly alter the shape of RamOBS (compared to RamINT) are present.
In other cases, however, the diffuse reflectance spectra do not exhibit sharp features and a simpler, λ-independent correction method may be used. This will occur when μa and μs, hence Rd, do not vary appreciably with wavelength. An example of interest is our Raman studies to measure blood analyte concentrations, which are conducted in the near infrared at 830-1000 nm [12]. In this case, the amplitude of RamOBS varies with sample-to-sample variations in turbidity but its spectral shape is not distorted. As a result, the λ-independent correction method can be employed in which the measured spectra are not corrected, but instead the reference concentrations are converted into the number of molecules measured in the same sampling volume as the measured Raman spectra. In the remainder of this paper we will implement IRS using the λ-independent method, and illustrate it with physical tissue models that are relevant to our blood analyte studies.
Raman scattering is proportional to the number of Raman scatterers sampled, N, which varies with optical sampling volume. However, most reference techniques measure local concentrations of analytes. We can thus consider a constant optical sampling volume such that the concentration, CREF, is equivalent to number of molecules, NREF. As a result of turbidity-induced variations in sampling volume, however, RamOBS measures a quantity NOBS rather than NREF. In order to correct for this, NOBS can be related to NREF in the same manner as RamOBS is related to RamINT:
| (2) |
For multivariate calibration, NOBS may then be used in conjunction with RamOBS to construct a calibration model that is used for prediction. The resulting predicted concentration value NPRED’, which gives the actual number of analyte molecules measured, may then be converted to NPRED, which is equivalent to the analyte concentration measured by a separate reference technique, using Eq. (2) as before.
A key advantage of this approach, as opposed to correcting the spectra themselves, is that reference concentration measurements are in principle more accurate than spectral data and therefore less likely to suffer from the amplification of minor noise or errors associated with the application of Eq. (1).
In summary, Raman spectra may be relied upon to accurately measure a given number of molecules, NOBS. Diffuse reflectance (Rd) and the extracted μt provide the necessary relationship between the sampling volume-dependent NOBS and the sampling volume-independent NREF. The λ-independent method corrects for intensity variations only, whereas the λ-dependent method corrects for both spectral and intensity variations.
3. Experimental
3.1 Apparatus
The experimental setup (Fig. 1) employs an 830 nm diode laser (Process Instruments) as the Raman excitation source and a tungsten-halogen lamp (Avantes AvaLight-HAL-S) as the diffuse reflectance excitation source. A laser line filter (Semrock MaxLine 830) is placed at the laser output to remove unwanted spontaneous emission that broadens the laser linewidth. An RG850 absorption filter is placed at the lamp output to reduce shorter wavelengths that may cause scatter within the spectrometer. The two beams are independently shuttered and combined using a MgF2 plate at 45°, with the laser being transmitted and the white light source being reflected and adjusted to be collinear. A photodiode placed at this intersection monitors the power of both sources. The light sources are focused through a small hole (4 mm dia.) in an off-axis, gold-coated, half-paraboloidal mirror (Perkin-Elmer, Inc.) and into a fused silica cuvette (1 cm pathlength) filled with the sample of interest. Achromatic doublet lenses are used to shape the beams. The beam diameters at the sample are approximately 1 mm, and the powers are 250 mW and 100 μW for the laser and white light source, respectively. The power level of the white light source is carefully controlled to prevent saturation of the CCD detector.
Fig. 1.
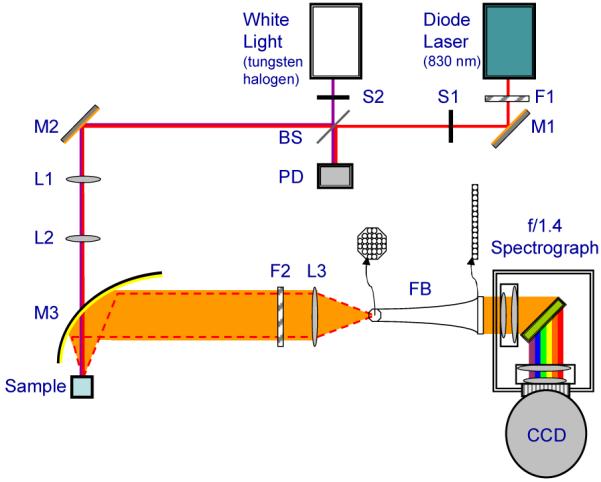
Schematic of experimental setup. F1: laser line filter (MaxLine, Semrock); S1-S2: shutters; BS: MgF2 plate used as beamsplitter; M1-M2: gold plated mirrors; L1-L2: beam shaping lenses; M3: paraboloidal mirror; F2: notch filter; L3: coupling lens; FB: fiber bundle.
Back-scattered Raman and diffusely reflected light are collected with the paraboloidal mirror and sent through a holographic notch filter (Kaiser Optical Systems, Inc.) to reduce the magnitude of the excitation peak. Specular reflection from the cuvette surface passes through the hole in the paraboloidal mirror and is significantly diminished. The collected light is then focused into a fiber bundle (RoMack, Inc.) that transforms the circular shape of the collected light (input end) into a vertical line (exit end, ∼400 μm×26 mm). The exit end of the fiber bundle serves as the entrance slit of a modified f/1.4 spectrometer (Kaiser Optical Systems, Inc.). The light is dispersed with a holographic grating onto a 1300×1340 pixel liquid nitrogen-cooled CCD detector (Princeton Instruments).
Raman spectra and diffuse reflectance (DRS) spectra were alternately acquired, with a total of 10 Raman spectra and 10 DRS spectra collected for each sample. The Raman spectra were accumulated with a 2s integration time and the DRS spectra were accumulated with a 0.5s integration time. Identical excitation-collection geometry was maintained throughout the experiment by fixing the cuvette position. Samples were replaced via pipette following a water rinse and two rinses of the sample of interest. Data were processed off-line for image curvature correction, summation, and removal of cosmic rays [13]. Spectra from 280-1700 cm-1 (850-966 nm) were used in all data analysis.
3.2 Materials and methods
Physical tissue models in which scattering, absorption and Raman scattering could be precisely varied throughout their respective relevant physiological ranges were used to test the applicability of intrinsic Raman spectroscopy. Two experiments were performed. In the first, elastic scattering and absorption were varied independently while Raman scatterer density was held constant. In the second, the Raman scatterer density was also simultaneously varied.
The physical tissue models were prepared in water solutions. The elastic scattering and absorption properties were varied over ranges similar to that found in human biological tissue [10]. The scattering coefficient, μs, was varied by altering the concentration of Intralipid (Baxter Healthcare), an anisotropic elastic scatterer commonly used to simulate tissue scattering. The value, g, for intralipid is between 0.8 and 0.9 [14], as is tissue. The absorption coefficient, μa, was varied by altering the concentration of India ink (Super Black India Ink, Speedball Art Products company), which possesses a nearly flat absorption profile in the near infrared region. For each specific mixture (sample), 10 Raman spectra were collected and averaged. Scattering and absorption coefficients at the excitation wavelength (830 nm) of representative physical tissue models were determined by integrating sphere measurements utilizing a look-up table based on Monte Carlo simulations performed by Enejder [15]. The value of g for each of the samples was taken to be fixed at 0.8.
In the first experiment, 49 samples were prepared following a 7×7 matrix in which μs was varied from 24 to 130 cm-1 (median 81.6 cm-1) at 830 nm, and μa was varied from 0.08 to 1.3 cm-1 (median 0.31 cm-1) at 830 nm. A constant 50 mM concentration of creatinine was included in each sample to serve as an indicator of an intrinsically constant Raman signal, the concentration of which may be measured incorrectly as a result of turbidity. The relatively high concentration of creatinine enabled higher absorption values to be studied while retaining a satisfactory signal-to-noise ratio.
In the second experiment, 50 samples were prepared with randomized concentration profiles and turbidities. In these samples, μs was randomly varied from 48.4 to 95.1 cm-1 (mean 73.4 cm-1) at 830, and μa was randomly varied from 0.09 to 0.18 cm-1 (mean 0.136 cm-1) at 830 nm. Two Raman scatterers, glucose and creatinine, were also present, with concentrations varying randomly from 5 to 30 mM (mean 16 mM).
3.3 Data analysis
Data were initially analyzed via ordinary least squares (OLS) [16] using a seven- or eight-component model. The model components include the Raman spectra of fused silica (cuvette), water, Intralipid, India ink, creatinine (as measured in water, with the background subtracted, glucose (second experiment only), fluorescence (from impurities in the cuvette - obtained by subtracting the tenth spectrum from the first spectrum for a representative sample), and a DC offset to account for the increased or decreased signal level due to scattering or absorption, respectively.
The OLS model components for the first experiment are shown in Fig. 2. Each spectrum was fit individually to account for varying levels of fluorescence and offset, and the creatinine fit coefficients for the 10 spectra in each set were averaged for each sample.
Fig. 2.
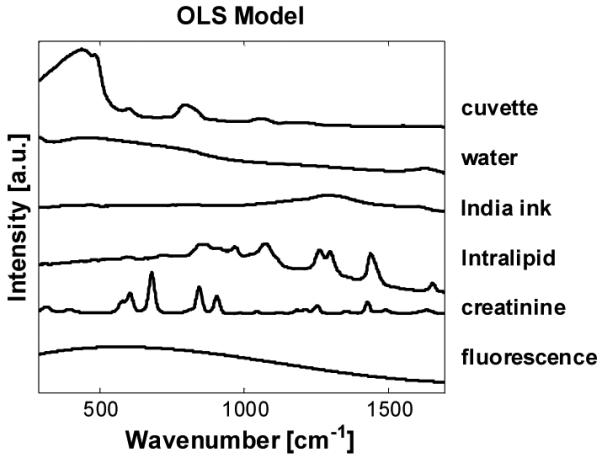
OLS model components for experiment 1.
Data from the second experiment were analyzed via OLS to obtain fit coefficients, as in the first experiment. Additionally, data from the second experiment were analyzed via partial least squares (PLS) [17], as described in detail below, to demonstrate the utility of the IRS correction method with an implicit multivariate calibration technique.
4. Results and discussion
We begin with the first experiment (fixed concentration Raman scatterers in all 49 samples.) The observed Raman spectra of the 49 samples are shown in Fig. 3(a). The associated DRS spectra are shown in Fig. 3(b). To better illustrate the relative change in the DRS spectra due to changing optical properties, the DRS spectra in Fig. 3(b) are normalized to 4% intralipid with no absorber added (μs = 73.6 cm-1). Note that any of the samples could in theory be chosen as the basis for normalization. The DRS spectra were not normalized to a white reflectance standard such as Spectralon (>99%, Labsphere) because the reference glucose and creatinine concentrations were measured in an aqueous solution. Thus, the “intrinsic” Raman spectrum includes the effects of water absorption and it was not necessary to correct the turbid samples for this effect. Because the DRS spectra were internally normalized, Rd is here the relative diffuse reflectance rather than the absolute diffuse reflectance. This difference has no effect on the functionality of the IRS correction method. As can be seen in Fig. 3(b), even though the intensity variations in this wavelength region (830-960 nm) are substantial, the spectral variations from sample to sample, owing to changes in Intralipid and water absorption, are minimal. Thus, we can use the -independent correction method.
Fig. 3.

(a). Raman spectra (b). Relative DRS spectra normalized to 4% intralipid with no absorber present.
For the λ-independent method, we compare the observed number of molecules measured (NOBS) for each sample to the actual reference concentration (NREF). For convenience, NOBS was obtained through OLS analysis, in which the measured Raman spectrum is fit by a linear combination of the spectra of each component in the physical tissue model (e.g., water, Intralipid, India ink, creatinine, fused silica cuvette, etc.). The fit coefficient for the (constant) concentration of the Raman scatterer, in this case creatinine, is NOBS. (It does not matter which Raman scatterer is used as the probe to calibrate f(Rd), as the shape of this parameter is not analyte-dependent.) In the absence of turbidity, the ratio NOBS/NREF should be 1. However, owing to the effects of absorption and scattering, this value varied significantly, from 0.34 to 1.14. NOBS/NREF for each of the 49 samples is displayed in Fig. 4 as a function of μa and μs.
Fig. 4.

The ratio of observed to actual values of an analyte at constant concentration showing significant deviation from 1 resulting from turbidity distortions, plotted as a function of μa and μs.
Following Eq. (2), values of (NOBS/NREF)·μt versus Rd are plotted in Fig. 5. Data collected from the two experiments are combined. The fit to the data is f(Rd), which for these experiments is best represented by an exponential function. Although the data collection periods were two weeks apart and the sample was replaced several times, it can be seen that f(Rd) remains constant, as would be expected for similar geometry and anisotropy, g.
Fig. 5.
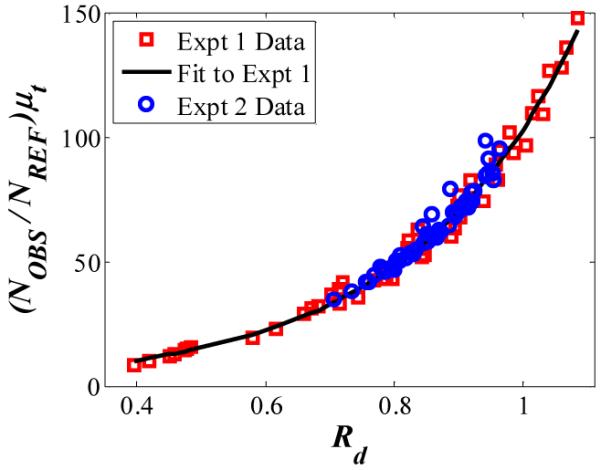
(NOBS/NREF)·μt versus Rd for experiments 1 and 2, showing reproducible curvature.
The noise in Fig. 5, i.e., data points that do not lie on the curve, is a result of experimental signal-to-noise. The creatinine signal is much smaller than the signals from most of the other constituents; therefore slight modeling errors and noise in the spectral data contribute to error in extracting the OLS fit coefficient for creatinine. As noted above, any Raman scatterer can be used to determine the IRS calibration curve, f(Rd). For example, intralipid itself may be used provided that its apparent concentration change due to varying optical properties is deconvolved from its real concentration change. Processed accordingly, use of intralipid data with its higher signal-to-noise ratio results in a tight f(Rd) curve equivalent to the curvature of Fig. 5, but with no appreciable spread. However, to prevent potential confusion on the part of the reader, we opted to present the analysis using the fixed-concentration Raman scatterer, creatinine.
Once f(Rd) was obtained, NOBS for each sample was multiplied by the corresponding μt of that sample and divided by f(Rd) to obtain NPRED, which may be compared to NREF for assessing prediction accuracy. The root-mean-square error of prediction (RMSEP) for the uncorrected data (NOBS) was 36%, versus an RMSEP for the IRS-corrected data (NPRED) of 6%. Thus, IRS significantly improved the prediction accuracy.
The second data set was designed to illustrate the effectiveness of the IRS correction method under an implicit calibration framework [18]. The data were analyzed twice, first using OLS (explicit calibration) and then via PLS (implicit calibration). Prior to implicit analysis, the 10 sequential Raman spectra for each sample were averaged and smoothed via a Savitsky-Golay function.
The Raman spectra were then split into calibration and validation sets following two different procedures. In the first procedure, the 50 sample spectra were randomly split into 36 calibration and 14 prediction sample spectra for 500 independent iterations. For comparison between the uncorrected analysis, in which sampling volume changes are ignored, and IRS, the reference values used in conjunction with the calibration data set to generate the regression vector [18] were either the actual reference values, NREF, or, for IRS application, NOBS, as calculated from Eq. (2). The number of PLS factors employed in either case was 6, which is in accordance with the number of known variables and the size of the calibration set. The resulting calibration algorithms were applied to the same validation set to compare predictive capabilities. In the case of IRS application, a final step was needed to convert the predicted values NPRED’ to NPRED, again via Eq. (2). The boxplot for the 500 RMSEP values for glucose generated with uncorrected and IRS-corrected data is shown in Fig. 6. The mean and standard deviation are 0.56 and 0.11 for the uncorrected data, and 0.43 and 0.08 for the corrected data, resulting in more than a 20% reduction in mean and standard deviation of prediction error by implementing IRS.
Fig. 6.
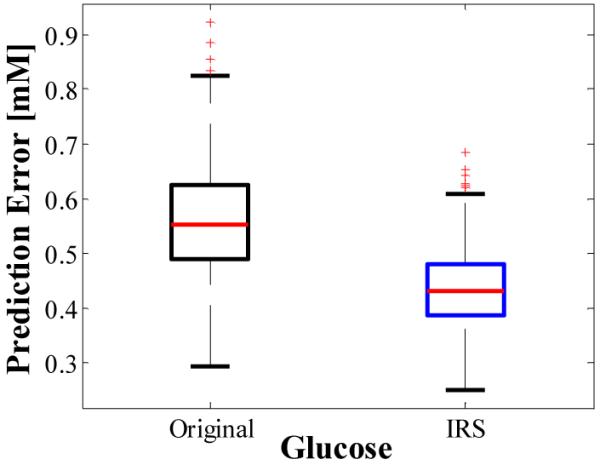
Boxplot showing reduction in mean and standard deviation of prediction error by application of IRS. Values were derived from 500 unique splittings of 50 samples into 36 calibration and 14 prediction sample sets.
In the second procedure the samples were split into calibration and validation sets according to the μs/μa value, where μs=μs(1-g) with g=0.8. Samples having μs/μa values less than 65 comprised the calibration set and samples having μs/μa values higher than 65 comprised the validation set (Fig. 7(a)). Such a splitting replicates the scenario of not incorporating the full range of optical property variability into the calibration set. Typically, this scenario results in a calibration algorithm that is not robust. In other words, applying the generated calibration algorithm to samples lying outside the range of the calibration set gives much larger prediction error.
Fig. 7.

(a). 50 samples split into 36 calibration and 14 prediction samples based on optical property values. (b). Standard error of cross-validation (SECV) and standard error of prediction (SEP) for samples split into calibration and prediction sets based on optical property values.
Indeed, for uncorrected analysis of the data (using NREF for the reference values as described above), the standard error of cross-validated (SECV) for the calibration set is 0.47 mM whereas the standard error of prediction (SEP) for the validation set is nearly twice as large, 0.86 mM. For the IRS analysis, the SECV is 0.41 mM and, in contrast to the uncorrected analysis, the SEP remains nearly level at 0.42 mM (Fig. 7(b)). This is an improvement of over 50% compared to the uncorrected RMSEP. This scenario illustrates the advantages of the IRS correction method. By replacing the reference values with the actual number of analyte molecules in the sampling-volume region NOBS, the multivariate calibration technique is better able to lock onto the signal from the analyte of interest, generating a more robust model and greatly reducing the prediction error.
5. Conclusions
We have demonstrated the principles of the intrinsic Raman correction method to obtain more accurate, quantitative results in turbid media. IRS provides a way to overcome inaccuracies caused by sampling volume variations as a result of absorption and scattering. This reduction in variability by use of IRS means fewer number of samples over a smaller optical property range may be used to form the calibration model. The resulting calibration algorithm is robust and provides improved accuracy in prospective studies.
We note that implementation of the IRS correction method requires knowledge of μt and the calibration function, f(Rd). In the reported tissue phantom studies, the optical property μt for each sample was determined with the aid of an integrating sphere. For experimental applications in which this is not possible, e.g., in vivo studies, the optical property information may be extracted from diffuse reflectance measurements. This is a well-developed research area and the reader is referred to a number of excellent references on this topic [19-23]. The calibration function, f(Rd), may be determined either experimentally or through Monte Carlo simulations. However, practical concerns in slight sampling size and excitation-collection geometry variations may justify the experimental approach over a Monte Carlo-based approach.
A related method of analysis to IRS in which variations in μa are considered to be insubstantial, and therefore knowledge of the optical properties is not required, will be the subject of a forthcoming publication.
Acknowledgment
This work was performed at the MIT Laser Biomedical Research Center and supported by the NIH National Center for Research Resources, grant P41-RR02594, and a grant from Bayer Health Care, LLC.
Footnotes
OCIS codes: (170.1470) Blood or tissue constituent monitoring; (170.3660) Light propagation in tissue; (170.5280) Photon migration; (170.5660) Raman spectroscopy; (170.7050) Turbid media.
References and links
- 1.Zhadin NN, Alfano RR. Correction of the internal absorption effect in fluorescence emission and excitation spectra from absorbing and highly scattering media: Theory and experiment. J. Biomed. Opt. 1998;3:171–186. doi: 10.1117/1.429874. [DOI] [PubMed] [Google Scholar]
- 2.Biswal NC, Gupta S, Ghosh N, Pradhan A. Recovery of turbidity free fluorescence from measured fluorescence: an experimental approach. Opt. Express. 2003;11:3320–3331. doi: 10.1364/oe.11.003320. [DOI] [PubMed] [Google Scholar]
- 3.Finlay JC, Foster TH. Recovery of hemoglobin oxygen saturation and intrinsic fluorescence with a forward-adjoint model. Appl. Opt. 2005;44:1917–1933. doi: 10.1364/ao.44.001917. [DOI] [PubMed] [Google Scholar]
- 4.Wu J, Feld MS, Rava RP. Analytical model for extracting intrinsic fluorescence in turbid media. Appl. Opt. 1993;32:3585–3595. doi: 10.1364/AO.32.003585. [DOI] [PubMed] [Google Scholar]
- 5.Muller MG, Georgakoudi I, Zhang QG, Wu J, Feld MS. Intrinsic fluorescence spectroscopy in turbid media: disentangling effects of scattering and absorption. Appl. Opt. 2001;40:4633–4646. doi: 10.1364/ao.40.004633. [DOI] [PubMed] [Google Scholar]
- 6.Kuba S, Knozinger H. Time-resolved in situ Raman spectroscopy of working catalysts: sulfated and tungstated zirconia. J. Raman Spectrosc. 2002;33:325–332. [Google Scholar]
- 7.Nijhuis TA, Tinnemans SJ, Visser T, Weckhuysen BM. Operando spectroscopic investigation of supported metal oxide catalysts by combined time-resolved UV-VIS/Raman/on-line mass spectrometry. Phys. Chem. Chem. Phys. 2003;5:4361–4365. [Google Scholar]
- 8.Aarnoutse PJ, Westerhuis JA. Quantitative Raman reaction monitoring using the solvent as internal standard. Anal. Chem. 2005;77:1228–1236. doi: 10.1021/ac0401523. [DOI] [PubMed] [Google Scholar]
- 9.Tinnemans SJ, Kox MHF, Nijhuis TA, Visser T, Weckhuysen BM. Real time quantitative Raman spectroscopy of supported metal oxide catalysts without the need of an internal standard. Phys. Chem. Chem. Phys. 2005;7:211–216. doi: 10.1039/b414427a. [DOI] [PubMed] [Google Scholar]
- 10.Tuchin VV. Tissue optics: light scattering methods and instruments for medical diagnosis. SPIE Press; Bellingham, Wash.: 2000. [Google Scholar]
- 11.Tsuboi M. Raman scattering anisotropy of biological systems. J. Biomed. Opt. 2002;7:435–441. doi: 10.1117/1.1482720. [DOI] [PubMed] [Google Scholar]
- 12.Enejder AMK, Scecina TG, Oh J, Hunter M, Shih WC, Sasic S, Horowitz GL, Feld MS. Raman spectroscopy for noninvasive glucose measurements. J. Biomed. Opt. 2005;10:031114. doi: 10.1117/1.1920212. [DOI] [PubMed] [Google Scholar]
- 13.Shih W-C, Bechtel KL, Feld MS. Quantitative biological Raman spectroscopy. In: Tuchin VV, Taylor, Francis, editors. Handbook of Optical Sensing of Glucose in biological fluids and tissues. 2008. Chap. 12. [Google Scholar]
- 14.Flock ST, Jacques SL, Wilson BC, Star WM, Vangemert MJC. Optical-Properties of Intralipid - a Phantom Medium for Light-Propagation Studies. Lasers Surg. Med. 1992;12:510–519. doi: 10.1002/lsm.1900120510. [DOI] [PubMed] [Google Scholar]
- 15.Enejder AMK. Doctoral Thesis. Department of Physics, Lund Institute of Technology; 1997. [Google Scholar]
- 16.Frank IE, Friedman JH. A Statistical View of Some Chemometrics Regression Tools. Technometrics. 1993;35:109–135. [Google Scholar]
- 17.Wold S, Martin H, Wold H. Lecture Notes in Mathematics. Springer-Verlag; Heidelberg: 1983. [Google Scholar]
- 18.Shih W-C, Bechtel KL, Feld MS. Constrained regularization: Hybrid method for multivariate calibration. Anal. Chem. 2007;79:234–239. doi: 10.1021/ac060732v. [DOI] [PubMed] [Google Scholar]
- 19.Zonios G, Perelman LT, Backman VM, Manoharan R, Fitzmaurice M, Van Dam J, Feld MS. Diffuse reflectance spectroscopy of human adenomatous colon polyps in vivo. Appl. Opt. 1999;38:6628–6637. doi: 10.1364/ao.38.006628. [DOI] [PubMed] [Google Scholar]
- 20.Farrell TJ, Patterson MS, Wilson B. A Diffusion-Theory Model of Spatially Resolved, Steady-State Diffuse Reflectance for the Noninvasive Determination of Tissue Optical-Properties Invivo. Med. Phys. 1992;19:879–888. doi: 10.1118/1.596777. [DOI] [PubMed] [Google Scholar]
- 21.Dam JS, Pedersen CB, Dalgaard T, Fabricius PE, Aruna P, Andersson-Engels S. Fiber-optic probe for noninvasive real-time determination of tissue optical properties at multiple wavelengths. Appl. Opt. 2001;40:1155–1164. doi: 10.1364/ao.40.001155. [DOI] [PubMed] [Google Scholar]
- 22.Doornbos RMP, Lang R, Aalders MC, Cross FW, Sterenborg HJCM. The determination of in vivo human tissue optical properties and absolute chromophore concentrations using spatially resolved steady-state diffuse reflectance spectroscopy. Phys. Med. Biol. 1999;44:967–981. doi: 10.1088/0031-9155/44/4/012. [DOI] [PubMed] [Google Scholar]
- 23.Nichols MG, Hull EL, Foster TH. Design and testing of a white-light, steady-state diffuse reflectance spectrometer for determination of optical properties of highly scattering systems. Appl. Opt. 1997;36:93–104. doi: 10.1364/ao.36.000093. [DOI] [PubMed] [Google Scholar]